1. Introduction
Pediatric Glioblastoma (pGBM) and Diffuse Intrinsic Pontine Glioma (DIPG), are amongst the most aggressive tumors of the central nervous system affecting children and young adults, for which there is no effective treatment [
1,
2]. Integrated molecular profiling has revealed that these neoplasms are characterized by recurrent specific mutations in histone genes together with aberrations in canonical oncogenic pathways associated with differences in tumor location, histopathological features, patient age distribution, and clinical outcome [
3,
4,
5,
6,
7]. The histone mutations involve the H3.3 (
H3F3A) and H3.1 (
HIST1H3B, HIST1H3C) variants, resulting in amino-acid substitutions (G34R/V and K27M) at the histone tail interfering with their natural function.
A significant degree of genetic and phenotypic intratumor heterogeneity has been recently identified in pGBM and DIPG, which may represent one of the most challenging aspect in the effort to develop new effective therapeutic strategies for these diseases [
8,
9,
10,
11,
12]. It has been shown that they are characterized by temporal and spatial intratumoral genomic heterogeneity, which rapidly evolves following surgical and chemotherapy treatment. Also, it has been recently demonstrated that pGBMs and DIPGs are characterized by a complex and heterogenous sub-clonal architecture, where distinct cell subpopulations coexist and co-operate, building a cellular network promoting tumorigenesis and responsible of the aggressive phenotype [
12]. Moreover, using single-cell RNA sequencing, it has been shown that these cells exhibit high plasticity in the transition between different cellular states and that it can be influenced by the microenvironment [
13]. Of note, evidence has clearly demonstrated the existence of direct interconnections between the neuronal compartment of the brain microenvironment wiring the glioma cells and vice versa [
14,
15].
Despite the increasing knowledge, questions remain to be answered. For example, what are the precise mechanisms that regulate the direct or indirect cell–cell communication within the heterogeneous glioma populations and with its microenvironment? How can we interfere with the mechanisms of crosstalk in order to weaken glioma growth and invasion? How do the heterogeneous subpopulations evolve over time upon therapeutic selective pressure?
In order to be able to study these mechanisms at cellular and molecular levels, we would need to identify and track individual cells and/or specific cell subpopulations.
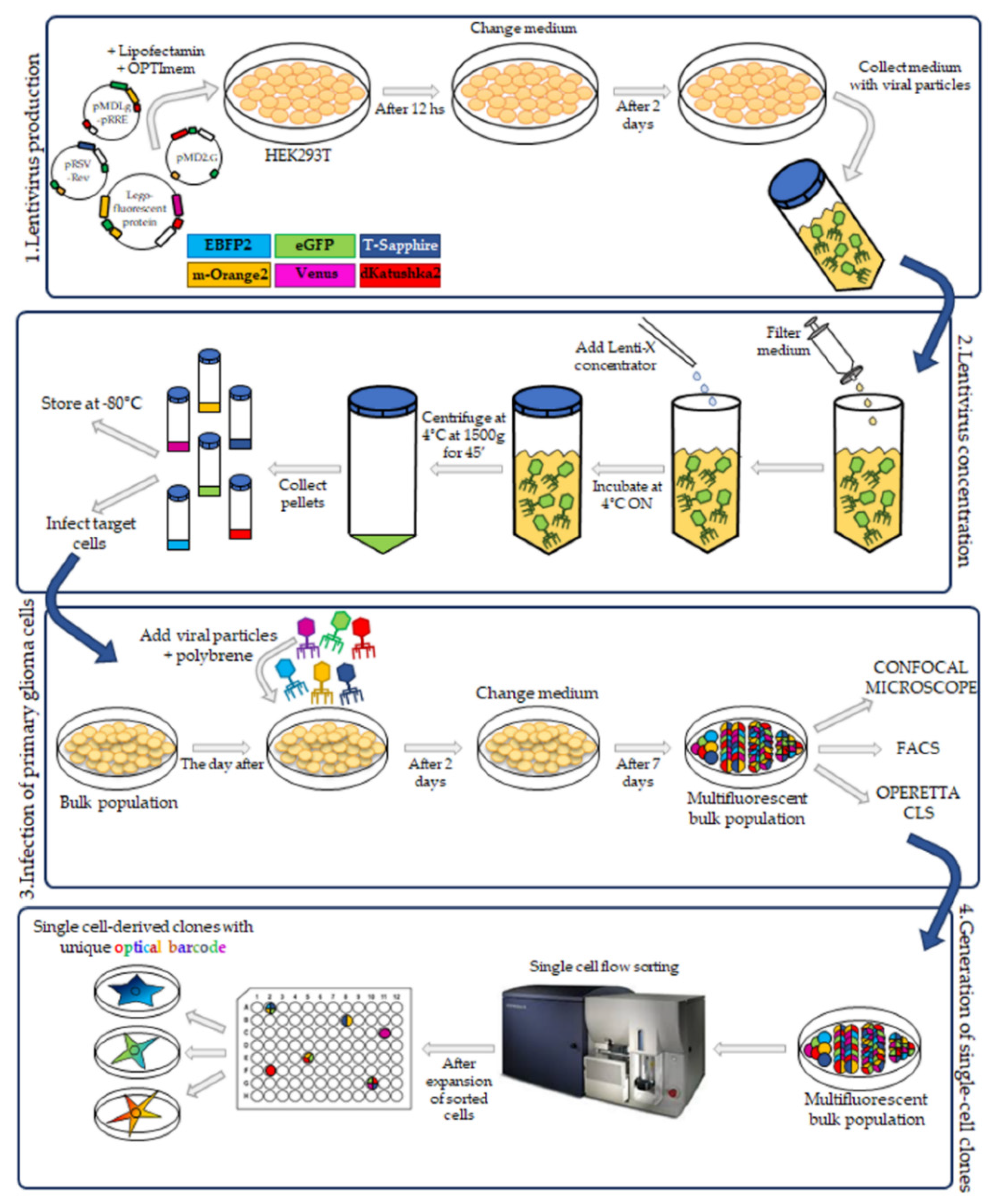
The RGB marking technology [
16] was originally developed for the simultaneous cell transduction of three lentiviral gene ontology (LeGO) vectors, encoding for red-green-blue fluorescent proteins. This technology has recently evolved with the use of up to six multiple vectors, each expressing fluorescent proteins with distinct excitation and emission properties, allowing the generation of multifluorescent cell populations, stable through cell divisions [
17]. The RGB marking approach has been used to assess the clonality of primary hepatocytes in the regeneration of injured livers in mice, to track the spatial and temporal fate of neural stem cells in the adult brain as well as to study tumor heterogeneity in terms of clonal expansion in vitro and in vivo [
16,
17,
18].
In this study we have applied the Multifluorescent Marking Technology adapted from the RGB marking approach [
16,
17], to pGBM and DIPG patient primary derived cell lines. We describe the generation of multifluorescent bulk cell lines, the derivation of stable optical barcoded single cell-derived clones and the assessment of the Multifluorescent Marking Technology in 2D and 3D in vitro/ex vivo culture systems. We focus on how we can integrate different fluorescence analysis platforms and identify their strengths and limitations, with the aim to establish the tools that will enable future more in-depth studies on the crosstalk between distinct heterogeneous subpopulations in pGBM and DIPG.
3. Discussion
The genetic and phenotypic intratumor heterogeneity may represent one of the most challenging obstacles in the development of effective therapies for cancer. Intratumor heterogeneity has been shown to be strictly associated to therapeutic resistance and, as such, may be a major cause of tumor progression and disease relapse [
26,
27,
28,
29].
pGBM and DIPG are known to be constituted by heterogeneous cell subpopulations coexisting within the same tumor and cooperating into a functional network, causing resistance to drug treatments, and increasing the aggressive phenotype [
12]. Moreover, a functional network is also present between the tumor and the normal brain microenvironment, in particular with its neuronal compartment, which actively contributes to promote glioma progression [
14,
15].
Understanding the mechanisms of cell–cell communication taking place within the tumor and with its microenvironment could lead to the identification of new, more effective therapeutic strategies to treat these devastating diseases.
We sought to use the Multifluorescent Marking Technology to establish the tools that will enable future investigations into the cellular and molecular mechanisms of intra-tumor heterogeneity in pGBM and DIPG.
Originally the RGB Marking technology was used to study the heterogeneity and the clonal dynamics in vitro and in vivo, in osteosarcoma [
30], pancreatic adenocarcinoma [
31], hepatocellular carcinoma [
32], mammary adenocarcinoma [
33], and neuroendocrine carcinoma [
16].
Here, we focused on the application of the Multifluorescent Marking Technology and on the integration of different fluorescence analysis platforms, identifying their strengths and limitations, with the goal to understand how we could apply this technology to further expand our studies on pGBM and DIPG heterogeneity and clonal dynamics.
After the establishment of four pHGG patient primary derived cell lines, we generated the bulk multifluorescent populations using the approach initially reported by Weber et al., for the RGB marking technique [
16,
19], and with the six LeGO vectors used for the optical barcoding described by Momhe et al. [
17].
To transduce our primary derived cell lines, we adapted the original protocol to accommodate the stem cell culture conditions, used for pGBM and DIPG cells. The neurosphere stem cell culture is generally accepted as the gold standard for primary glioma stem cells [
34,
35]. Recently, two studies have employed a reliable protocol for lentiviral cell transduction of primary DIPG cell lines cultured as neurospheres, by exposing the cells to FBS for a short period of time [
36,
37]. They have demonstrated that the short-term exposure of the cells to serum and a rapid return to serum-free conditions, improve the neurosphere transduction efficiency without inducing a change in their stem cell proprieties. However, in our case, we decided not to expose our primary derived cells to serum and to perform the lentiviral transduction on laminin-adherent stem cell culture condition [
20]. This allowed us to obtain a good enough lentiviral cell transduction efficiency and at the same time, as the cells grew adherent, we could easily assess the different fluorescences during the initial stages of the multifluorescent bulk cell expansion, and during the processes of the generation and expansion of the single cell-colonies. It is important to note that these are rare tumors and often the primary cultures are established by small biopsy samples. Any type of culture will exert a selective pressure on the expanded cell lines. For this reason, we transduced our cells after passage 10 in order to work with cell lines more stable in their sub-clonal composition. We believe this has been maintained over time and over passages also after cell transduction as demonstrated by the phenotypic features (e.g., morphology, migration, invasion pattern) that the bulk cell lines and the single-cell-derived clones display.
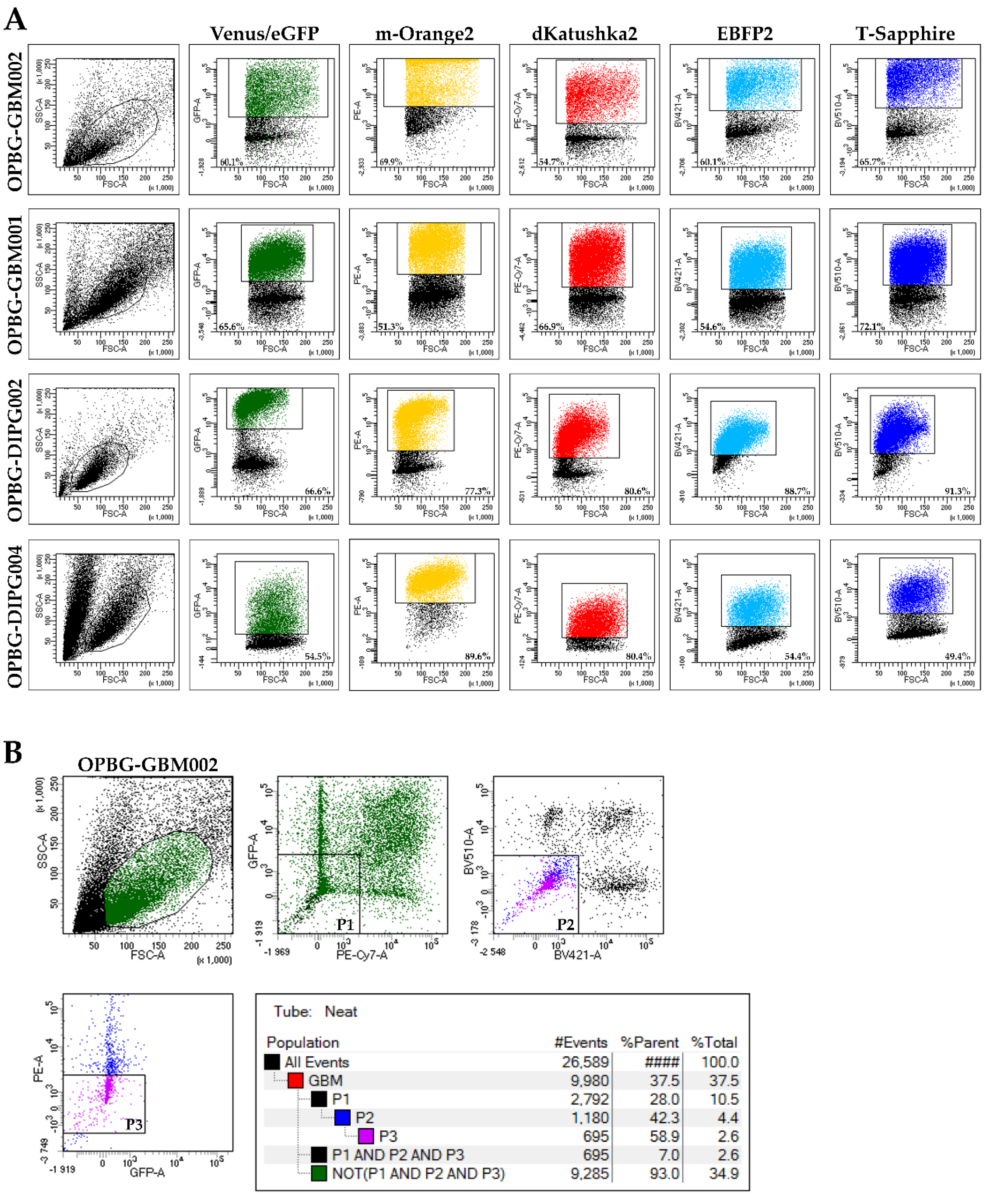
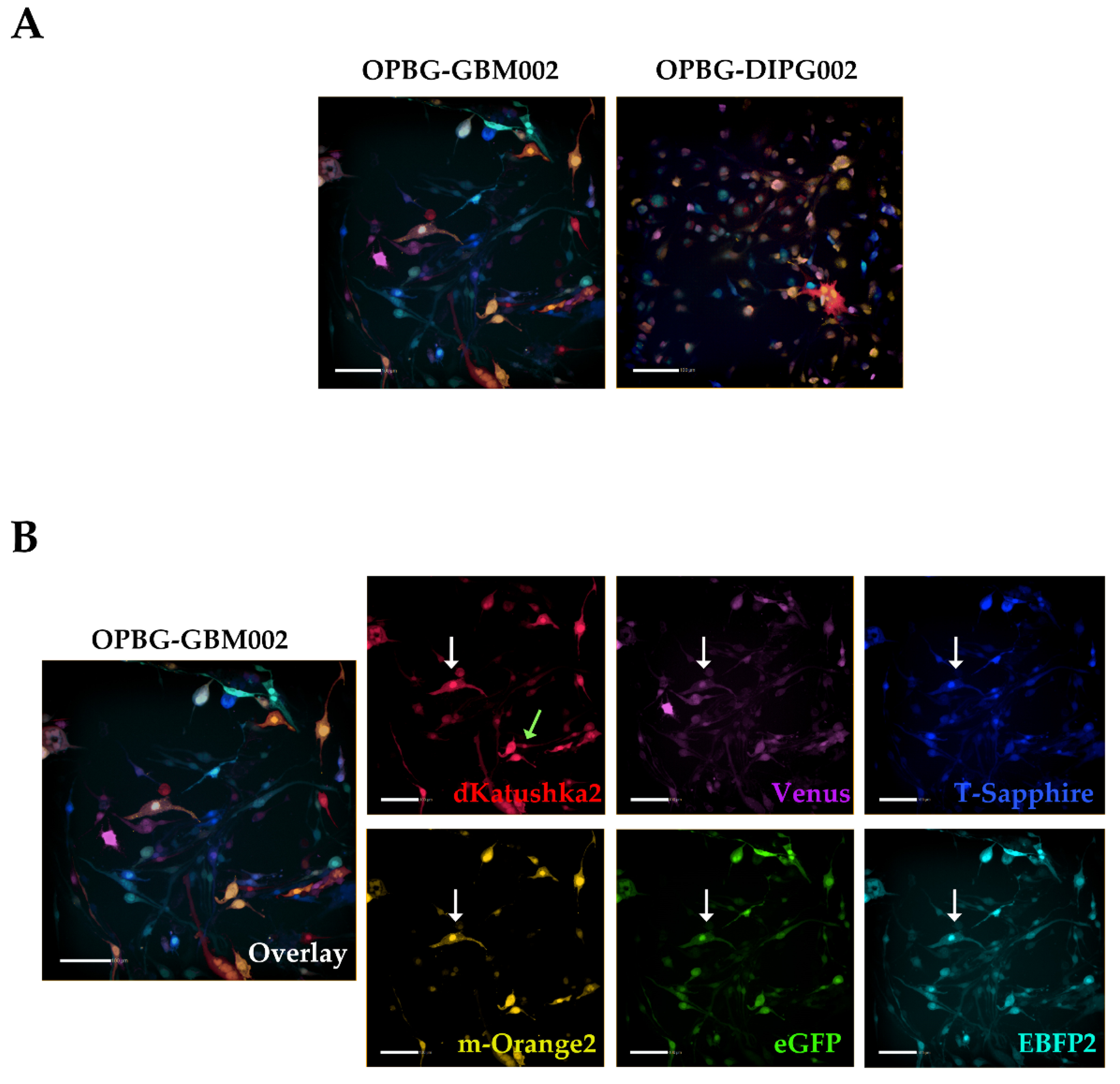
To evaluate the transduction efficiency of the bulk cell lines, we used the flow cytometry. Our challenge has been to separate the six different fluorescences, which have distinct but, in some cases, overlapping excitation and emission wavelengths. Given the filter configuration of our flow cytometer (
Table 2) and the close range of the wavelengths, we were able to discriminate only four out of the six fluorescent proteins. For each cell lines, we could determine the percentage of positive cells and the fluorescent intensity level for m-Orange2, dKatushka2, EBFP2, and T-Sapphire. However, we were unable to distinguish Venus from eGFP due to the strong overlap of their emission spectra, thus, preventing us from determining the transduction efficiency for these two fluorescent proteins. Our results are partially in contrast with what has been previously reported by Mohme et al., who have been able to separate the six different fluorescent proteins and perform the analysis of the clonal composition of the established glioblastoma cell line by flow cytometry [
17]. The reason of the discrepancy between these results is the use, by Mohme et al., of a specific customized filter for Venus detection, which has been fundamental for their analysis and subsequent assignment of the optical barcodes for the cells and the derived clones.
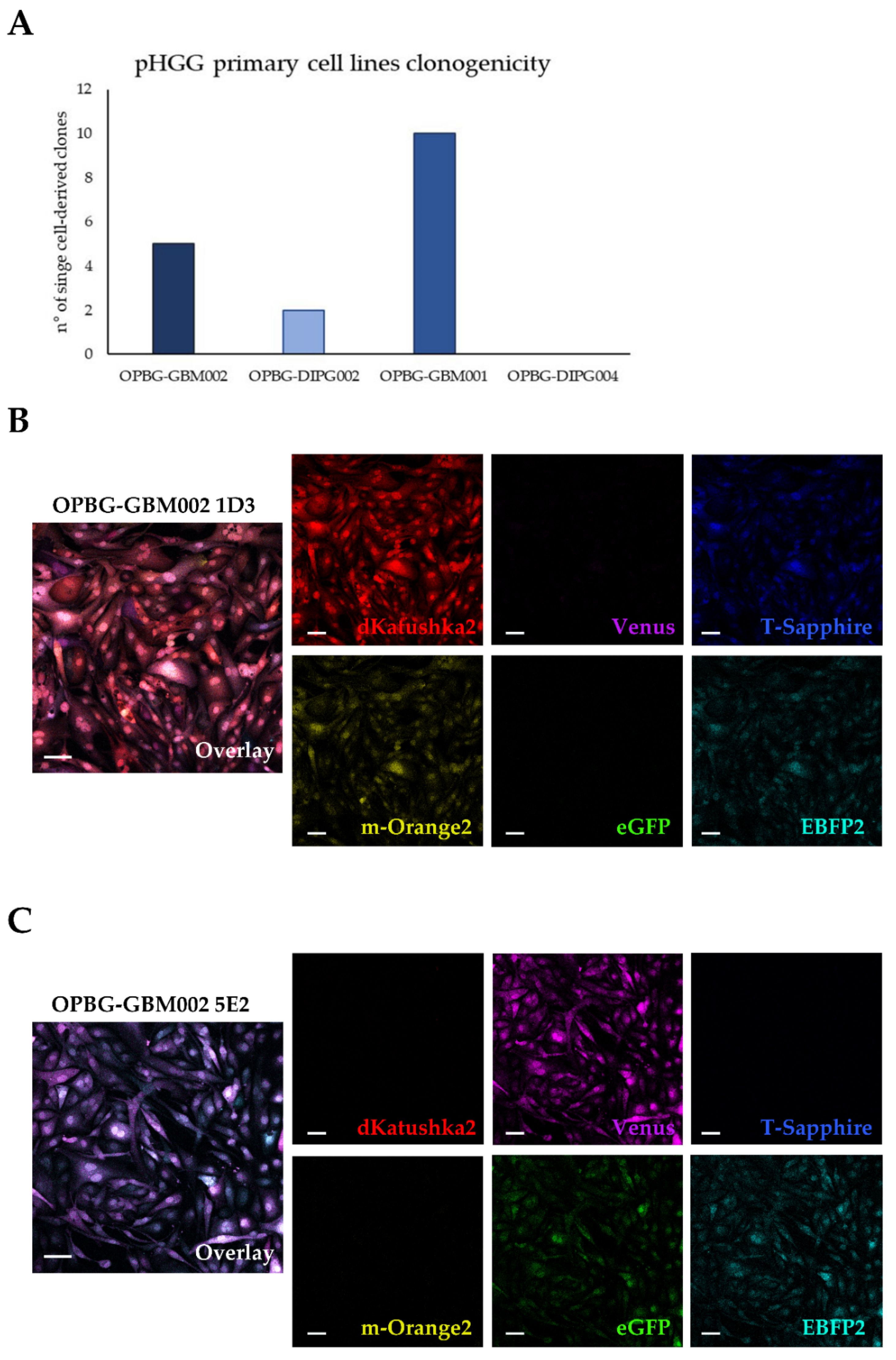
Although we were not able to discriminate two out of the six fluorescent proteins, the flow cytometer has been essential in our study for the generation of the single cell-derived clones. Using a precise cell sorting strategy, we obtained single cell-derived clones positive for any of the six proteins as further confirmed with the acquisition of the cell images by confocal microscopy.
Moreover, differently from Mohme et al., where the flow cytometry was their main approach used to assign optical barcodes, our major interest was to visualize the multifluorescent bulk cells and the derived clones and based on that, set up the right models that would enable us to study the mechanisms of cell–cell interactions at different levels of complexity, over a long period of time.
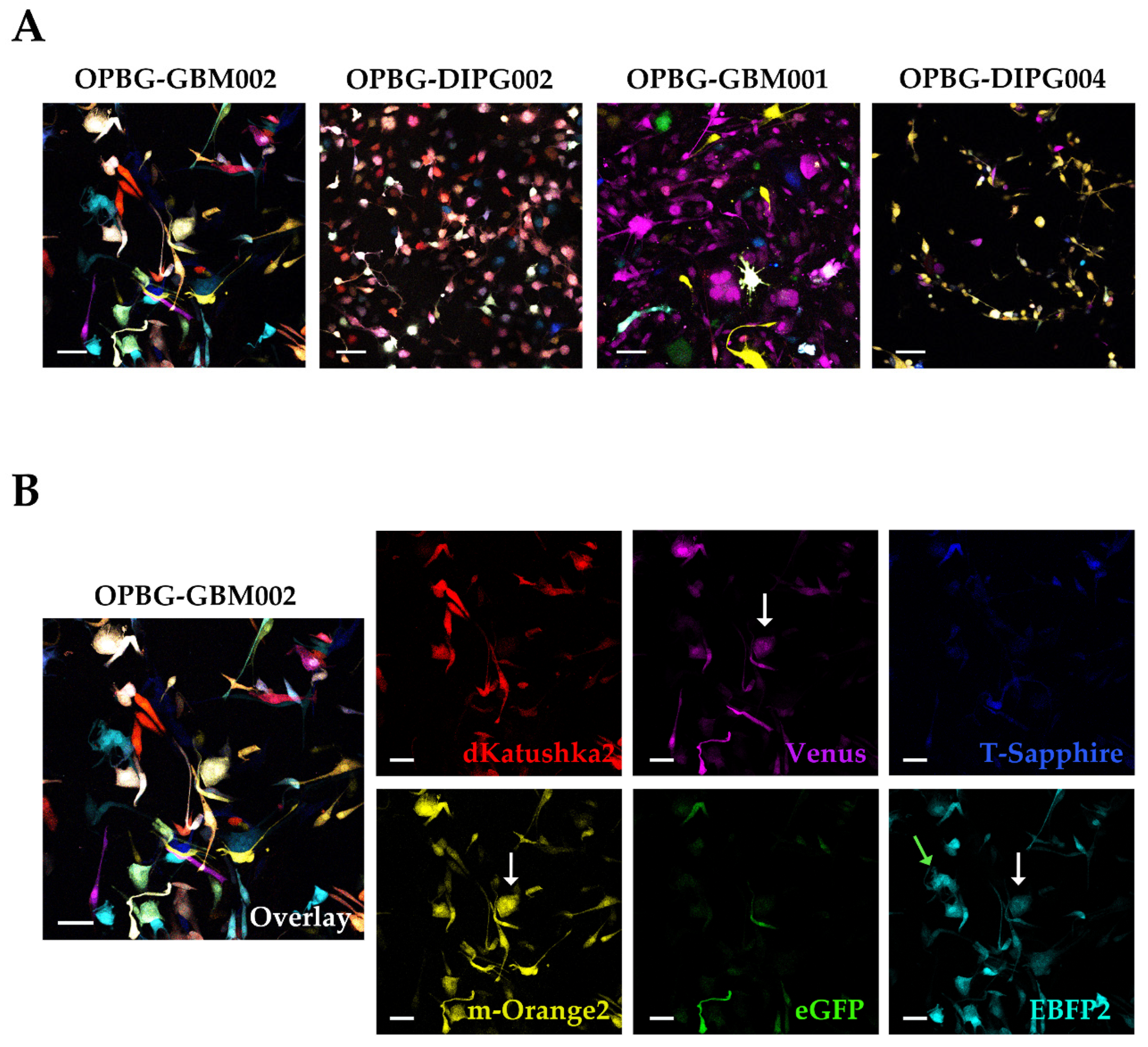
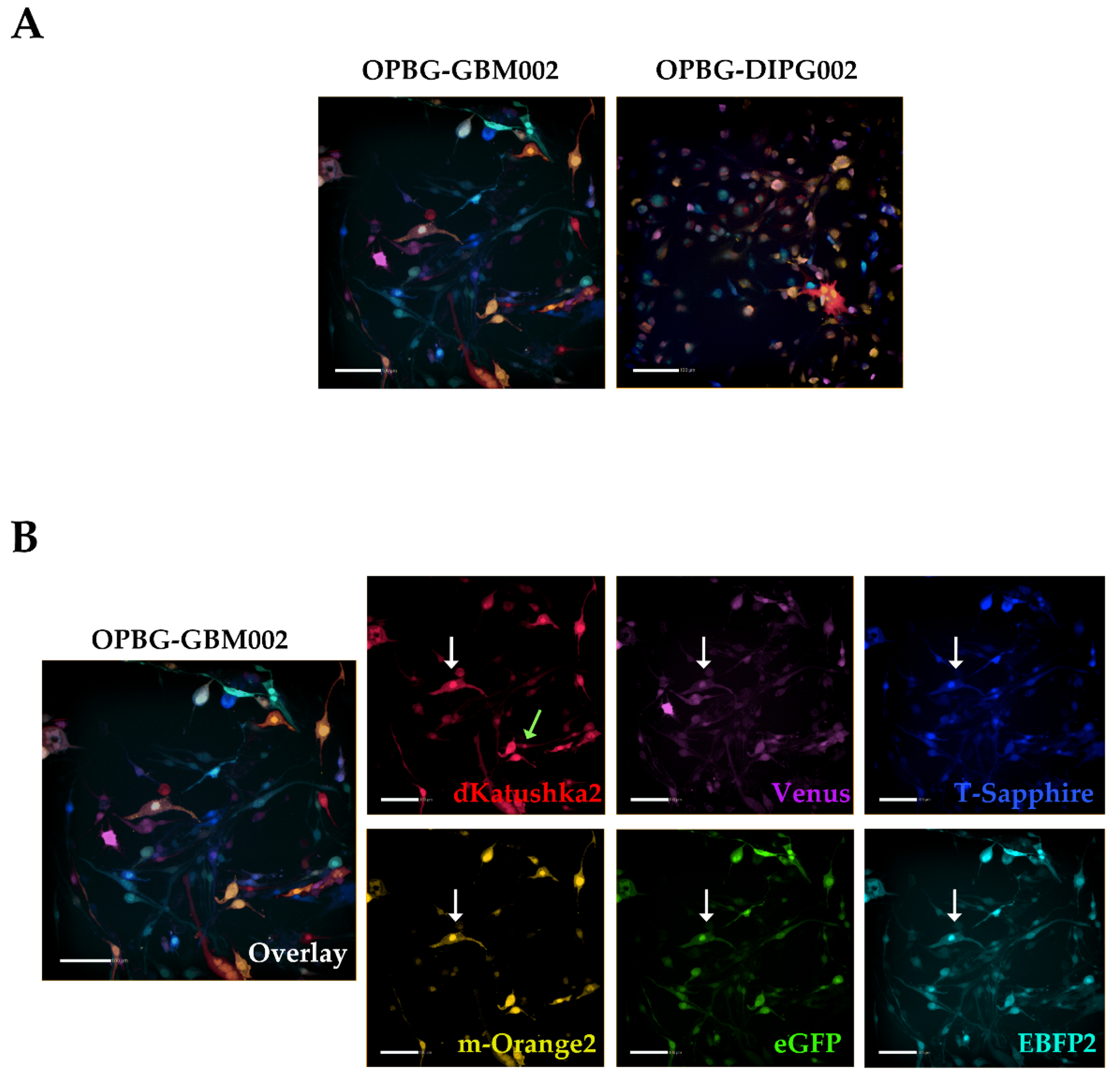
To achieve this, we focused our image analysis using a freely tunable confocal microscope, the Leica TCS AOBS-SP8X laser scanning confocal microscope. This platform is equipped with a tunable white light-laser (WLL) and a tunable beam splitter, which enabled us to precisely distinguish each individual fluorescent protein, for all the four multifluorescent bulk cell lines. This has been possible based on the pulsed nature of the WLL that allows to specify any excitation wavelength between 470–670 nm, together with a fast beam splitting device. The AOBS can select simultaneously up to eight discrete laser lines, even with narrow distances between excitation lines, with a 1nm precision. Furthermore, we used the WLL-AOBS system in conjunction with hybrid detectors, thus obtaining gated removal of autofluorescence and reflected light. All these devices allowed us to reach maximum detection sensitivity results.
In addition, to clearly characterize the multifluorescent pGBM and DIPG primary cell lines, the WLL-AOBS confocal microscope has been fundamental for determining the OB of the single cell-derived clones.
Of note, the OBs could be further refined taking in consideration not just the combination of fluorescences, but also the fluorescent intensity of each of the protein expressed. As clearly shown by Gomez-Nicola D. et al., cells are randomly transduced by one or more lentiviral vectors. Also, the viruses may integrate at different sites leading to different expression levels. In addition, multiple hits from the same vector may lead to additional expression variation [
38]. The combination of these factors determines the unique hues associated with each clone. An important point to note is that the metabolic state and the cell cycle phase can also affect the fluorescent protein expression level leading to variations in fluorescence intensity in cells from the same clone [
39].
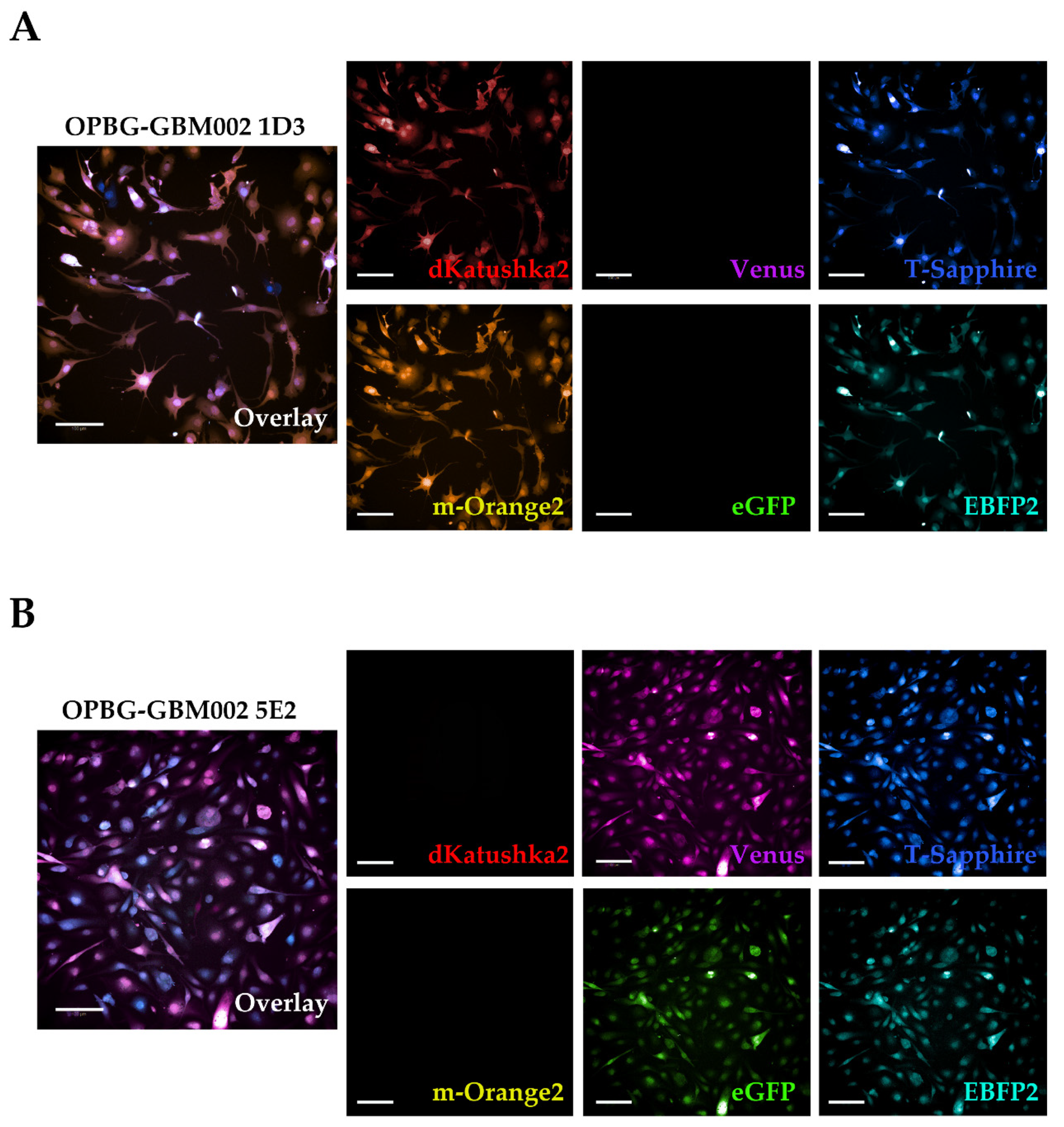
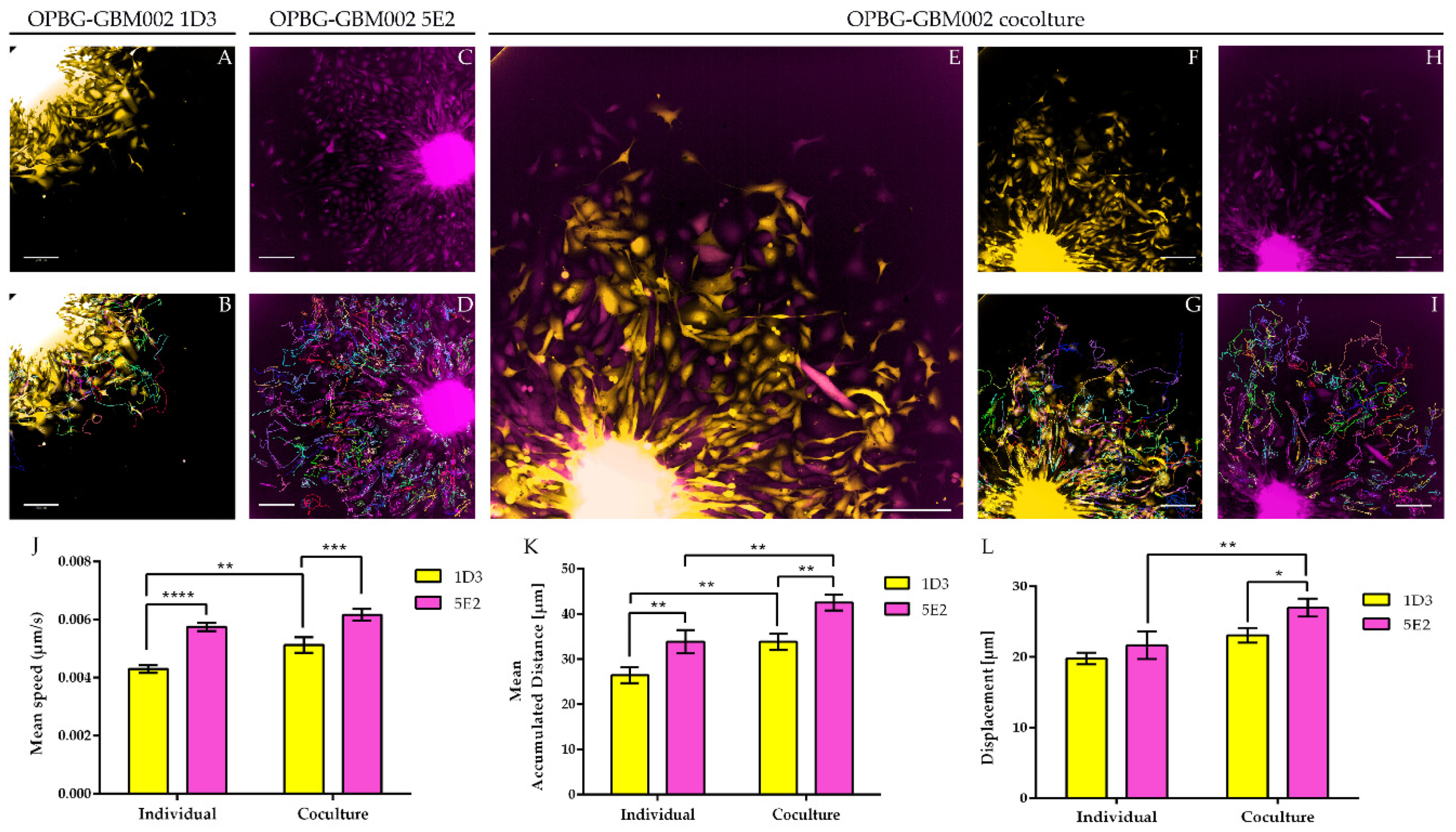
Next, we used the Operetta CLS developed by PerkinElmer. The Operetta CLS is an automated microscope for high-content/high-throughput image acquisition and analysis. It can acquire and analyze fluorescence, brightfield and digital phase images as well as be used in direct or confocal mode. Also, it allows the acquisition of fixed and live images. It could represent an ideal instrument to study cell–cell interaction, communication, migration, single cell tracking and perform time laps experiments in a high-content manner. Although this platform may be equipped with different LED sources and as many combinations of excitation and emission band range selection filters, there are not very restrictive filters making the Operetta a not very versatile platform in the separation of multiple fluorescences as tested in our study. With the Operetta CLS, we could identify m-Orange2 and dKatushka2, but we were not able to discern Venus from eGFP and EBFP2 from T-Sapphire. This inability to discriminate some of the fluorescences, does not make the Operetta CLS suitable for the analysis of the multifluorescent bulk population of cells neither for the identification and assignment of unique OB for the single cell-derived clones. Since the Operetta CLS is a highly automated microscopy station, it does not have the same versatility and possibility of tuning the spectrum wavelengths as the SP8 AOBS platform, useful to perform both a fine separation of the emission spectra of the different fluorescent proteins and for the high imaging resolution. Therefore, a freely tunable or a spectral confocal microscope platform remains the ideal imaging platform to characterize the multifluorescent pGBM and DIPG bulk cell lines and to determine the OB of individual clones. On the other hand, a confocal microscope would not be ideal for a multi-well format (96-384 well plates), large scale experiments, where, for example, different combination of clone co-cultures are tested, or drug treatments are applied to different clones in co-cultures. Based on this, we believe that despite its limitations, an automated high-content imaging platform such as the Operetta CLS can still be very useful for studying their phenotypic characterization and acquiring a large amount of data. Moreover, although some fluorescences may not be well separated, using the exclusive expression of one or another fluorescent protein of the OBs, clones in co-culture could still be discriminated. All of this has been clearly exemplified in the single cell-tracking experiments performed with the two single cell-derived clones in mono- and co-culture. The live imaging and analysis performed on the Operetta CLS has readily provided insights into the phenotypic heterogeneity that characterize these cell populations in particular in terms of their motile capability and how this can be modulated by their direct cell–cell interactions.
Following the assessment in 2D culture, we exploited the application of the Multifluorescent Marking Technology on two different 3D invasion models, in vitro and ex vivo, and evaluated the suitability of our assays with the imaging capability of our systems.
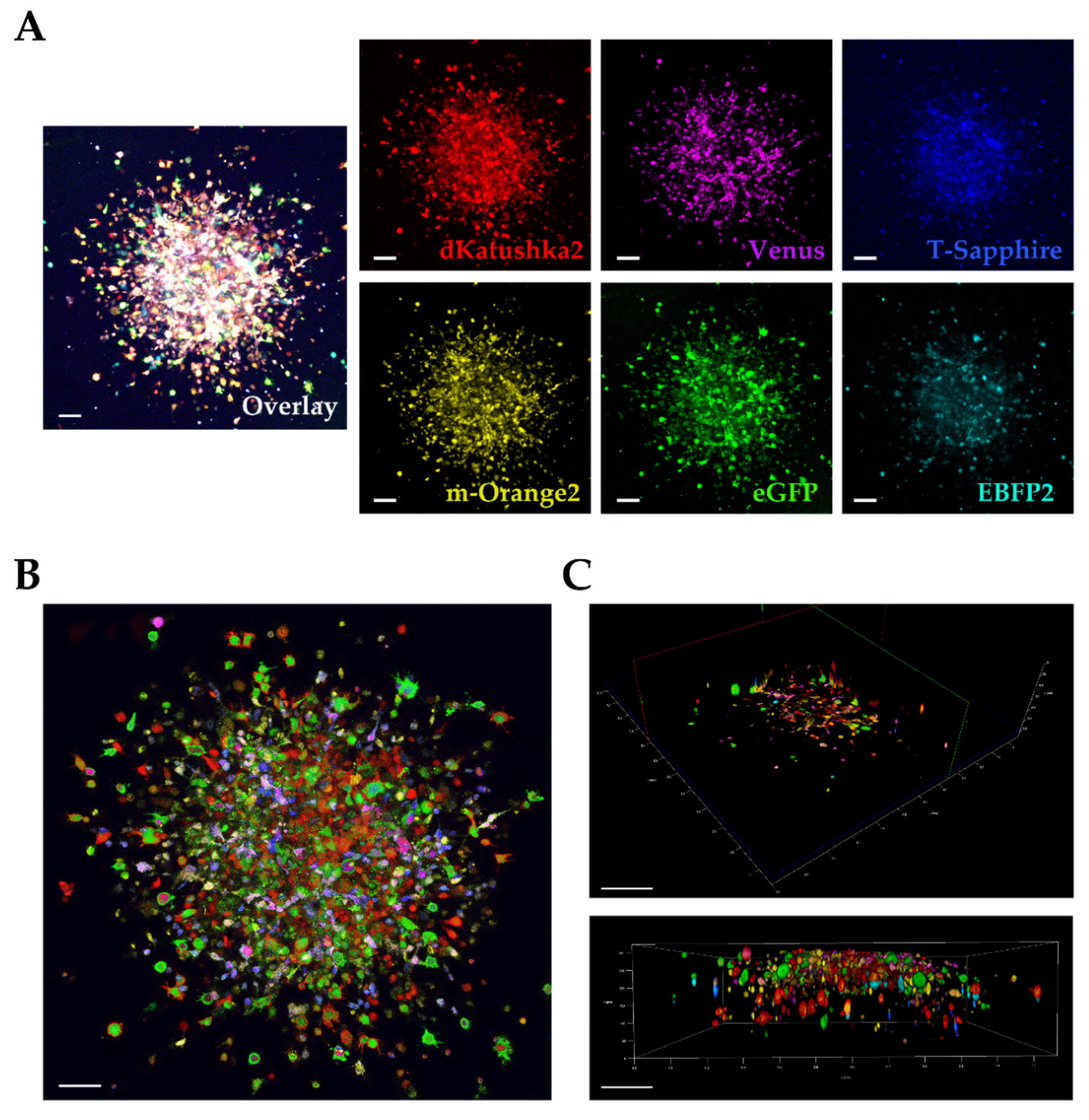
We used a multifluorescent bulk DIPG cell line in vitro for the 3D invasion into Matrigel [
21,
22] and at the invasion assay end point, the images were acquired and analyzed on our Leica TCS AOBS-SP8X laser scanning confocal microscope. We were satisfied that despite the dense, packed 3D cell invasion, we could successfully distinguish each invading cell from the others, not only at the periphery but also in the more central area of the tumor invasion, discriminating the individual fluorescences. In particular, acquiring in Z-stack and performing a deconvolution analysis for the 3D rendering, allowed us a very clear detection of the individual fluorescent cells in the thick Matrigel sample.
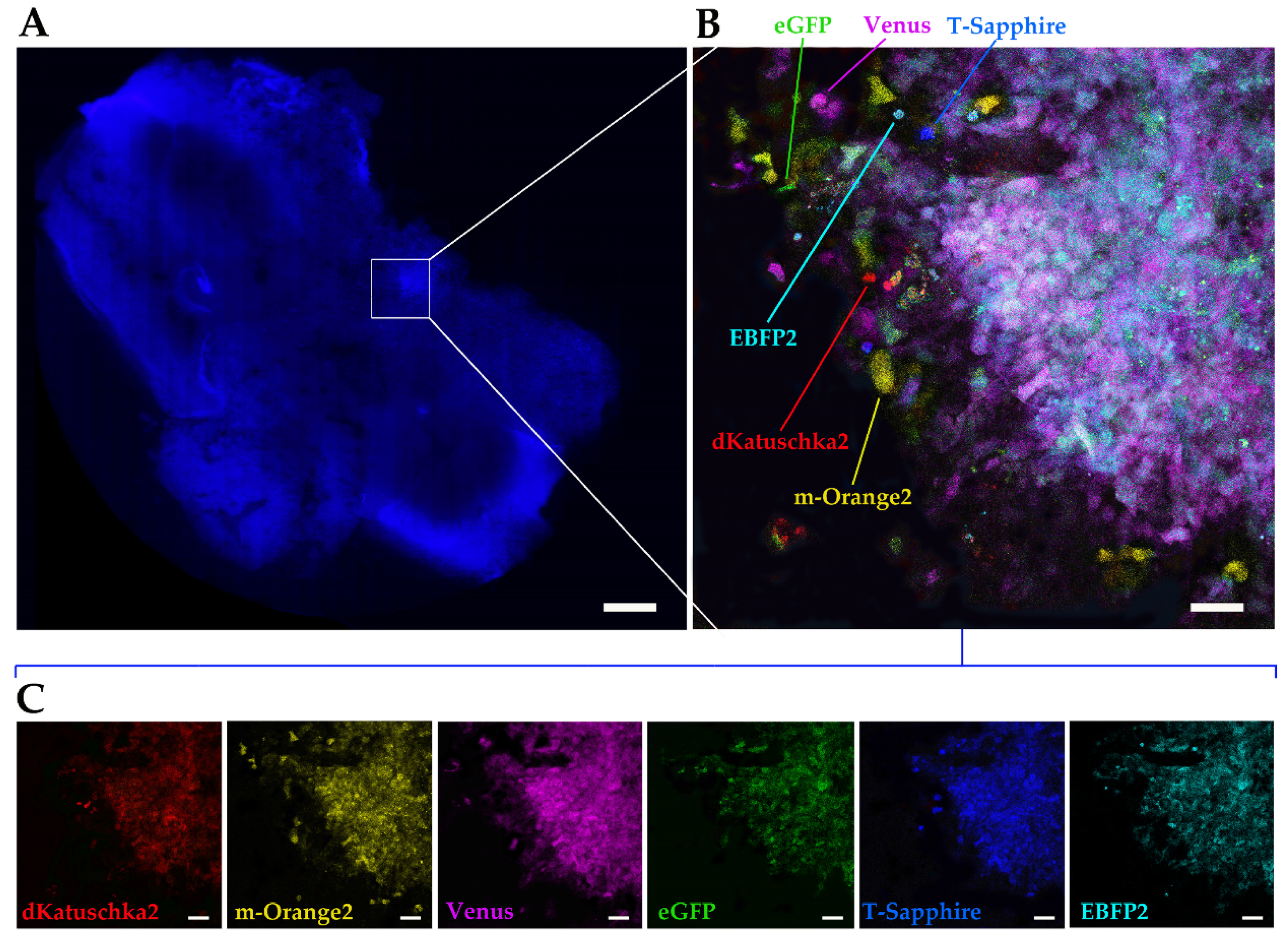
Finally, we utilized the ex vivo OBS model [
23] to provide a more physiological brain-like microenvironmental context for our DIPG cells to invade in. The same multifluorescent DIPG cell line used for the 3D invasion into matrigel, was implanted on OBSs. The OBSs themselves can determine some autofluorescence, which, together with the multicolor cells and the thickness of the slices, can make more challenging the separation of the different fluorescences, even using our confocal microscope. To achieve that and obtain a clearer view of the implanted, infiltrated multifluorescent DIPG cells, we performed tissue clearing, which enabled to distinguish all the six fluorescences.
Overall, our study demonstrates the applicability of the Multifluorescent Marking Technology to patient primary cultures of pGBM and DIPG, from 2D to more complex 3D environments. We have explored and integrated multiple fluorescent analysis platforms, highlighting their strengths and limitations in the analysis of such technology. In conclusion, with the specific platforms we had at our disposal, we have successfully used our Leica TCS AOBS-SP8X laser-scanning confocal microscope to perform high resolution imaging of the multifluorescent bulk cell lines, in 2D and 3D, as well as precisely define the OB of the single cell-derived clones. The BD FacsAriaTM III flow cytometer, despite its limitations in the separation of 2 out of 6 fluorescent proteins, has been critical with the adopted cell sorting strategy, for the generation of the single cell-derived clones. Finally, the Operetta CLS has also shown limitations in the separation of the 6 fluorescent proteins, but its fluorescence, automated, confocal, high-content imaging capability, has demonstrated its applicability for an experiment of phenotypic characterization of the single-cell-derived clones and their direct cell–cell interactions.
Our study has contributed on establishing the tools to study further the intratumor heterogeneity and the interclonal interactions in pGBM and DIPG.
4. Materials and Methods
4.1. Cell Cultures
pGBM and DIPG primary patient-derived cell lines were established either immediately after collection (biopsy or resection) or from live cryopreserved tissue. Cell lines were established from fresh tissue, first minced with a sterile scalpel followed by enzymatic dissociation with LiberaseTL (Roche Life Science, Penzberg, Germany) for 20 min at 37 °C in the presence of 1 U/mL DNase I (Thermofisher Scientific, Waltham, MA, USA) (shaking every 5 min). After enzyme neutralization and centrifugation, minced tissue was resuspended first in 1 mL HBSS 1× for further mechanical dissociation, then in 5 mL. After that, the minced tissue was filtered through a 70 μm filter. Cell cultures were established under stem cell conditions as two-dimensional (2D) adherent cultures on laminin (10 μg/mL) (Millipore, Burlington, MA, USA) and/or three-dimensional (3D) as neurospheres (NS). Hemispheric pGBM cell lines OPBG-GBM002 (histone WT), OPBG-GBM001 (H3.3 G34R), and DIPGs OPBG-DIPG002 (H3.3 K27M) and OPBG-DIPG004 (H3.1 K27M) were cultured in a serum-free medium designated as “Tumor Stem Media (TSM)”, consisting of 1:1 Neurobasal(-A) (Invitrogen, Carslsbad, CA, USA), and DMEM: F12 (Invitrogen), supplemented with Anti-mycotic/Anti-biotic, HEPES, NEAA, GlutamaX, Sodium Pyruvate (Invitrogen) and B27(-A) (Invitrogen, Carlsbad, CA, USA), human bFGF (20 ng/mL), human EGF (20 ng/mL), human PDGF-AA (10 ng/mL) and PDGF-BB (10 ng/mL) (Peprotech, Rocky Hill, NJ, USA) and heparin (2 ng/mL) (Stem Cell Technologies, Vancouver, BC, Canada). The cell authenticity was verified using short tandem repeat (STR) DNA fingerprinting by Eurofins Genomics (
Table 5) and certified mycoplasma-free.
HEK293T cells (ATCC, Manassas, VA, USA) were used for lentiviral particle production and cultured in DMEM high glucose (Euroclone, Pero, Italy) including 10% Fetal Bovine Serum (FBS) (Invitrogen, Carlsbad, CA, USA) and 1% pen-strep (Euroclone, Pero, Italy).
All patient samples were collected under full Research Ethics Committee approval.
4.2. DNA Extraction and Sanger Sequencing
DNA was extracted from multifluorescent primary cell pellets following the DNeasy Blood & Tissue kit protocol (Qiagen, Hilden, Germany) and was measured on the Nanodrop 2000 (Thermofisher, Waltham, MA, USA). PCR for H3F3A and HISTI1H3B was carried out using primers obtained from Sigma Aldrich (St. Louis, MO, USA). Products were purified using NucleoSpin Gel and PCR Clean-up (Macherey-Nagel, Dueren, Germany), quantified on a Nanodrop 2000 (Thermo Scientific, Waltham, MA, USA) and subjected to bidirectional sequencing using BigDye Terminator v3.1 (Applied Biosystem™, Foster City, CA, USA). After purification with Nucleoseq (Macherey Nagel, Dueren, Germany), capillary sequencing was done on a 3500 Genetic analyzer (Applied Biosytem™). Sequences were analyzed using the Mutation Surveyor (SoftGenetics, State College, PA, USA) and manually with FinchTV (Geospiza, Seattle, WA, USA).
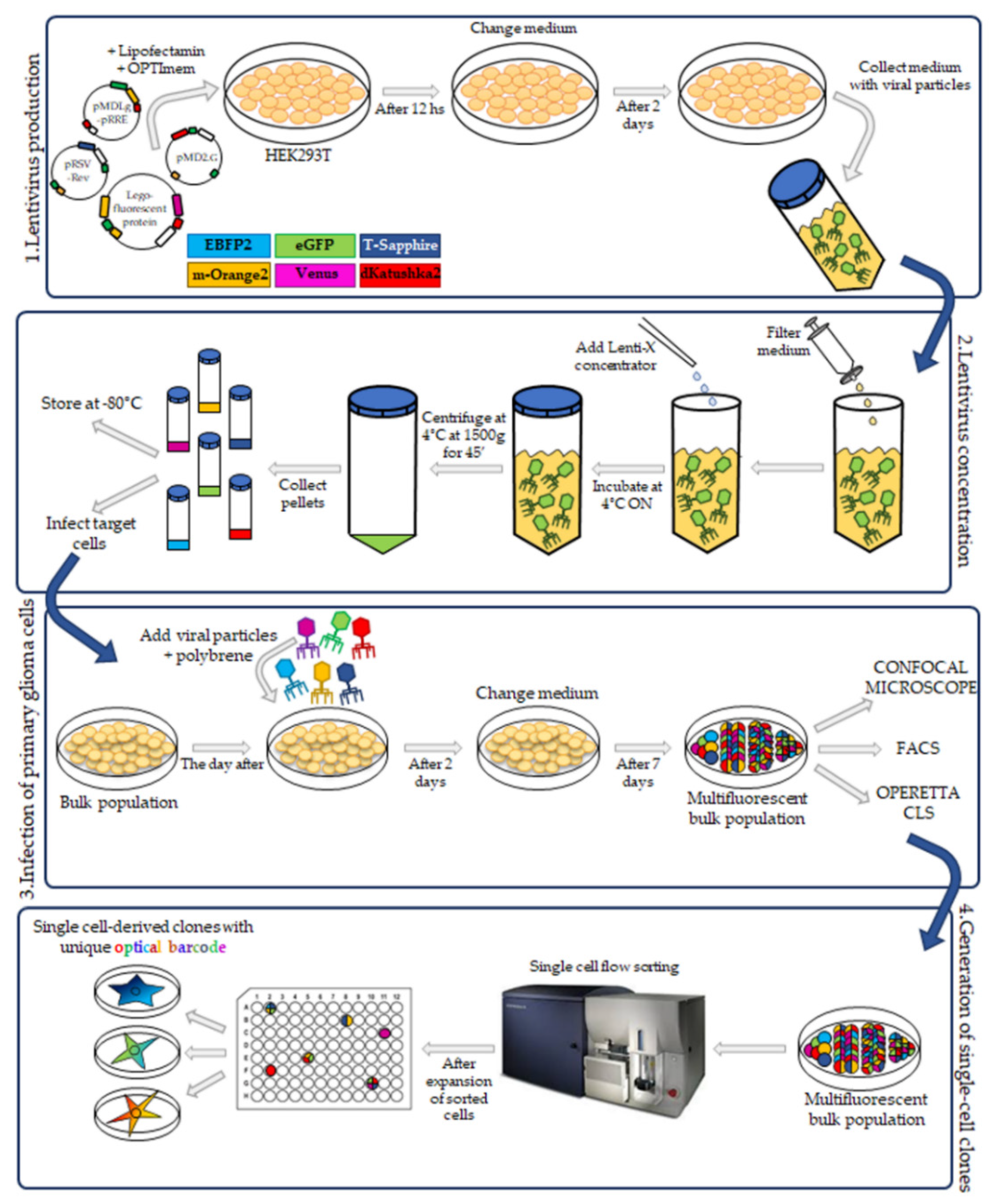
4.3. Lentivirus Particles Production
The third-generation HIV-1-derived self-inactivating lentiviral gene ontology vectors (LeGOs) were used for stable transduction of pGBM and DIPG cells. The vectors were previously described by Mohme et al. [
17]: LeGO-G2 (expressing eGFP, plasmid 25917; Addgene, Watertown, MA, USA), LeGO-V2 (expressing Venus, plasmid 27340; Addgene, Watertown, MA, USA), LeGO-EBFP2 (expressing EBFP2, plasmid 85213; Addgene), LeGO-S2 (expressing T-Sapphire, plasmid 85211; Addgene), LeGO-mOrange2 (expressing mOrange2, plasmid 85212; Addgene) and LeGO-dKatushka2 (expressing dKatushka2, plasmid 85214; AddgeneLentivirus particle production was performed as previously described [
19], with some modifications. Briefly, HEK293T cells were seeded, and transient transfection, with Lipofectamine 2000 (Invitrogen) in according to the manufacturer’s instructions, was performed using separately each one of the LeGO plasmids and the third-generation packaging plasmids pMDLg/pRRE (plasmid 12251; Addgene), pRSV-Rev (plasmid 12253; Addgene) and pMD2.G (plasmid 12259; Addgene). Cells were incubated for 12 h and then the medium was changed. After 48 h, the supernatant with the lentivirus particles was collected.
4.4. Lentivirus Concentration
Lentiviral supernatants were filtered with 0.22 µm Steriflip-GV (Millipore). After that, the lentiviral particles were concentrated using the Lenti-X concentrator (TakaraBio, Shiga, Japan) according to the manufacturer’s protocol. 1 volume of Lenti-X concentrator was combined with 3 volumes of supernatant and then mixed by gentle inversion. The mixture was incubated overnight at 4 °C. The day after, the mixture was centrifuged at 1500× g for 45 min at 4 °C. After centrifugation, the supernatant was carefully removed, and the lentiviral particle pellet was resuspended in 1/10 of the original volume in Phosphate-Buffered Saline (PBS) (Euroclone, Pero, Italy).
4.5. Titration and Titer Calculation of Lentivirus Vector
The viral particle titration was performed as previously described [
19]. Briefly, 5 × 10
4 HEK293T cells were seeded in 24-well plate and following cell attachment, the polybrene (8 μg/mL) (Sigma-Aldrich, St. Louis, MO, USA) was added to the medium to increase the transduction efficiency. For each lentivirus, different lentiviral particle amounts (1; 10 and 100 μL) were added in three different wells, the plate was centrifuged for 1 h at 1000×
g at 25 °C and then placed in an incubator at 37 °C, 5% CO
2 for 72 h.
For the titer calculation, the transduced HEK293T cells were analyzed at the flow cytometer and then the titer calculation was done following the previously described method [
19].
4.6. Transduction of Primary Patient-Derived pGBM and DIPG Cell Lines
The pGBM and DIPGs primary cell transduction was performed as previously described [
19], with some modifications. All cell lines were transduced between passage 10–14. 5 × 10
4 target cells were seeded in a laminin pre-coated 24-well plate and incubated at 37 °C and 5% CO
2. The day after, the media was changed, and the polybrene (8 μg/mL) was added. The lentivirus particles from the six vectors were added to the cells altogether as well as individually. As already reported for primary cells [
19], our primary cultures were difficult to transduce. For this, the amount of lentivirus used was 100× more concentrated than the titer calculated for our cells. After adding the lentivirus particles, the plate was centrifuged for 1 h at 1000×
g at 25 °C, incubated at a 37 °C, 5% CO
2 and 48 h later, the medium was changed. During the following week, the fluorescence associated with a successful cell transduction was checked at a Leica DMi8 (Leica Microsystems, Wetzlar, Germany) fluorescence microscope. The bulk cell lines were then expanded.
4.7. FACS Analysis and Sorting of Multifluorescent Bulk Population Primary Cell Lines
To obtain a bulk cell line composed of only transduced cells, the non-marked cells were sorted out using BD FacsAria
TM III (BD Bioscience, San Jose, CA, USA) flow cytometer with cell-sorting capability. To analyze the transduction efficiency of multifluorescent bulk DIPG and pGBM cells, we used the BD FacsAria
TM III (BD Bioscience, USA) flow cytometer with a specific filter configuration (
Table 2). The percentage of fluorescently positive cell was determined with FACSDiva software 8.0 (BD Bioscience).
4.8. Confocal Microscopy
Confocal microscopy was performed on a Leica TCS AOBS-SP8X laser-scanning confocal microscope (Leica Microsystems) equipped with tunable white light laser (WLL, 470–670 nm of wavelength) source, 405 nm diode laser, 3 photomultiplier tubes, 2 HyD detectors and an acousto-optical beam splitter (AOBS) that allowed the separation of multiple fluorescences. Sequential confocal images were acquired using HC PL APO CS 10X/0.40 or HC PL APO CS2 20X/0.75 objectives (Leica Microsystems) with a 1024 × 1024 format, scan speed 400–600 Hz, and z-step size of 1 µm. Fluorochromes unmixing was performed by the acquisition of an automated-sequential collection of multi-channel images to reduce spectral crosstalk between channels.
Maximum intensity projection (MIP) of z-series and complete mosaic image using Tile Scan function were performed by LASX (Leica Microsystems) software; deconvolution analysis (HyVolution2 software, Leica Microsystems) was applied to z-stacks to improve contrast and resolution of confocal raw images, then deconvoluted images were imported into LASX 3D (Leica Microsystems) software to obtain their surface 3D rendering. Tables of images were processed using Adobe Photoshop CS4 software (Adobe Systems Inc., San Jose, CA, USA).
4.9. Operetta CLS Image Acquisition
The expression of the six different fluorescent proteins in the bulk cell lines and in the single cell-derived clones was performed on an Operetta CLS (PerkinElmer, Waltham, MA, USA), equipped with eight emission LED filters with the emission/excitation configuration described in
Table 4. Image acquisitions were performed using the 20× objective (numerical aperture 0.4).
4.10. Establishment of Single Cell-Derived Clones
Bulk multifluorescent cell lines were single cell-flow sorted using a BD FacsAria
TM III instrument (BD Bioscience) where single cells were sorted unbiased by any marker expression directly into the inner 60 wells of 5 laminin (Millipore) pre-coated flat-bottom 96-well plates (PerkinElmer). Single cells were dropped in 100 μL/well of the same medium as described above. The outer 16 wells were filled in with 200 μL/well of PBS to avoid evaporation of medium. After single cell-flow sorting, the plates were incubated at 37 °C, 5% CO
2, and colonies monitored as previously described [
12]. Once weekly cells were refed with 25 μL of medium/well and plates scanned on a CeligoS cytometer (Nexcelom Bioscience, Lawrence, MA, USA) using the Confluence application, to evaluate colony growth. Single cell-derived colonies were detached using Accutase (Euroclone, Pero, Italy) when they achieved approximately 60–80% confluency and collected for further expansion to stably derive single cell-derived clones.
4.11. Invasion Assay
3D invasion assays were performed as previously described [
12,
21,
22], with some modifications. The bulk cells were detached and counted. For single NS, 1000 cells/well in 100 μL were dispensed into ultra-low attachment (ULA) 96-well round-bottom plates (Corning, New York, NY, USA) using a multichannel pipette. The plates were transferred to an incubator (37 °C, 5% CO
2) and three to four days later, when the neurospheres reached a size of 300–350 μm in diameter, the invasion assay was performed. A total of 50 μL medium was removed from each well and then, using ice-cold tips, 50 μL of Matrigel was gently dispensed into the ULA plates, which were then incubated at 37 °C, 5% CO
2. 96 h later, the invasions were fixed with 4% paraformaldehyde (PFA) overnight at 4 °C. The day after, the fixed neurospheres were washed 3 times with PBS for 30 min and then images acquired at the confocal microscope. Images were processed using Adobe Photoshop CS4 software (Adobe Systems Inc.).
3D migration assays were performed as previously described [
12,
22] with some modifications. The 5E2 and 1D3 single cell-derived clone cells from the OPBG-GBM002 multifluorescent bulk cell line, were seeded into 96-well round-bottom ULA plates (Corning) at 1000 cells/well either in monoculture or in co-culture (50:50), and allowed to form a single NS per well. When the NS reached a size of 250–3000 μm in diameter, the migration assay was performed. Briefly, flat-bottomed 96-well plates (PerkinElmer) were coated for 2 h at RT with 50μL/well of 125μg/mL Matrigel (Corning) in culture medium in the absence of growth factors. Once coating was completed, a total of 200 μL/well of culture medium was added to each well. A total of 50μL medium was removed from ULA 96-well round-bottom plates containing NS, the remaining medium including the NS were transferred onto the matrigel pre-coated plates and cell let migrate for 48h. For live imaging experiments of the single-cell tracking, automated fluorescent image acquisition was performed at the Operetta CLS (PerkinElmer, Waltham, MA, USA) every 30 min for 96 time points, starting from 24 h after the migration assay was set up. In order to clearly distinguish the two clones, based on their optical barcodes, the fluorescent signal for m-orange was acquired for 1D3 and Venus for 5E2. The Harmony software (PerkinElmer) on the Operetta was used to calculate the mean speed, mean accumulated distance, and mean displacement from
n = 3. Two independent experiments were performed.
4.12. Whole Brain Organotypic Slice Preparation and Co-Culture with Multifluorescent DIPG NS
Whole brain organotypic slices (OBSc)—encompassing pons and medulla—were prepared from CD1 mice pups (postnatal day 6–7) (Charles River, Wilmington, MA, USA) as previously described [
23,
24], with some modifications. In brief, mice were decapitated, and brains rapidly dissected and placed in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 3.5 KCl, 1.2 NaH
2PO
4, 1.2 MgCl
2, 2 CaCl
2, 25 NaHCO
3 and 11 glucose (pH 7.3), saturated with 95% O
2 and 5% CO
2. The brain was then embedded in 3% SeaPlaque™ agarose (Lonza, Basel, Switzerland) in PBS and 300 µm thick sagittal slices were cut on a vibrating microtome (Campden Instruments, Sileby, UK), constantly cooled and oxygenated. We obtained around six slices, complete of pons and medulla, from each brain. Each slice was transferred onto a porous membrane (0.45 µm pore size, Millipore) placed on a Millipore culture insert (Millipore), inserted into six-well plates with 1.2 mL of pGBM/DIPG culture medium/well, where the inserts were placed. The slices were incubated at 35 °C, 5% CO
2 for 7 days before the experiments to allow the inflammatory reaction following the mechanical procedure to subside. Following the first day of culture, the medium was replaced with fresh medium and, from that time, changed twice a week.
Some slices were processed immediately at the day of preparation (day 0) or two weeks after the preparation (day 14) for immunofluorescence for the cytoarchitecture characterization: brain slices were fixed with 4% PFA for 1 h at room temperature (RT) and rinsed twice with PBS. Slices were then permeabilized with 1% Triton in PBS for 90 min, blocked with 10% normal goat serum (NGS) + 1% bovine serum albumin (BSA) + 0.1% Triton in PBS for 1 h at RT, incubated with AffiniPure F(ab’)2 fragment goat anti-mouse IgG (H+L) 10 µg/mL (Jackson ImmunoResearch, West Grove, PA, USA) in PBS for 2 h and a half at RT and incubated with the primary antibodies over night at 4 °C. The antibodies were diluted in 2% NGS + 1% BSA in PBS at the following concentrations: rabbit anti-Glial Fibrillary Acidic Protein 1:500 (Z0334, Dako, Jena, Germany), mouse anti-CNPase 1:400 (clone 11-5B, Millipore, USA), rabbit anti-Iba1 1:1000 (FUJIFILM Wako Pure Chemical Corporation, Japan), mouse anti-NeuN 1:500 (clone A60, MAB#377, Millipore).
The day after, the slices were rinsed three times with 0.1% Triton in PBS and incubated over night at 4 °C with the secondary antibodies diluted in 2% NGS + 1% BSA in PBS at the following concentrations: goat anti-rabbit IgG (H+L) Alexa Fluor® 488—conjugated and goat anti-mouse IgG (H+L) Alexa Fluor® 555—conjugated, 1:500 (ThermoFisher Scientific). Slices were rinsed three times with 0.1% Triton in PBS, and nuclei were counterstained with Hoechst33342 (Invitrogen) in PBS for 45 min at RT, rinsed again with PBS and mounted on a slide. Images of the whole slices (4×) were taken on the Operetta CLS (PerkinElmer) while images of the hippocampal region at higher magnification (25×) were taken at Leica TCS-SP8X laser-scanning confocal microscope (Leica Microsystems).
7 days after slices were sectioned, OPBG-DIPG002 NS (1 neurosphere per slide) were implanted on the pontine area, and following 7 days of co-culture, during which DIPG cells had invaded, the brain slices were fixed with 4% PFA for 1 h at RT and rinsed twice with PBS. Slices were then permeabilized with 1% Triton in PBS for 90 min and incubated with Hoechst33342 (Invitrogen) 1:10,000 in PBS for 45 min at RT.
All animal procedures were under the European Communities Council Directive N. 2010/63/EU and the Italian Ministry of Health guidelines (DL 26/2014) and approved by the Italian Ministry of Health and by the local Institutional Animal Care and Use Committee (IACUC) at Istituto Superiore di Sanità (Rome, Italy; protocol n. D9997.N.BYG, 2019).
4.13. Tissue Clearing
To reduce brain tissue autofluorescence, whole-brain OSs at the end point of the co-culture with DIPG NS, were fixed over night at 4 °C in 4% PFA, washed three times in PBS, then embedded overnight at 4 °C in hydrogel monomer solution composed by 4% acrylamide (Sigma-Aldrich) supplemented with 0.25% of 2,2,-Azobis [2-(2-imidazolin-2-yl) propane] dihydrochloride initiator (VA-044, Fujifilm Wako Chemicals GmbH, Neuss, Germany) in PBS, as previously reported [
25]. Sample-hydrogel polymerization was achieved by heating the samples at 37 °C for 3 h, then after PBS washes, samples were incubated with 4% SDS in 200mM boric acid in distilled water, pH 8.5 at 37 °C for 1 day, to passively remove membrane lipids. After clearing, samples were rinsed in PBS, mounted with PBS/glycerol 1:1, and imaged for high power-magnification of the multifluorescent invaded area, using a confocal microscope (Leica AOBS TCS-SP8X, Leica MicrosystemsAfter confocal imaging, samples were incubated in PBS to remove coverslips, then nuclei were counterstained with Hoechst33342 (Invitrogen) 1:10,000 in PBS for 45 min at RT. Finally, samples were mounted with PBS/glycerol 1:1 and acquired at the Nanozoomer S60 (Hamamatsu, Shizuoka, Japan) digital slide scanner platform.
4.14. Images Analysis of Fluorescence Intensity
The Mean Fluorescence Intensity and Fluorescence Intensity/single cell for each fluorescent protein were analyzed using ImageJ software (NIH, downloadable at
http://rsbweb.nih.gov/ij/download.html). The analysis was carried out using 8-bit format digital images (TIFF format) acquired at the TCS AOBS-SP8X confocal microscope and at the Operetta CLS. For mean fluorescence intensity
n = 4 randomly selected images per each cell line and clones were used. For Fluorescent intensity,
n = 100 single cells were analyzed per each cell line and clones.
4.15. Statistical Analysis
All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad software Inc., San Diego, CA, USA). The data are presented as mean ± SD (bar-plot) and as single value (dot-box-plot). p-value < 0.05 was considered to be statistically significant. Statistical analysis was performed using 2way ANOVA multiple comparison test.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}