HCV Glycoprotein Structure and Implications for B-Cell Vaccine Development

Abstract
:1. Introduction
2. The E1 Glycoprotein
3. The E2 Glycoprotein
3.1. Hypervariable Regions (HVRs)
3.2. E2 Structures
3.3. Epitope I
3.4. Epitope II
3.5. The CD81-Binding Loop
3.6. The Front Layer Epitope
4. The E1E2 Heterodimer
5. Implications for a Structure-Based Design of a B Cell Vaccine
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Naggie, S.; Muir, A.J. Oral Combination therapies for hepatitis C Virus infection: Successes, Challenges, and unmet needs. Annu. Rev. Med. 2017, 68, 345–358. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Hepatitis Report; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Enkelmann, J.; Gassowski, M.; Nielsen, S.; Wenz, B.; Ross, S.; Marcus, U.; Bremer, V.; Zimmermann, R.; DRUCK Study Group. High prevalence of hepatitis C virus infection and low level of awareness among people who recently started injecting drugs in a cross-sectional study in Germany, 2011–2014: Missed opportunities for hepatitis C testing. Harm. Reduct. J. 2020, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Marino, Z.; Perello, C.; Inarrairaegui, M.; Ribeiro, A.; Lens, S.; Diaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanduzzi-Zamparelli, M.; Boix, L.; Leal, C.; Reig, M. Hepatocellular carcinoma recurrence in HCV patients treated with direct antiviral agents. Viruses 2019, 11, 406. [Google Scholar] [CrossRef] [Green Version]
- El Kassas, M.; Tawheed, A.; Eltabbakh, M.; Kaseb, A. Hepatitis C antiviral therapy in patients with successfully treated hepatocellular carcinoma: Dancing With wolves. J. Hepatocell Carcinoma 2019, 6, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Islam, N.; Krajden, M.; Shoveller, J.; Gustafson, P.; Gilbert, M.; Buxton, J.A.; Wong, J.; Tyndall, M.W.; Janjua, N.Z. British Columbia Hepatitis testers cohort, t., incidence, risk factors, and prevention of hepatitis C reinfection: A population-based cohort study. Lancet Gastroenterol. Hepatol. 2017, 2, 200–210. [Google Scholar] [CrossRef]
- Swadling, L.; Klenerman, P.; Barnes, E. Ever closer to a prophylactic vaccine for HCV. Expert Opin. Biol. Ther. 2013, 13, 1109–1124. [Google Scholar] [CrossRef] [Green Version]
- Ball, J.K.; Tarr, A.W.; McKeating, J.A. The past, present and future of neutralizing antibodies for hepatitis C virus. Antiviral Res. 2014, 105, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Stamataki, Z.; Coates, S.; Evans, M.J.; Wininger, M.; Crawford, K.; Dong, C.; Fong, Y.L.; Chien, D.; Abrignani, S.; Balfe, P.; et al. Hepatitis C virus envelope glycoprotein immunization of rodents elicits cross-reactive neutralizing antibodies. Vaccine 2007, 25, 7773–7784. [Google Scholar] [CrossRef]
- Meunier, J.C.; Gottwein, J.M.; Houghton, M.; Russell, R.S.; Emerson, S.U.; Bukh, J.; Purcell, R.H. Vaccine-induced cross-genotype reactive neutralizing antibodies against hepatitis C virus. J. Infect. Dis. 2011, 204, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Choo, Q.L.; Kuo, G.; Ralston, R.; Weiner, A.; Chien, D.; van Nest, G.; Han, J.; Berger, K.; Thudium, K.; Kuo, C.; et al. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl. Acad. Sci. USA 1994, 91, 1294–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, J.L.; Chen, C.; Wong, J.; Hockman, D.; Santer, D.M.; Frey, S.E.; Belshe, R.B.; Wakita, T.; Bukh, J.; Jones, C.T.; et al. A hepatitis C virus (HCV) vaccine comprising envelope glycoproteins gpE1/gpE2 derived from a single isolate elicits broad cross-genotype neutralizing antibodies in humans. PLoS ONE 2013, 8, e59776. [Google Scholar] [CrossRef] [PubMed]
- Logan, M.; Law, J.; Wong, J.A.J.; Hockman, D.; Landi, A.; Chen, C.; Crawford, K.; Kundu, J.; Baldwin, L.; Johnson, J.; et al. Native folding of a recombinant gpE1/gpE2 heterodimer vaccine antigen from a precursor protein fused with Fc IgG. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukh, J.; Engle, R.E.; Faulk, K.; Wang, R.Y.; Farci, P.; Alter, H.J.; Purcell, R.H. Immunoglobulin with High-titer in vitro cross-neutralizing hepatitis C virus antibodies passively protects chimpanzees from homologous, but not heterologous, challenge. J. Virol. 2015, 89, 9128–9132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanwolleghem, T.; Bukh, J.; Meuleman, P.; Desombere, I.; Meunier, J.C.; Alter, H.; Purcell, R.H.; Leroux-Roels, G. Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology 2008, 47, 1846–1855. [Google Scholar] [CrossRef]
- Giang, E.; Dorner, M.; Prentoe, J.C.; Dreux, M.; Evans, M.J.; Bukh, J.; Rice, C.M.; Ploss, A.; Burton, D.R.; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6205–6210. [Google Scholar] [CrossRef] [Green Version]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef]
- Osburn, W.O.; Fisher, B.E.; Dowd, K.A.; Urban, G.; Liu, L.; Ray, S.C.; Thomas, D.L.; Cox, A.L. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 2010, 138, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Vieyres, G.; Dubuisson, J.; Patel, A.H. Characterization of antibody-mediated neutralization directed against the hypervariable region 1 of hepatitis C virus E2 glycoprotein. J. Gen. Virol. 2011, 92, 494–506. [Google Scholar] [CrossRef]
- Helle, F.; Goffard, A.; Morel, V.; Duverlie, G.; McKeating, J.; Keck, Z.Y.; Foung, S.; Penin, F.; Dubuisson, J.; Voisset, C. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 2007, 81, 8101–8111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavie, M.; Hanoulle, X.; Dubuisson, J. Glycan Shielding and modulation of hepatitis C virus neutralizing antibodies. Front. Immunol. 2018, 9, 910. [Google Scholar] [CrossRef]
- Stroh, L.J.; Nagarathinam, K.; Krey, T. Conformational flexibility in the CD81-binding site of the hepatitis C virus glycoprotein E2. Front. Immunol. 2018, 9, 1396. [Google Scholar] [CrossRef] [Green Version]
- Logvinoff, C.; Major, M.E.; Oldach, D.; Heyward, S.; Talal, A.; Balfe, P.; Feinstone, S.M.; Alter, H.; Rice, C.M.; McKeating, J.A. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 10149–10154. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Keck, Z.Y.; Foung, S.K. Neutralizing antibody response to hepatitis C virus. Viruses 2011, 3, 2127–2145. [Google Scholar] [CrossRef] [PubMed]
- Cashman, S.B.; Marsden, B.D.; Dustin, L.B. The Humoral Immune response to HCV: Understanding is key to vaccine development. Front. Immunol. 2014, 5, 550. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.R.; Leung, P.; Eltahla, A.A.; Underwood, A.; Abayasingam, A.; Brasher, N.A.; Li, H.; Wu, B.R.; Maher, L.; Luciani, F.; et al. Clearance of hepatitis C virus is associated with early and potent but narrowly-directed, Envelope-specific antibodies. Sci. Rep. 2019, 9, 13300. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Fofana, I.; Heydmann, L.; Barth, H.; Soulier, E.; Habersetzer, F.; Doffoel, M.; Bukh, J.; Patel, A.H.; Zeisel, M.B.; et al. Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog. 2014, 10, e1004128. [Google Scholar] [CrossRef] [Green Version]
- Timpe, J.M.; Stamataki, Z.; Jennings, A.; Hu, K.; Farquhar, M.J.; Harris, H.J.; Schwarz, A.; Desombere, I.; Roels, G.L.; Balfe, P.; et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 2008, 47, 17–24. [Google Scholar] [CrossRef]
- Yi, M.; Nakamoto, Y.; Kaneko, S.; Yamashita, T.; Murakami, S. Delineation of regions important for heteromeric association of hepatitis C virus E1 and E2. Virology 1997, 231, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Op De Beeck, A.; Montserret, R.; Duvet, S.; Cocquerel, L.; Cacan, R.; Barberot, B.; le Maire, M.; Penin, F.; Dubuisson, J. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J. Biol. Chem. 2000, 275, 31428–31437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douam, F.; Dao Thi, V.L.; Maurin, G.; Fresquet, J.; Mompelat, D.; Zeisel, M.B.; Baumert, T.F.; Cosset, F.L.; Lavillette, D. Critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 2014, 59, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.C.; Fournillier, A.; Choukhi, A.; Cahour, A.; Cocquerel, L.; Dubuisson, J.; Wychowski, C. Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J. Gen. Virol. 1999, 80, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Lavillette, D.; Li, Q.; Zhong, J. Role of hepatitis C Virus envelope glycoprotein E1 in virus entry and assembly. Front. Immunol. 2018, 9, 1411. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.Y.; Sung, V.M.; Perkins, S.; Rowe, J.; Paul, S.; Liang, T.J.; Lai, M.M.; Foung, S.K. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J. Virol. 2004, 78, 7257–7263. [Google Scholar] [CrossRef] [Green Version]
- Meunier, J.C.; Russell, R.S.; Goossens, V.; Priem, S.; Walter, H.; Depla, E.; Union, A.; Faulk, K.N.; Bukh, J.; Emerson, S.U.; et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J. Virol. 2008, 82, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Kadam, R.U.; Giang, E.; Ruwona, T.B.; Nieusma, T.; Culhane, J.C.; Stanfield, R.L.; Dawson, P.E.; Wilson, I.A.; Law, M. Structure of hepatitis C virus envelope glycoprotein E1 antigenic site 314-324 in Complex with antibody IGH526. J. Mol. Biol. 2015, 427, 2617–2628. [Google Scholar] [CrossRef] [Green Version]
- El Omari, K.; Iourin, O.; Kadlec, J.; Sutton, G.; Harlos, K.; Grimes, J.M.; Stuart, D.I. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat. Commun. 2014, 5, 4874. [Google Scholar] [CrossRef]
- Michalak, J.P.; Wychowski, C.; Choukhi, A.; Meunier, J.C.; Ung, S.; Rice, C.M.; Dubuisson, J. Characterization of truncated forms of hepatitis C virus glycoproteins. J. Gen. Virol. 1997, 78, 2299–2306. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flyak, A.I.; Ruiz, S.; Colbert, M.D.; Luong, T.; Crowe, J.E., Jr.; Bailey, J.R.; Bjorkman, P.J. HCV Broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe 2018, 24, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Tzarum, N.; Giang, E.; Kong, L.; He, L.; Prentoe, J.; Augestad, E.; Hua, Y.; Castillo, S.; Lauer, G.M.; Bukh, J.; et al. Genetic and structural insights into broad neutralization of hepatitis C virus by human VH1-69 antibodies. Sci. Adv. 2019, 5, eaav1882. [Google Scholar] [CrossRef] [Green Version]
- Flyak, A.I.; Ruiz, S.E.; Salas, J.; Rho, S.; Bailey, J.R.; Bjorkman, P.J. An ultralong CDRH2 in HCV neutralizing antibody demonstrates structural plasticity of antibodies against E2 glycoprotein. Elife 2020, 9, e53169. [Google Scholar] [CrossRef]
- Duan, H.; Kachko, A.; Zhong, L.; Struble, E.; Pandey, S.; Yan, H.; Harman, C.; Virata-Theimer, M.L.; Deng, L.; Zhao, Z.; et al. Amino acid residue-specific neutralization and nonneutralization of hepatitis C virus by monoclonal antibodies to the E2 protein. J. Virol. 2012, 86, 12686–12694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasiliauskaite, I.; Owsianka, A.; England, P.; Khan, A.G.; Cole, S.; Bankwitz, D.; Foung, S.K.H.; Pietschmann, T.; Marcotrigiano, J.; Rey, F.A.; et al. Conformational Flexibility in the immunoglobulin-like domain of the hepatitis C virus glycoprotein E2. MBio 2017, 8, e00382-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Ma, L.; Virata-Theimer, M.L.; Zhong, L.; Yan, H.; Zhao, Z.; Struble, E.; Feinstone, S.; Alter, H.; Zhang, P. Discrete conformations of epitope II on the hepatitis C virus E2 protein for antibody-mediated neutralization and nonneutralization. Proc. Natl. Acad. Sci. USA 2014, 111, 10690–10695. [Google Scholar] [CrossRef] [Green Version]
- Prentoe, J.; Velazquez-Moctezuma, R.; Augestad, E.H.; Galli, A.; Wang, R.; Law, M.; Alter, H.; Bukh, J. Hypervariable region 1 and N-linked glycans of hepatitis C regulate virion neutralization by modulating envelope conformations. Proc. Natl. Acad. Sci. USA 2019, 116, 10039–10047. [Google Scholar] [CrossRef] [Green Version]
- Bankwitz, D.; Steinmann, E.; Bitzegeio, J.; Ciesek, S.; Friesland, M.; Herrmann, E.; Zeisel, M.B.; Baumert, T.F.; Keck, Z.Y.; Foung, S.K.; et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 2010, 84, 5751–5763. [Google Scholar] [CrossRef] [Green Version]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.; Scheel, T.K.; Leroux-Roels, G.; Gottwein, J.M.; Bukh, J. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 2011, 85, 2224–2234. [Google Scholar] [CrossRef] [Green Version]
- Forns, X.; Thimme, R.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Chisari, F.V.; Bukh, J. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl. Acad. Sci. USA 2000, 97, 13318–13323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, N.; Sakamoto, N.; Kurosaki, M.; Marumo, F.; Sato, C. The hypervariable region of the HCV genome changes sequentially during the progression of acute HCV infection to chronic hepatitis. J. Hepatol. 1993, 17, 415–416. [Google Scholar] [CrossRef]
- Sakamoto, N.; Enomoto, N.; Kurosaki, M.; Marumo, F.; Sato, C. Sequential change of the hypervariable region of the hepatitis C virus genome in acute infection. J. Med. Virol. 1994, 42, 103–108. [Google Scholar] [CrossRef]
- Kurosaki, M.; Enomoto, N.; Marumo, F.; Sato, C. Rapid sequence variation of the hypervariable region of hepatitis C virus during the course of chronic infection. Hepatology 1993, 18, 1293–1299. [Google Scholar] [CrossRef]
- Jackson, P.; Petrik, J.; Alexander, G.J.; Pearson, G.; Allain, J.P. Reactivity of synthetic peptides representing selected sections of hepatitis C virus core and envelope proteins with a panel of hepatitis C virus-seropositive human plasma. J. Med. Virol. 1997, 51, 67–79. [Google Scholar] [CrossRef]
- Farci, P.; Shimoda, A.; Wong, D.; Cabezon, T.; de Gioannis, D.; Strazzera, A.; Shimizu, Y.; Shapiro, M.; Alter, H.J.; Purcell, R.H. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. USA 1996, 93, 15394–15399. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar] [CrossRef] [Green Version]
- Prentoe, J.; Velazquez-Moctezuma, R.; Foung, S.K.; Law, M.; Bukh, J. Hypervariable region 1 shielding of hepatitis C virus is a main contributor to genotypic differences in neutralization sensitivity. Hepatology 2016, 64, 1881–1892. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Girard-Blanc, C.; Wang, W.; Lau, P.; Zuiani, A.; Rey, F.A.; Krey, T.; Diamond, M.S.; Foung, S.K. Antibody response to hypervariable region 1 interferes with broadly neutralizing antibodies to hepatitis C virus. J. Virol. 2016, 90, 3112–3122. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.; Meyer, K.; Banerjee, A.; Basu, A.; Coates, S.; Abrignani, S.; Houghton, M.; Frey, S.E.; Belshe, R.B. Characterization of antibodies induced by vaccination with hepatitis C virus envelope glycoproteins. J. Infect. Dis. 2010, 202, 862–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, J.L.M.; Logan, M.; Wong, J.; Kundu, J.; Hockman, D.; Landi, A.; Chen, C.; Crawford, K.; Wininger, M.; Johnson, J.; et al. Role of the E2 hypervariable region (HVR1) in the immunogenicity of a recombinant hepatitis C virus vaccine. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffard, A.; Dubuisson, J. Glycosylation of hepatitis C virus envelope proteins. Biochimie 2003, 85, 295–301. [Google Scholar] [CrossRef]
- Helle, F.; Vieyres, G.; Elkrief, L.; Popescu, C.I.; Wychowski, C.; Descamps, V.; Castelain, S.; Roingeard, P.; Duverlie, G.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 2010, 84, 11905–11915. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.; Freedman, H.; Logan, M.; Wong, J.A.J.; Hockman, D.; Chen, C.; He, J.; Beard, M.R.; Eyre, N.S.; Baumert, T.F.; et al. A recombinant hepatitis C virus genotype 1a E1/E2 Envelope glycoprotein vaccine elicits antibodies that differentially neutralize closely related 2a Strains through interactions of the N-terminal hypervariable region 1 of E2 with scavenger receptor B1. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Bartosch, B.; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 2005, 79, 8217–8229. [Google Scholar] [CrossRef] [Green Version]
- Dreux, M.; Pietschmann, T.; Granier, C.; Voisset, C.; Ricard-Blum, S.; Mangeot, P.E.; Keck, Z.; Foung, S.; Vu-Dac, N.; Dubuisson, J.; et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 2006, 281, 18285–18295. [Google Scholar] [CrossRef] [Green Version]
- Voisset, C.; Op de Beeck, A.; Horellou, P.; Dreux, M.; Gustot, T.; Duverlie, G.; Cosset, F.L.; Vu-Dac, N.; Dubuisson, J. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J. Gen. Virol. 2006, 87, 2577–2581. [Google Scholar] [CrossRef]
- Keck, Z.; Wang, W.; Wang, Y.; Lau, P.; Carlsen, T.H.; Prentoe, J.; Xia, J.; Patel, A.H.; Bukh, J.; Foung, S.K. Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J. Virol. 2013, 87, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Pierce, B.G.; Wang, Q.; Keck, Z.Y.; Fuerst, T.R.; Foung, S.K.; Mariuzza, R.A. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J. Biol. Chem. 2015, 290, 10117–10125. [Google Scholar] [CrossRef] [Green Version]
- Anjum, S.; Wahid, A.; Afzal, M.S.; Albecka, A.; Alsaleh, K.; Ahmad, T.; Baumert, T.F.; Wychowski, C.; Qadri, I.; Penin, F.; et al. Additional glycosylation within a specific hypervariable region of subtype 3a of hepatitis C virus protects against virus neutralization. J. Infect. Dis. 2013, 208, 1888–1897. [Google Scholar] [CrossRef]
- Alhammad, Y.; Gu, J.; Boo, I.; Harrison, D.; McCaffrey, K.; Vietheer, P.T.; Edwards, S.; Quinn, C.; Coulibaly, F.; Poumbourios, P.; et al. Monoclonal antibodies directed toward the hepatitis C virus glycoprotein E2 detect antigenic differences modulated by the N-Terminal hypervariable region 1 (HVR1), HVR2, and intergenotypic variable region. J. Virol. 2015, 89, 12245–12261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffrey, K.; Gouklani, H.; Boo, I.; Poumbourios, P.; Drummer, H.E. The variable regions of hepatitis C virus glycoprotein E2 have an essential structural role in glycoprotein assembly and virion infectivity. J. Gen. Virol. 2011, 92, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, L.; Lees, W.D.; Moss, D.S.; Palor, M.; Bingham, R.J.; Shepherd, A.J.; Grove, J. Flexibility and intrinsic disorder are conserved features of hepatitis C virus E2 glycoprotein. PLoS Comput. Biol. 2020, 16, e1007710. [Google Scholar] [CrossRef]
- McCaffrey, K.; Boo, I.; Tewierek, K.; Edmunds, M.L.; Poumbourios, P.; Drummer, H.E. Role of conserved cysteine residues in hepatitis C virus glycoprotein e2 folding and function. J. Virol. 2012, 86, 3961–3974. [Google Scholar] [CrossRef] [Green Version]
- Vietheer, P.T.; Boo, I.; Gu, J.; McCaffrey, K.; Edwards, S.; Owczarek, C.; Hardy, M.P.; Fabri, L.; Center, R.J.; Poumbourios, P.; et al. The core domain of hepatitis C virus glycoprotein E2 generates potent cross-neutralizing antibodies in guinea pigs. Hepatology 2017, 65, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Castelli, M.; Clementi, N.; Sautto, G.A.; Pfaff, J.; Kahle, K.M.; Barnes, T.; Doranz, B.J.; Dal Peraro, M.; Clementi, M.; Burioni, R.; et al. HCV E2 core structures and mAbs: Something is still missing. Drug Discov. Today 2014, 19, 1964–1970. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.; Boo, I.; Poumbourios, P.; Drummer, H.E. Hepatitis C virus (HCV) envelope glycoproteins E1 and E2 contain reduced cysteine residues essential for virus entry. J. Biol. Chem. 2011, 286, 31984–31992. [Google Scholar] [CrossRef] [Green Version]
- Tzarum, N.; Wilson, I.A.; Law, M. The neutralizing face of hepatitis C virus E2 envelope glycoprotein. Front. Immunol. 2018, 9, 1315. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Giang, E.; Nieusma, T.; Robbins, J.B.; Deller, M.C.; Stanfield, R.L.; Wilson, I.A.; Law, M. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J. Virol. 2012, 86, 13085–13088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Giang, E.; Robbins, J.B.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A.; Law, M. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. USA 2012, 109, 9499–9504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantua, H.; Diao, J.; Ultsch, M.; Hazen, M.; Mathieu, M.; McCutcheon, K.; Takeda, K.; Date, S.; Cheung, T.K.; Phung, Q.; et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 2013, 425, 1899–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, J.A.; Owsianka, A.M.; Jeffery, N.; Matthews, D.J.; Keck, Z.Y.; Lau, P.; Foung, S.K.; Taylor, G.L.; Patel, A.H. Toward a hepatitis C virus vaccine: The structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J. Virol. 2012, 86, 12923–12932. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Hardy, J.; Boo, I.; Vietheer, P.; McCaffrey, K.; Alhammad, Y.; Chopra, A.; Gaudieri, S.; Poumbourios, P.; Coulibaly, F.; et al. Escape of hepatitis C virus from epitope I neutralization increases sensitivity of other neutralization epitopes. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meola, A.; Tarr, A.W.; England, P.; Meredith, L.W.; McClure, C.P.; Foung, S.K.; McKeating, J.A.; Ball, J.K.; Rey, F.A.; Krey, T. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J. Virol. 2015, 89, 2170–2181. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Lee, D.E.; Kadam, R.U.; Liu, T.; Giang, E.; Nieusma, T.; Garces, F.; Tzarum, N.; Woods, V.L., Jr.; Ward, A.B.; et al. Structural flexibility at a major conserved antibody target on hepatitis C virus E2 antigen. Proc. Natl. Acad. Sci. USA 2016, 113, 12768–12773. [Google Scholar] [CrossRef] [Green Version]
- Tarr, A.W.; Owsianka, A.M.; Jayaraj, D.; Brown, R.J.; Hickling, T.P.; Irving, W.L.; Patel, A.H.; Ball, J.K. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J. Gen. Virol. 2007, 88, 2991–3001. [Google Scholar] [CrossRef]
- Krey, T.; Meola, A.; Keck, Z.Y.; Damier-Piolle, L.; Foung, S.K.; Rey, F.A. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 2013, 9, e1003364. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Wang, Y.; Lau, P.; Lund, G.; Rangarajan, S.; Fauvelle, C.; Liao, G.C.; Holtsberg, F.W.; Warfield, K.L.; Aman, M.J.; et al. Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute hepatitis C virus infection in mice. Hepatology 2016, 64, 1922–1933. [Google Scholar] [CrossRef]
- Deng, L.; Zhong, L.; Struble, E.; Duan, H.; Ma, L.; Harman, C.; Yan, H.; Virata-Theimer, M.L.; Zhao, Z.; Feinstone, S.; et al. Structural evidence for a bifurcated mode of action in the antibody-mediated neutralization of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7418–7422. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.R.; Flyak, A.I.; Cohen, V.J.; Li, H.; Wasilewski, L.N.; Snider, A.E.; Wang, S.; Learn, G.H.; Kose, N.; Loerinc, L.; et al. Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI Insight. 2017, 2, e92872. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Hadlock, K.G.; Foung, S.K.; Levy, S. V(H)1-69 gene is preferentially used by hepatitis C virus-associated B cell lymphomas and by normal B cells responding to the E2 viral antigen. Blood 2001, 97, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Falson, P.; Bartosch, B.; Alsaleh, K.; Tews, B.A.; Loquet, A.; Ciczora, Y.; Riva, L.; Montigny, C.; Montpellier, C.; Duverlie, G.; et al. Hepatitis C virus envelope glycoprotein E1 forms trimers at the surface of the virion. J. Virol. 2015, 89, 10333–10346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, M.; Clementi, N.; Pfaff, J.; Sautto, G.A.; Diotti, R.A.; Burioni, R.; Doranz, B.J.; Dal Peraro, M.; Clementi, M.; Mancini, N. A biologically-validated HCV E1E2 heterodimer structural model. Sci Rep. 2017, 7, 214. [Google Scholar] [CrossRef]
- Freedman, H.; Logan, M.R.; Hockman, D.; Koehler Leman, J.; Law, J.L.; Houghton, M. Computational Prediction of the heterodimeric and higher-order structure of gpE1/gpE2 envelope glycoproteins encoded by hepatitis C virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Nayak, A.; Pattabiraman, N.; Fadra, N.; Goldman, R.; Kosakovsky Pond, S.L.; Mazumder, R. Structure-function analysis of hepatitis C virus envelope glycoproteins E1 and E2. J. Biomol. Struct. Dyn. 2015, 33, 1682–1694. [Google Scholar] [CrossRef]
- Cao, L.; Yu, B.; Kong, D.; Cong, Q.; Yu, T.; Chen, Z.; Hu, Z.; Chang, H.; Zhong, J.; Baker, D.; et al. Functional expression and characterization of the envelope glycoprotein E1E2 heterodimer of hepatitis C virus. PLoS Pathog. 2019, 15, e1007759. [Google Scholar] [CrossRef] [Green Version]
- Guest, J.D.; Pierce, B.G. Computational modeling of hepatitis C Virus envelope glycoprotein structure and recognition. Front. Immunol. 2018, 9, 1117. [Google Scholar] [CrossRef]
- Vieyres, G.; Thomas, X.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 2010, 84, 10159–10168. [Google Scholar] [CrossRef] [Green Version]
- Jusoh, S.A.; Welsch, C.; Siu, S.W.; Bockmann, R.A.; Helms, V. Contribution of charged and polar residues for the formation of the E1-E2 heterodimer from hepatitis C virus. J. Mol. Model. 2010, 16, 1625–1637. [Google Scholar] [CrossRef]
- Ciczora, Y.; Callens, N.; Penin, F.; Pecheur, E.I.; Dubuisson, J. Transmembrane domains of hepatitis C virus envelope glycoproteins: Residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J. Virol. 2007, 81, 2372–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Donato, G.; Acosta-Rivero, N.; Morales-Grillo, J.; Musacchio, A.; Vina, A.; Alvarez, C.; Figueroa, N.; Guerra, I.; Garcia, J.; Varas, L.; et al. Expression and processing of hepatitis C virus structural proteins in Pichia pastoris yeast. Biochem. Biophys. Res. Commun. 2006, 342, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Tello, D.; Rodriguez-Rodriguez, M.; Yelamos, B.; Gomez-Gutierrez, J.; Ortega, S.; Pacheco, B.; Peterson, D.L.; Gavilanes, F. Expression and structural properties of a chimeric protein based on the ectodomains of E1 and E2 hepatitis C virus envelope glycoproteins. Protein Expr. Purif. 2010, 71, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazzoli, M.; Helenius, A.; Foung, S.K.; Houghton, M.; Abrignani, S.; Merola, M. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology 2005, 332, 438–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzyb, K.; Czarnota, A.; Brzozowska, A.; Cieslik, A.; Rabalski, L.; Tyborowska, J.; Bienkowska-Szewczyk, K. Immunogenicity and functional characterization of Leishmania-derived hepatitis C virus envelope glycoprotein complex. Sci. Rep. 2016, 6, 30627. [Google Scholar] [CrossRef]
- Xiang, Z.H.; Cai, W.J.; Zhao, P.; Kong, L.B.; Ye, L.B.; Wu, Z.H. Purification and application of bacterially expressed chimeric protein E1E2 of hepatitis C virus. Protein Expr. Purif. 2006, 49, 95–101. [Google Scholar] [CrossRef]
- Tello, D.; Rodriguez-Rodriguez, M.; Yelamos, B.; Gomez-Gutierrez, J.; Peterson, D.L.; Gavilanes, F. High-yield production of a chimeric glycoprotein based on permuted E1 and E2 HCV envelope ectodomains. J. Virol. Methods 2015, 213, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Ruwona, T.B.; Giang, E.; Nieusma, T.; Law, M. Fine mapping of murine antibody responses to immunization with a novel soluble form of hepatitis C virus envelope glycoprotein complex. J. Virol. 2014, 88, 10459–10471. [Google Scholar] [CrossRef] [Green Version]
- Fauvelle, C.; Colpitts, C.C.; Keck, Z.Y.; Pierce, B.G.; Foung, S.K.; Baumert, T.F. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert Rev. Vaccines 2016, 15, 1535–1544. [Google Scholar] [CrossRef]
- Guo, X.; Zhong, J.Y.; Li, J.W. Hepatitis C Virus infection and vaccine development. J. Clin. Exp. Hepatol. 2018, 8, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, J.R.; Barnes, E.; Cox, A.L. Approaches, progress, and challenges to hepatitis C vaccine development. Gastroenterology 2019, 156, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, E.; Patient, R.; Hourioux, C.; Dimier-Poisson, I.; Roingeard, P. Chimeric hepatitis B virus/hepatitis C virus envelope proteins elicit broadly neutralizing antibodies and constitute a potential bivalent prophylactic vaccine. Hepatology 2013, 57, 1303–1313. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Nagy, K.; Chavez, D.; Willis, S.; McBride, R.; Giang, E.; Honda, A.; Bukh, J.; Ordoukhanian, P.; Zhu, J.; et al. Antibody responses to immunization with HCV Envelope glycoproteins as a baseline for B-cell-based vaccine development. Gastroenterology 2020, 158, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Khera, T.; Behrendt, P.; Bankwitz, D.; Brown, R.J.P.; Todt, D.; Doepke, M.; Khan, A.G.; Schulze, K.; Law, J.; Logan, M.; et al. Functional and immunogenic characterization of diverse HCV glycoprotein E2 variants. J. Hepatol. 2019, 70, 593–602. [Google Scholar] [CrossRef]
- Torrents de la Pena, A.; Julien, J.P.; de Taeye, S.W.; Garces, F.; Guttman, M.; Ozorowski, G.; Pritchard, L.K.; Behrens, A.J.; Go, E.P.; Burger, J.A.; et al. Improving the immunogenicity of native-like HIV-1 envelope trimers by hyperstabilization. Cell Rep. 2017, 20, 1805–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Taeye, S.W.; Ozorowski, G.; Torrents de la Pena, A.; Guttman, M.; Julien, J.P.; van den Kerkhof, T.L.; Burger, J.A.; Pritchard, L.K.; Pugach, P.; Yasmeen, A.; et al. Immunogenicity of stabilized HIV-1 envelope trimers with reduced exposure of non-neutralizing epitopes. Cell 2015, 163, 1702–1715. [Google Scholar] [CrossRef] [Green Version]
- Urbanowicz, R.A.; McClure, C.P.; Brown, R.J.; Tsoleridis, T.; Persson, M.A.; Krey, T.; Irving, W.L.; Ball, J.K.; Tarr, A.W. A diverse panel of hepatitis C virus glycoproteins for use in vaccine research reveals extremes of monoclonal antibody neutralization resistance. J. Virol. 2015, 90, 3288–3301. [Google Scholar] [CrossRef] [Green Version]
- Wasilewski, L.N.; Ray, S.C.; Bailey, J.R. Hepatitis C virus resistance to broadly neutralizing antibodies measured using replication-competent virus and pseudoparticles. J. Gen. Virol. 2016, 97, 2883–2893. [Google Scholar] [CrossRef]
- Bailey, J.R.; Wasilewski, L.N.; Snider, A.E.; El-Diwany, R.; Osburn, W.O.; Keck, Z.; Foung, S.K.; Ray, S.C. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J. Clin. Invest. 2015, 125, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. Scaffolding to build a rational vaccine design strategy. Proc. Natl. Acad. Sci. USA 2010, 107, 17859–17860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawood, R.M.; Moustafa, R.I.; Abdelhafez, T.H.; El-Shenawy, R.; El-Abd, Y.; Bader El Din, N.G.; Dubuisson, J.; El Awady, M.K. A multiepitope peptide vaccine against HCV stimulates neutralizing humoral and persistent cellular responses in mice. BMC Infect. Dis. 2019, 19, 932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yassine, H.M.; Boyington, J.C.; McTamney, P.M.; Wei, C.J.; Kanekiyo, M.; Kong, W.P.; Gallagher, J.R.; Wang, L.; Zhang, Y.; Joyce, M.G. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 2015, 21, 1065–1070. [Google Scholar] [CrossRef]
- He, L.; Cheng, Y.; Kong, L.; Azadnia, P.; Giang, E.; Kim, J.; Wood, M.R.; Wilson, I.A.; Law, M.; Zhu, J. Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding. Sci. Rep. 2015, 5, 12501. [Google Scholar] [CrossRef]
- Sandomenico, A.; Leonardi, A.; Berisio, R.; Sanguigno, L.; Foca, G.; Foca, A.; Ruggiero, A.; Doti, N.; Muscariello, L.; Barone, D.; et al. Generation and characterization of monoclonal antibodies against a cyclic variant of hepatitis C virus E2 epitope 412–422. J. Virol. 2016, 90, 3745–3759. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.G.; Boucher, E.N.; Piepenbrink, K.H.; Ejemel, M.; Rapp, C.A.; Thomas, W.D., Jr.; Sundberg, E.J.; Weng, Z.; Wang, Y. Structure-based design of hepatitis C virus vaccines that elicit neutralizing antibody responses to a conserved epitope. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Owsianka, A.; Fadda, V.; Potter, J.A.; Cowton, V.M.; Jeffery, N.; di Lorenzo, C.; Ortega-Prieto, A.M.; Taylor, G.L.; Dorner, M.D.; Patel, A.H. Anti-idiotype vaccine that mimics a broadly neutralizing HCV E2 Epitope. In Proceedings of the 24th International Symposium on Hepatitis C Virus and Related Viruses, Cape Cod, MA, USA, 25–28 September 2017. [Google Scholar]
- Masavuli, M.G.; Wijesundara, D.K.; Torresi, J.; Gowans, E.J.; Grubor-Bauk, B. Preclinical development and production of virus-like particles as vaccine candidates for hepatitis, C. Front. Microbiol. 2017, 8, 2413. [Google Scholar] [CrossRef]
- Masavuli, M.G.; Wijesundara, D.K.; Underwood, A.; Christiansen, D.; Earnest-Silveira, L.; Bull, R.; Torresi, J.; Gowans, E.J.; Grubor-Bauk, B. A hepatitis C virus DNA vaccine encoding a secreted, oligomerized form of envelope proteins is highly immunogenic and elicits neutralizing antibodies in vaccinated mice. Front. Immunol. 2019, 10, 1145. [Google Scholar] [CrossRef] [Green Version]
- Elmowalid, G.A.; Qiao, M.; Jeong, S.H.; Borg, B.B.; Baumert, T.F.; Sapp, R.K.; Hu, Z.; Murthy, K.; Liang, T.J. Immunization with hepatitis C virus-like particles results in control of hepatitis C virus infection in chimpanzees. Proc. Natl. Acad. Sci. USA 2007, 104, 8427–8432. [Google Scholar] [CrossRef] [Green Version]
- Baumert, T.F.; Vergalla, J.; Satoi, J.; Thomson, M.; Lechmann, M.; Herion, D.; Greenberg, H.B.; Ito, S.; Liang, T.J. Hepatitis C virus-like particles synthesized in insect cells as a potential vaccine candidate. Gastroenterology 1999, 117, 1397–1407. [Google Scholar] [CrossRef]
- Patient, R.; Hourioux, C.; Vaudin, P.; Pages, J.C.; Roingeard, P. Chimeric hepatitis B and C viruses envelope proteins can form subviral particles: Implications for the design of new vaccine strategies. N. Biotechnol. 2009, 25, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaumont, E.; Roch, E.; Chopin, L.; Roingeard, P. Hepatitis C virus E1 and E2 proteins used as separate immunogens induce neutralizing antibodies with additive properties. PLoS ONE 2016, 11, e0151626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihailova, M.; Boos, M.; Petrovskis, I.; Ose, V.; Skrastina, D.; Fiedler, M.; Sominskaya, I.; Ross, S.; Pumpens, P.; Roggendorf, M.; et al. Recombinant virus-like particles as a carrier of B- and T-cell epitopes of hepatitis C virus (HCV). Vaccine 2006, 24, 4369–4377. [Google Scholar] [CrossRef]
- Sominskaya, I.; Skrastina, D.; Dislers, A.; Vasiljev, D.; Mihailova, M.; Ose, V.; Dreilina, D.; Pumpens, P. Construction and immunological evaluation of multivalent hepatitis B virus (HBV) core virus-like particles carrying HBV and HCV epitopes. Clin. Vaccine Immunol. 2010, 17, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, A.; Tanaka, T.; Hoshi, Y.; Kato, N.; Tachibana, K.; Iizuka, H.; Machida, A.; Okamoto, H.; Yamasaki, M.; Miyakawa, Y.; et al. Chimeric hepatitis B virus core particles with parts or copies of the hepatitis C virus core protein. J. Virol. 1993, 67, 6064–6070. [Google Scholar] [CrossRef] [Green Version]
- Denis, J.; Majeau, N.; Acosta-Ramirez, E.; Savard, C.; Bedard, M.C.; Simard, S.; Lecours, K.; Bolduc, M.; Pare, C.; Willems, B.; et al. Immunogenicity of papaya mosaic virus-like particles fused to a hepatitis C virus epitope: Evidence for the critical function of multimerization. Virology 2007, 363, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Wang, X.; Lou, P.; Hu, Z.; Qu, P.; Li, D.; Li, Q.; Xu, Y.; Niu, J.; He, Y.; et al. A nanoparticle-based hepatitis C virus vaccine with enhanced potency. J. Infect. Dis. 2020, 221, 1304–1314. [Google Scholar]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell 2019, 176, 1420–1431. [Google Scholar] [CrossRef] [Green Version]
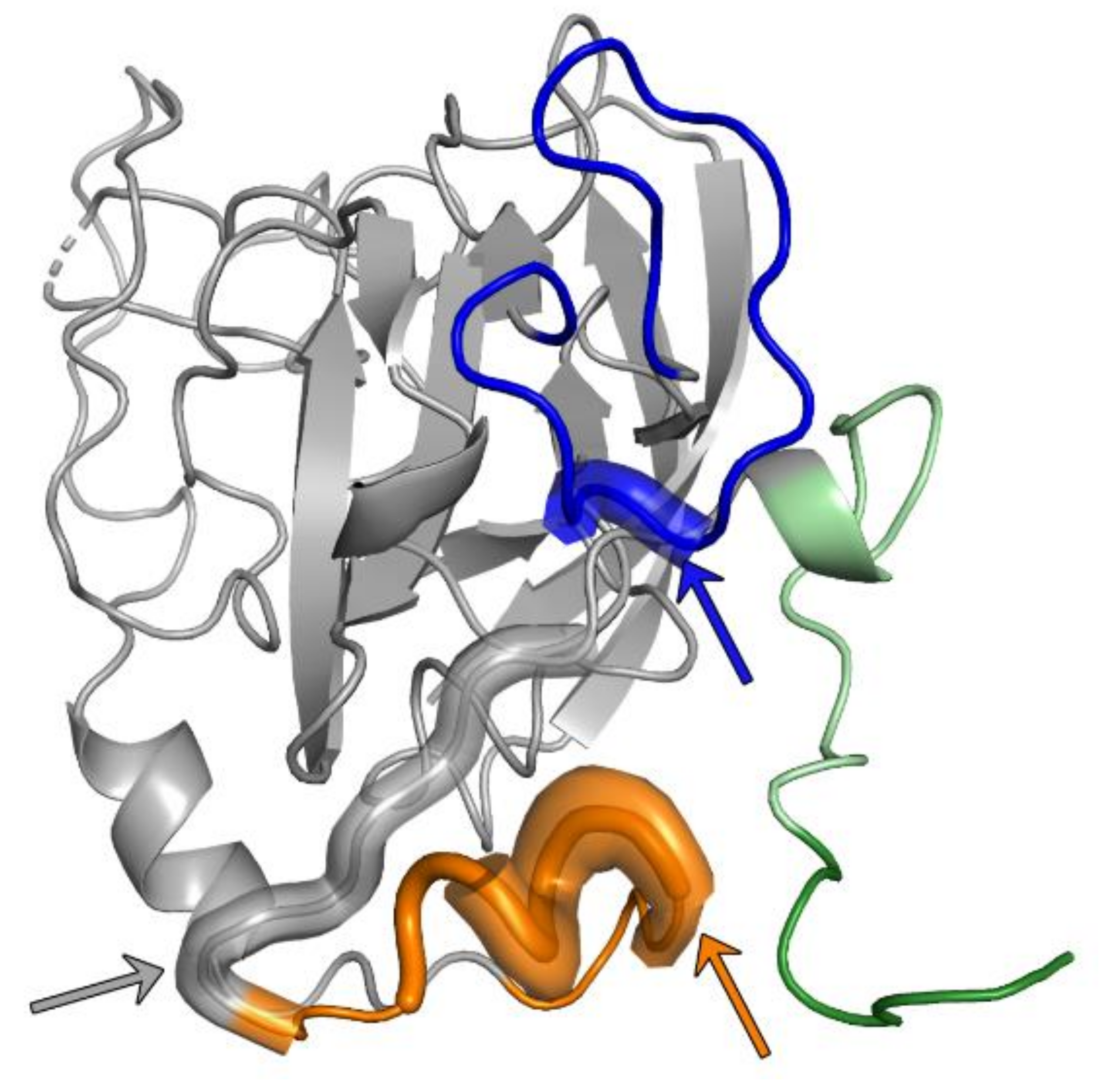

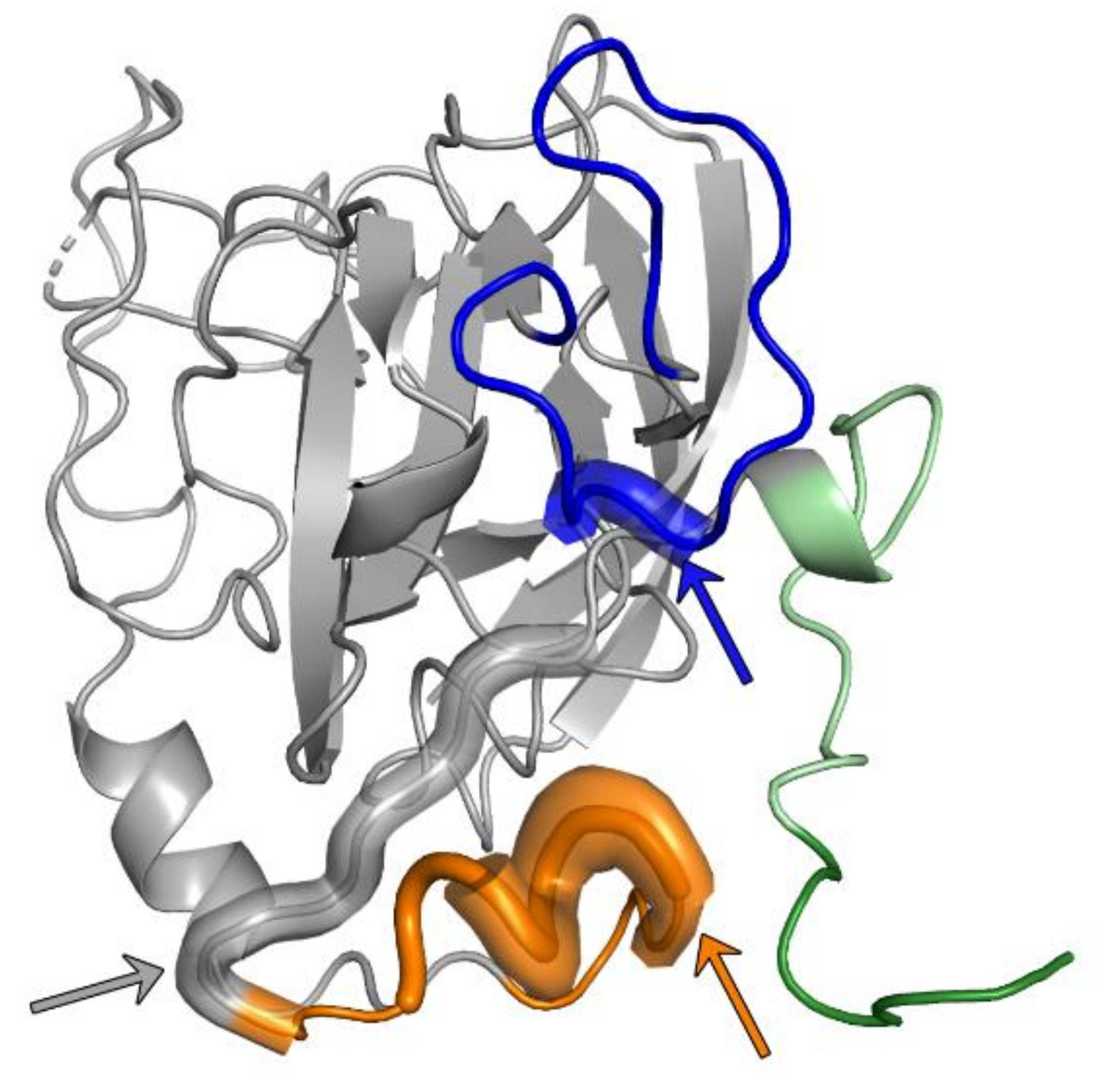{kind=link}
| Disulfide Bond | E2 Core 1a53 | E2 Core H77 | E2 Core J6 | E2 Core GT6a | E2 Core GT6a | E2 Core GT6a | E2 Ecto 1b09 | E2 Ecto 1b09 | E2 Ecto 1a53 | E2 Ecto 1b09 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB | 6MEK | 4MWF | 4WEB | 6BKD | 6BKB | 6BKC | 6MEI | 6MEH | 6MEJ | 6URH | |
| Complexed Fab | HEPC3, HEPC46 | AR3C | 2A12 | AR3D | AR3A | AR3B | HEPC3 | HEPC74 | HEPC3, HEPC46 | AR3X | |
| 429–503 | + | + | § C429 | + | + | + | + | + | + | + | |
| 452–620 | + | + | - | disordered | + | disordered | + | + | + | + | |
| 459–486 | disordered | disordered | - | disordered | disordered | disordered | + | + | + | disordered | |
| 486–620 | - | - | + | disordered | disordered | disordered | - | - | - | disordered | |
| 494–564 | + | + | + | + | + | + | + | + | + | + | |
| 508–552 | + | + | + | + | + | + | + | + | + | + | |
| 569–581 | - | + | - | § | § | § | - | - | - | - | |
| 569–597 | + | - | + | § | § | § | + | + | + | + | |
| 581–585 | - | - | disordered | § | § | § | + | + | + | disordered | |
| 585–597 | - | + | - | § | § | § | - | - | - | - | |
| 607–644 | + | + | + | + | + | + | + | + | + | + | |
| Reference | [44] | [43] | [78] | [45] | [45] | [45] | [44] | [44] | [44] | [46] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ströh, L.J.; Krey, T. HCV Glycoprotein Structure and Implications for B-Cell Vaccine Development. Int. J. Mol. Sci. 2020, 21, 6781. https://doi.org/10.3390/ijms21186781
Ströh LJ, Krey T. HCV Glycoprotein Structure and Implications for B-Cell Vaccine Development. International Journal of Molecular Sciences. 2020; 21(18):6781. https://doi.org/10.3390/ijms21186781
Chicago/Turabian StyleStröh, Luisa J., and Thomas Krey. 2020. "HCV Glycoprotein Structure and Implications for B-Cell Vaccine Development" International Journal of Molecular Sciences 21, no. 18: 6781. https://doi.org/10.3390/ijms21186781
APA StyleStröh, L. J., & Krey, T. (2020). HCV Glycoprotein Structure and Implications for B-Cell Vaccine Development. International Journal of Molecular Sciences, 21(18), 6781. https://doi.org/10.3390/ijms21186781