RNA-Binding Proteins as Regulators of Migration, Invasion and Metastasis in Oral Squamous Cell Carcinoma

 , and
, and
Abstract
:1. Introduction
2. General Functions and Mechanisms of RNA-Binding Proteins
3. RBPs Associated with Migration, Invasion, and Metastasis in OSCC
3.1. ADAR1
3.2. DDX3
3.3. ELAVL1/HuR
3.4. ESRP1 and ESRP2
3.5. IGF2BP3
3.6. LIN28B
3.7. METTL3-METTL14 Complex
4. Therapeutic Targeting Options for RBPs Involved in Oral Cancer Progression
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Adenosine |
| ADAR1 | Adenosine deaminase acting on RNA 1 |
| ALDH1 | Aldehyde dehydrogenase 1 |
| AML | Acute myeloid leukemia |
| ARE | (AU)-rich element |
| ARID3B | AT-rich interaction domain molecule 3B |
| ATF4 | Activating transcription factor 4 |
| BIRC5 | Baculoviral IAP Repeat Containing 5 |
| BMI1 | B lymphoma Mo-MLV insertion region 1 homolog |
| BRD4 | Bromodomain-containing protein 4 |
| CBC | Cap-binding complex |
| CD | Cluster of differentiation |
| CDK | Cyclin-dependent kinase |
| CDKN2A | Cyclin dependent kinase inhibitor 2A |
| c-FOS | cellular oncogene fos |
| circRNA | circular RNA |
| c-MYC | Avian myelocytomatosis virus oncogene cellular homolog |
| COX | Cyclooxygenase |
| CREBBP | CREB-binding protein |
| CSC | Cancer stem cell |
| DDX3 | DEAD Box Protein 3 |
| EBSE | ESRP binding splicing enhancer |
| EBSI | ESRP binding splicing inhibitor |
| EGF | Epidermal growth factor |
| EGFR | Epithelial growth factor receptor |
| eIF3 | eukaryotic initiation factor 3 |
| ELAVL | Embryonic lethal and abnormal vision gene |
| EMT | Epithelial-to-mesenchymal transition |
| ESRP | Epithelial splicing regulatory protein |
| FGF | Fibroblast growth factor |
| HMGA2 | High-mobility group AT-hook 2 |
| hnRNP | heterogeneous nuclear ribonucleoprotein |
| HNRNPL | Heterogeneous nuclear ribonucleoprotein L |
| HNS | HuR nucleocytoplasmic shuttling sequence |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papilloma virus |
| HuB | Hu antigen B |
| HuC | Hu antigen C |
| HuD | Hu antigen D |
| HuR | Human antigen R |
| I | Inosine |
| IFN | Interferone |
| IGF2BP | Insulin-like growth factor 2 mRNA binding protein |
| IL-6 | Interleukin 6 |
| JAK | Janus kinase |
| KH | K homology |
| La | Small RNA binding exonuclease protection factor La |
| lncRNA | long non-coding RNA |
| M6A | N6-Methyladenosine |
| MDM2 | Mouse double minutes 2 |
| MeRIP-seq | Methylated RNA immunoprecipitation sequencing |
| METTL | Methyltransferase-like |
| miRNA | microRNA |
| mRNA | messenger RNA |
| MSI1 | Musashi1 |
| mTOR | mammalian target of rapamycin |
| ncRNA | non-coding RNA |
| ORF | Open reading frame |
| OSCC | Oral squamous cell carcinoma |
| PDPN | Podoplanin |
| PI3K | Phosphatidylinositol-3-kinase |
| POU5F1 | POU class 5 homeobox 1 |
| PRC1 | Polycomb repressive complex 1 |
| PROTAC | Proteolysis Targeting Chimeras |
| QKI | Quaking |
| RB1 | Retinoblastoma-associated protein 1 |
| RBD | RNA-binding domain |
| RBL1/2 | Retinoblastoma-like protein 1 |
| RBP | RNA-binding protein |
| RING | Really Interesting New Gene |
| RISC | RNA-induced silencing complex |
| RRM | RNA recognition motifs |
| SNAI1 | Snail family transcriptional repressor 1 |
| SOCS | Suppressor of cytokine signaling |
| SOX2 | SRY-Box transcription factor 2 |
| SRSF3 | Serine and arginine rich splicing factor 3 |
| STAT | Signal transducer and activator of transcription |
| TCGA | The Cancer Genome Atlas |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| TP53 | Tumor protein 53 |
| TTP | Tristetraprolin |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor |
| WTAP | Wilms’ tumor 1-associated protein |
| YTHDF1 | YTH domain family member |
| ZEB | Zinc finger E-box binding homeobox |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Filho, A.; Bray, F. Global patterns and trends in cancers of the lip, tongue and mouth. Oral. Oncol. 2020, 102, 104551. [Google Scholar] [CrossRef] [PubMed]
- Melchers, L.J.; Schuuring, E.; van Dijk, B.A.; de Bock, G.H.; Witjes, M.J.; van der Laan, B.F.; van der Wal, J.E.; Roodenburg, J.L. Tumour infiltration depth ≥4 mm is an indication for an elective neck dissection in pT1cN0 oral squamous cell carcinoma. Oral. Oncol. 2012, 48, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.H.; Scognamiglio, T.; Gudas, L.J. Basal stem cells contribute to squamous cell carcinomas in the oral cavity. Carcinogenesis 2013, 34, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Vig, N.; Mackenzie, I.C.; Biddle, A. Phenotypic plasticity and epithelial-to-mesenchymal transition in the behaviour and therapeutic response of oral squamous cell carcinoma. J. Oral. Pathol. Med. 2015, 44, 649–655. [Google Scholar] [CrossRef] [PubMed]
- de Camargo Cancela, M.; de Souza, D.L.; Curado, M.P. International incidence of oropharyngeal cancer: A population-based study. Oral. Oncol. 2012, 48, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.; Dayal, H.; Rohrer, T.; Swanson, M.; Sultz, H.; Shedd, D.; Fischman, S. Dentition, diet, tobacco, and alcohol in the epidemiology of oral cancer. J. Nat. Cancer Inst. 1977, 59, 1611–1618. [Google Scholar] [CrossRef]
- Muttagi, S.S.; Chaturvedi, P.; Gaikwad, R.; Singh, B.; Pawar, P. Head and neck squamous cell carcinoma in chronic areca nut chewing Indian women: Case series and review of literature. Ind. J. Med. Paediatr. Oncol. 2012, 33, 32–35. [Google Scholar] [CrossRef] [Green Version]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Kiess, A.; Chung, C.H. Emerging biomarkers in head and neck cancer in the era of genomics. Nat. Rev. Clin. Oncol. 2015, 12, 11–26. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasurya, R.; Sathyan, K.M.; Lakshminarayanan, K.; Abraham, T.; Nalinakumari, K.R.; Abraham, E.K.; Nair, M.K.; Kannan, S. Phenotypic alterations in Rb pathway have more prognostic influence than p53 pathway proteins in oral carcinoma. Mod. Pathol. 2005, 18, 1056–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Do, S.I.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Investig. 2016, 96, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcheri, C.; Meisel, C.T.; Mitsiadis, T. Multifactorial Contribution of Notch Signaling in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessions, D.G.; Spector, G.J.; Lenox, J.; Parriott, S.; Haughey, B.; Chao, C.; Marks, J.; Perez, C. Analysis of treatment results for floor-of-mouth cancer. Laryngoscope 2000, 110, 1764–1772. [Google Scholar] [CrossRef]
- Sim, Y.C.; Hwang, J.H.; Ahn, K.M. Overall and disease-specific survival outcomes following primary surgery for oral squamous cell carcinoma: Analysis of consecutive 67 patients. J. Korean Assoc. Oral Maxillofac. Surg. 2019, 45, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Ries, L.K.G.; Hankey, B.F.; Miller, B.A.; Harras, A.; Edwards, B.K. SEER Cancer Statistics Review, 1973–1994; US Department of Health and Human Services, National Institutes of Health, Eds.; Bethesda: Rockville, MD, USA, 1997; Volume NIH publication no. 97-2789. [Google Scholar]
- Duprez, F.; Berwouts, D.; De Neve, W.; Bonte, K.; Boterberg, T.; Deron, P.; Huvenne, W.; Rottey, S.; Mareel, M. Distant metastases in head and neck cancer. Head Neck 2017, 39, 1733–1743. [Google Scholar] [CrossRef]
- Noguti, J.; De Moura, C.F.; De Jesus, G.P.; Da Silva, V.H.; Hossaka, T.A.; Oshima, C.T.; Ribeiro, D.A. Metastasis from oral cancer: An overview. Cancer Genom. Proteom. 2012, 9, 329–335. [Google Scholar]
- Okura, M.; Aikawa, T.; Sawai, N.Y.; Iida, S.; Kogo, M. Decision analysis and treatment threshold in a management for the N0 neck of the oral cavity carcinoma. Oral. Oncol. 2009, 45, 908–911. [Google Scholar] [CrossRef]
- Greenberg, J.S.; Fowler, R.; Gomez, J.; Mo, V.; Roberts, D.; El Naggar, A.K.; Myers, J.N. Extent of extracapsular spread: A critical prognosticator in oral tongue cancer. Cancer 2003, 97, 1464–1470. [Google Scholar] [CrossRef]
- Leemans, C.R.; Tiwari, R.; Nauta, J.J.; van der Waal, I.; Snow, G.B. Regional lymph node involvement and its significance in the development of distant metastases in head and neck carcinoma. Cancer 1993, 71, 452–456. [Google Scholar] [CrossRef]
- Gowen, G.F.; Desuto-Nagy, G. The incidence and sites of distant metastases in head and neck carcinoma. Surg. Gynecol. Obstet. 1963, 116, 603–607. [Google Scholar] [PubMed]
- Kotwall, C.; Sako, K.; Razack, M.S.; Rao, U.; Bakamjian, V.; Shedd, D.P. Metastatic patterns in squamous cell cancer of the head and neck. Am. J. Surg. 1987, 154, 439–442. [Google Scholar] [CrossRef]
- O’Brien, P.H.; Carlson, R.; Steubner, E.A., Jr.; Staley, C.T. Distant metastases in epidermoid cell carcinoma of the head and neck. Cancer 1971, 27, 304–307. [Google Scholar] [CrossRef]
- Adel, M.; Kao, H.K.; Hsu, C.L.; Huang, J.J.; Lee, L.Y.; Huang, Y.; Browne, T.; Tsang, N.M.; Chang, Y.L.; Chang, K.P. Evaluation of Lymphatic and Vascular Invasion in Relation to Clinicopathological Factors and Treatment Outcome in Oral Cavity Squamous Cell Carcinoma. Med. Baltim. 2015, 94, e1510. [Google Scholar] [CrossRef]
- Jardim, J.F.; Francisco, A.L.; Gondak, R.; Damascena, A.; Kowalski, L.P. Prognostic impact of perineural invasion and lymphovascular invasion in advanced stage oral squamous cell carcinoma. Int. J. Oral. Maxillofac. Surg. 2015, 44, 23–28. [Google Scholar] [CrossRef]
- Rahima, B.; Shingaki, S.; Nagata, M.; Saito, C. Prognostic significance of perineural invasion in oral and oropharyngeal carcinoma. Oral. Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2004, 97, 423–431. [Google Scholar] [CrossRef]
- Varsha, B.K.; Radhika, M.B.; Makarla, S.; Kuriakose, M.A.; Kiran, G.S.; Padmalatha, G.V. Perineural invasion in oral squamous cell carcinoma: Case series and review of literature. J. Oral Maxillofac. Pathol. 2015, 19, 335–341. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- LaFave, L.M.; Kartha, V.K.; Ma, S.; Meli, K.; Del Priore, I.; Lareau, C.; Naranjo, S.; Westcott, P.M.K.; Duarte, F.M.; Sankar, V.; et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020, 38, 212–228. [Google Scholar] [CrossRef]
- Marjanovic, N.D.; Hofree, M.; Chan, J.E.; Canner, D.; Wu, K.; Trakala, M.; Hartmann, G.G.; Smith, O.C.; Kim, J.Y.; Evans, K.V.; et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020, 38, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Jayanthi, P.; Varun, B.R.; Selvaraj, J. Epithelial-mesenchymal transition in oral squamous cell carcinoma: An insight into molecular mechanisms and clinical implications. J. Oral Maxillofac. Pathol. 2020, 24, 189. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Campbell, K.; Casanova, J. A common framework for EMT and collective cell migration. Development 2016, 143, 4291–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, L.A.; Abella, V.; Valladares, M.; Figueroa, A. Posttranscriptional regulation by RNA-binding proteins during epithelial-to-mesenchymal transition. Cell Mol. Life Sci. 2013, 70, 4463–4477. [Google Scholar] [CrossRef] [Green Version]
- Bebee, T.W.; Cieply, B.W.; Carstens, R.P. Genome-wide activities of RNA binding proteins that regulate cellular changes in the epithelial to mesenchymal transition (EMT). Adv. Exp. Med. Biol. 2014, 825, 267–302. [Google Scholar] [CrossRef]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Dhamija, S.; Diederichs, S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int. J. Cancer 2016, 139, 269–280. [Google Scholar] [CrossRef]
- Dorn, A.; Glass, M.; Neu, C.T.; Heydel, B.; Huttelmaier, S.; Gutschner, T.; Haemmerle, M. LINC00261 Is Differentially Expressed in Pancreatic Cancer Subtypes and Regulates a Pro-Epithelial Cell Identity. Cancers Basel 2020, 12, 1227. [Google Scholar] [CrossRef]
- Lai, X.N.; Li, J.; Tang, L.B.; Chen, W.T.; Zhang, L.; Xiong, L.X. MiRNAs and LncRNAs: Dual Roles in TGF-beta Signaling-Regulated Metastasis in Lung Cancer. Int. J. Mol. Sci. 2020, 21, 1193. [Google Scholar] [CrossRef]
- Di Stefano, B.; Luo, E.C.; Haggerty, C.; Aigner, S.; Charlton, J.; Brumbaugh, J.; Ji, F.; Rabano Jimenez, I.; Clowers, K.J.; Huebner, A.J.; et al. The RNA Helicase DDX6 Controls Cellular Plasticity by Modulating P-Body Homeostasis. Cell Stem Cell 2019, 25, 622–638. [Google Scholar] [CrossRef] [Green Version]
- Hodson, D.J.; Screen, M.; Turner, M. RNA-binding proteins in hematopoiesis and hematological malignancy. Blood 2019, 133, 2365–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.; Diaz-Munoz, M.D. RNA-binding proteins control gene expression and cell fate in the immune system. Nat. Immunol. 2018, 19, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jung, Y.; Hyun, J.; Friedersdorf, M.; Oh, S.H.; Kim, J.; Premont, R.T.; Keene, J.D.; Diehl, A.M. RNA Binding Proteins Control Transdifferentiation of Hepatic Stellate Cells into Myofibroblasts. Cell Physiol. Biochem. 2018, 48, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Pazaitis, N.; Bley, N.; Fiskin, E.; Uckelmann, H.; Heim, A.; Grobeta, M.; Hofmann, N.; Geffers, R.; et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology 2014, 59, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Gutschner, T.; Reches, A.; Diederichs, S.; Mandelboim, O. RNA-binding proteins regulate the expression of the immune activating ligand MICB. Nat. Commun. 2014, 5, 4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, S.; Bley, N.; Busch, B.; Glass, M.; Lederer, M.; Misiak, C.; Fuchs, T.; Wedler, A.; Haase, J.; Bertoldo, J.B.; et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020, 48, 8576–8590. [Google Scholar] [CrossRef]
- Muller, S.; Bley, N.; Glass, M.; Busch, B.; Rousseau, V.; Misiak, D.; Fuchs, T.; Lederer, M.; Huttelmaier, S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018, 46, 6285–6303. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [Green Version]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.C.; Yi, H.; Eichelbaum, K.; Fohr, S.; Fischer, B.; You, K.T.; Castello, A.; Krijgsveld, J.; Hentze, M.W.; Kim, V.N. The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvelin, A.I.; Noerenberg, M.; Davis, I.; Castello, A. The new (dis)order in RNA regulation. Cell Commun. Signal. 2016, 14, 9. [Google Scholar] [CrossRef] [Green Version]
- Castello, A.; Hentze, M.W.; Preiss, T. Metabolic Enzymes Enjoying New Partnerships as RNA-Binding Proteins. Trends Endocrinol. Metab. 2015, 26, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Brannan, K.W.; Jin, W.; Huelga, S.C.; Banks, C.A.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Van Nostrand, E.L.; Pratt, G.A.; Schwinn, M.K.; et al. SONAR Discovers RNA-Binding Proteins from Analysis of Large-Scale Protein-Protein Interactomes. Mol. Cell 2016, 64, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, C.O.; Friedersdorf, M.; Keene, J.D. Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA 2017, 23, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Hammerle, M.; Gutschner, T.; Uckelmann, H.; Ozgur, S.; Fiskin, E.; Gross, M.; Skawran, B.; Geffers, R.; Longerich, T.; Breuhahn, K.; et al. Posttranscriptional destabilization of the liver-specific long noncoding RNA HULC by the IGF2 mRNA-binding protein 1 (IGF2BP1). Hepatology 2013, 58, 1703–1712. [Google Scholar] [CrossRef]
- Hu, W.L.; Jin, L.; Xu, A.; Wang, Y.F.; Thorne, R.F.; Zhang, X.D.; Wu, M. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat. Cell Biol. 2018, 20, 492–502. [Google Scholar] [CrossRef]
- Jonas, K.; Calin, G.A.; Pichler, M. RNA-Binding Proteins as Important Regulators of Long Non-Coding RNAs in Cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P.; et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017, 24, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, A.M.; Garcia, J.T.; Hung, T.; Flynn, R.A.; Shen, Y.; Qu, K.; Payumo, A.Y.; Peres-da-Silva, A.; Broz, D.K.; Baum, R.; et al. An inducible long noncoding RNA amplifies DNA damage signaling. Nat. Genet. 2016, 48, 1370–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Nussbacher, J.K.; Yeo, G.W. Systematic Discovery of RNA Binding Proteins that Regulate MicroRNA Levels. Mol. Cell 2018, 69, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Treiber, T.; Treiber, N.; Meister, G. Identification of microRNA Precursor-Associated Proteins. Methods Mol. Biol. 2018, 1823, 103–114. [Google Scholar] [CrossRef]
- Busch, B.; Bley, N.; Muller, S.; Glass, M.; Misiak, D.; Lederer, M.; Vetter, M.; Strauss, H.G.; Thomssen, C.; Huttelmaier, S. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016, 44, 3845–3864. [Google Scholar] [CrossRef]
- Jing, Q.; Huang, S.; Guth, S.; Zarubin, T.; Motoyama, A.; Chen, J.; Di Padova, F.; Lin, S.C.; Gram, H.; Han, J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 2005, 120, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiber, A.; Doring, K.; Friedrich, U.; Klann, K.; Merker, D.; Zedan, M.; Tippmann, F.; Kramer, G.; Bukau, B. Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature 2018, 561, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C. Protein complexes assemble as they are being made. Nature 2018, 561, 186–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Mayr, C. Gain of Additional BIRC3 Protein Functions through 3′-UTR-Mediated Protein Complex Formation. Mol. Cell 2019, 74, 701–712. [Google Scholar] [CrossRef]
- Ma, W.; Mayr, C. A Membraneless Organelle Associated with the Endoplasmic Reticulum Enables 3′UTR-Mediated Protein-Protein Interactions. Cell 2018, 175, 1492–1506. [Google Scholar] [CrossRef] [Green Version]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.L.; Hagemann, S.; Holien, J.K.; Liu, T.; Nagy, Z.; Schulte, J.H.; Misiak, D.; Huttelmaier, S. Identification of RNA-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma. Int. J. Mol. Sci. 2020, 21, 5098. [Google Scholar] [CrossRef]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef]
- Glass, M.; Michl, P.; Huttelmaier, A.S. RNA Binding Proteins as Drivers and Therapeutic Target Candidates in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 4190. [Google Scholar] [CrossRef]
- Kechavarzi, B.; Janga, S.C. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014, 15, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libner, C.D.; Salapa, H.E.; Levin, M.C. The Potential Contribution of Dysfunctional RNA-Binding Proteins to the Pathogenesis of Neurodegeneration in Multiple Sclerosis and Relevant Models. Int. J. Mol. Sci 2020, 21, 4571. [Google Scholar] [CrossRef] [PubMed]
- Salem, E.S.B.; Vonberg, A.D.; Borra, V.J.; Gill, R.K.; Nakamura, T. RNAs and RNA-Binding Proteins in Immuno-Metabolic Homeostasis and Diseases. Front. Cardiovasc. Med. 2019, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Schuschel, K.; Helwig, M.; Huttelmaier, S.; Heckl, D.; Klusmann, J.H.; Hoell, J.I. RNA-Binding Proteins in Acute Leukemias. Int. J. Mol. Sci. 2020, 21, 3409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Brown, R.D.; Stenmark, K.R.; Hu, C.J. RNA-Binding Proteins in Pulmonary Hypertension. Int. J. Mol. Sci. 2020, 21, 3757. [Google Scholar] [CrossRef]
- Fritzell, K.; Xu, L.D.; Lagergren, J.; Ohman, M. ADARs and editing: The role of A-to-I RNA modification in cancer progression. Semin. Cell Dev. Biol. 2018, 79, 123–130. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Song, Y.; Shi, X.; Liu, J.; Xiong, S.; Chen, W.; Fu, Q.; Huang, Z.; Gu, N.; Zhang, R. The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res. 2018, 28, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Nemlich, Y.; Greenberg, E.; Ortenberg, R.; Besser, M.J.; Barshack, I.; Jacob-Hirsch, J.; Jacoby, E.; Eyal, E.; Rivkin, L.; Prieto, V.G.; et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J. Clin. Investig. 2013, 123, 2703–2718. [Google Scholar] [CrossRef] [Green Version]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Fu, Y.; Huang, J.; Wu, M.; Zhang, Z.; Xu, R.; Zhang, P.; Zhao, S.; Liu, L.; Jiang, H. ADAR1 promotes the epithelial-to-mesenchymal transition and stem-like cell phenotype of oral cancer by facilitating oncogenic microRNA maturation. J. Exp. Clin. Cancer Res. 2019, 38, 315. [Google Scholar] [CrossRef] [PubMed]
- Caponio, V.C.A.; Troiano, G.; Botti, G.; Pedicillo, M.C.; Lo Russo, L.; Mastrangelo, F.; Ciavarella, D.; Losito, N.S.; Aquino, G.; Nocini, R.; et al. Overexpression of ADAR1 into the cytoplasm correlates with a better prognosis of patients with oral squamous cells carcinoma. J. Oral Pathol. Med. 2019, 48, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front. Genet. 2014, 5, 423. [Google Scholar] [CrossRef] [Green Version]
- Kukhanova, M.K.; Karpenko, I.L.; Ivanov, A.V. DEAD-box RNA Helicase DDX3: Functional Properties and Development of DDX3 Inhibitors as Antiviral and Anticancer Drugs. Mol. Basel Switz. 2020, 25, 1015. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Yu, H.I.; Tarn, W.Y. DDX3 Modulates Neurite Development via Translationally Activating an RNA Regulon Involved in Rac1 Activation. J. Neurosci. 2016, 36, 9792–9804. [Google Scholar] [CrossRef]
- Chen, H.H.; Yu, H.I.; Yang, M.H.; Tarn, W.Y. DDX3 Activates CBC-eIF3-Mediated Translation of uORF-Containing Oncogenic mRNAs to Promote Metastasis in HNSCC. Cancer Res. 2018, 78, 4512–4523. [Google Scholar] [CrossRef] [Green Version]
- Soto-Rifo, R.; Rubilar, P.S.; Limousin, T.; de Breyne, S.; Decimo, D.; Ohlmann, T. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J. 2012, 31, 3745–3756. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Dias, A.P.; Jedrychowski, M.; Patel, A.H.; Hsu, J.L.; Reed, R. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 2008, 36, 4708–4718. [Google Scholar] [CrossRef] [Green Version]
- Shih, J.W.; Tsai, T.Y.; Chao, C.H.; Wu Lee, Y.H. Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene 2008, 27, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Bol, G.M.; Xie, M.; Raman, V. DDX3, a potential target for cancer treatment. Mol. Cancer 2015, 14, 188. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Yu, H.I.; Cho, W.C.; Tarn, W.Y. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene 2015, 34, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Good, P.J. A conserved family of elav-like genes in vertebrates. Proc. Natl. Acad. Sci. USA 1995, 92, 4557–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, C.W.; Preet, R.; Dhir, T.; Dixon, D.A.; Brody, J.R. Understanding and targeting the disease-related RNA binding protein human antigen R (HuR). Wiley Interdiscip. Rev. RNA 2020, 11, e1581. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Elemento, O.; Zhang, J.; Zhuang, Z.W.; Simons, M.; Hla, T. ELAVL1 regulates alternative splicing of eIF4E transporter to promote postnatal angiogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 18309–18314. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, M.; Chakrama, F.Z.; Combe, E.; Desmet, F.O.; Mortada, H.; Espinoza, M.P.; Gratadou, L.; Auboeuf, D. A recently evolved class of alternative 3′-terminal exons involved in cell cycle regulation by topoisomerase inhibitors. Nat. Commun. 2014, 5, 3395. [Google Scholar] [CrossRef]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhausser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [Green Version]
- Ripin, N.; Boudet, J.; Duszczyk, M.M.; Hinniger, A.; Faller, M.; Krepl, M.; Gadi, A.; Schneider, R.J.; Sponer, J.; Meisner-Kober, N.C.; et al. Molecular basis for AU-rich element recognition and dimerization by the HuR C-terminal RRM. Proc. Natl. Acad. Sci. USA 2019, 116, 2935–2944. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.C.; Steitz, J.A. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef] [Green Version]
- Dixon, D.A.; Tolley, N.D.; King, P.H.; Nabors, L.B.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Investig. 2001, 108, 1657–1665. [Google Scholar] [CrossRef]
- Nabors, L.B.; Gillespie, G.Y.; Harkins, L.; King, P.H. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001, 61, 2154–2161. [Google Scholar] [PubMed]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. Landmark Ed. 2012, 17, 189–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Furuuchi, N.; Aslanukova, L.; Huang, Y.H.; Brown, S.Z.; Jiang, W.; Addya, S.; Vishwakarma, V.; Peters, E.; Brody, J.R.; et al. Elevated HuR in Pancreas Promotes a Pancreatitis-Like Inflammatory Microenvironment That Facilitates Tumor Development. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, H.; Kakuguchi, W.; Kuroshima, T.; Kitamura, T.; Tanaka, S.; Kitagawa, Y.; Totsuka, Y.; Shindoh, M.; Higashino, F. HuR is exported to the cytoplasm in oral cancer cells in a different manner from that of normal cells. Br. J. Cancer 2009, 100, 1943–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakuguchi, W.; Kitamura, T.; Kuroshima, T.; Ishikawa, M.; Kitagawa, Y.; Totsuka, Y.; Shindoh, M.; Higashino, F. HuR knockdown changes the oncogenic potential of oral cancer cells. Mol. Cancer Res. 2010, 8, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.D.; Li, S.; Cha, I.H. Association between expression of embryonic lethal abnormal vision-like protein HuR and cyclooxygenase-2 in oral squamous cell carcinoma. Head Neck 2011, 33, 627–637. [Google Scholar] [CrossRef]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Cherry, S.; Lynch, K.W. Alternative splicing and cancer: Insights, opportunities, and challenges from an expanding view of the transcriptome. Genes Dev. 2020, 34, 1005–1016. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Fiszbein, A.; Kornblihtt, A.R. Alternative splicing switches: Important players in cell differentiation. Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Jiang, P.; Amirikian, K.; Dittmar, K.A.; Lu, H.; Shen, S.; Guo, W.; Xing, Y.; Carstens, R.P. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 2010, 29, 3286–3300. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Sato, T.K.; Nabet, B.; Hogenesch, J.B.; Carstens, R.P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 2009, 33, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Warzecha, C.C.; Shen, S.; Xing, Y.; Carstens, R.P. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009, 6, 546–562. [Google Scholar] [CrossRef] [Green Version]
- Shirakihara, T.; Horiguchi, K.; Miyazawa, K.; Ehata, S.; Shibata, T.; Morita, I.; Miyazono, K.; Saitoh, M. TGF-beta regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J. 2011, 30, 783–795. [Google Scholar] [CrossRef]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Ionov, Y. RNA-binding motif protein 35A is a novel tumor suppressor for colorectal cancer. Cell Cycle 2009, 8, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef] [Green Version]
- Ishii, H.; Saitoh, M.; Sakamoto, K.; Kondo, T.; Katoh, R.; Tanaka, S.; Motizuki, M.; Masuyama, K.; Miyazawa, K. Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J. Biol. Chem. 2014, 289, 27386–27399. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Cui, Y.; Liu, L.; Qi, X.; Liu, J.; Ma, S.; Hu, X.; Zhang, Z.; Wang, Y.; Li, H.; et al. Splicing factor derived circular RNA circUHRF1 accelerates oral squamous cell carcinoma tumorigenesis via feedback loop. Cell Death Differ. 2020, 27, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.L.; Wachter, K.; Muhleck, B.; Pazaitis, N.; Kohn, M.; Lederer, M.; Huttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [Green Version]
- Wachter, K.; Kohn, M.; Stohr, N.; Huttelmaier, S. Subcellular localization and RNP formation of IGF2BPs (IGF2 mRNA-binding proteins) is modulated by distinct RNA-binding domains. Biol. Chem. 2013, 394, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Lederer, M.; Bley, N.; Schleifer, C.; Huttelmaier, S. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin. Cancer Biol. 2014, 29, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Clauditz, T.S.; Wang, C.J.; Gontarewicz, A.; Blessmann, M.; Tennstedt, P.; Borgmann, K.; Tribius, S.; Sauter, G.; Dalchow, C.; Knecht, R.; et al. Expression of insulin-like growth factor II mRNA-binding protein 3 in squamous cell carcinomas of the head and neck. J. Oral Pathol. Med. 2013, 42, 125–132. [Google Scholar] [CrossRef]
- Hwang, Y.S.; Xianglan, Z.; Park, K.K.; Chung, W.Y. Functional invadopodia formation through stabilization of the PDPN transcript by IMP-3 and cancer-stromal crosstalk for PDPN expression. Carcinogenesis 2012, 33, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.Y.; Cha, I.H. A novel algorithm for lymph node status prediction of oral cancer before surgery. Oral Oncol. 2011, 47, 1069–1073. [Google Scholar] [CrossRef]
- Kim, K.Y.; Cha, I.H. Risk stratification of oral cancer patients using a combined prognostic factor including lymph node density and biomarker. J. Cancer Res. Clin. Oncol. 2012, 138, 483–490. [Google Scholar] [CrossRef]
- Kim, K.Y.; Li, S.; Cha, J.D.; Zhang, X.; Cha, I.H. Significance of molecular markers in survival prediction of oral squamous cell carcinoma. Head Neck 2012, 34, 929–936. [Google Scholar] [CrossRef]
- Li, S.; Cha, J.; Kim, J.; Kim, K.Y.; Kim, H.J.; Nam, W.; Cha, I.H. Insulin-like growth factor II mRNA-binding protein 3: A novel prognostic biomarker for oral squamous cell carcinoma. Head Neck 2011, 33, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Chen, S.T.; Jeng, Y.M.; Yeh, C.C.; Chou, H.Y.; Deng, Y.T.; Chang, C.C.; Kuo, M.Y. Insulin-like growth factor II mRNA-binding protein 3 expression promotes tumor formation and invasion and predicts poor prognosis in oral squamous cell carcinoma. J. Oral Pathol. Med. 2011, 40, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.S.; Park, K.K.; Cha, I.H.; Kim, J.; Chung, W.Y. Role of insulin-like growth factor-II mRNA-binding protein-3 in invadopodia formation and the growth of oral squamous cell carcinoma in athymic nude mice. Head Neck 2012, 34, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.S.; Ahn, S.Y.; Moon, S.; Zheng, Z.; Cha, I.H.; Kim, J.; Zhang, X. Insulin-like growth factor-II mRNA binding protein-3 and podoplanin expression are associated with bone invasion and prognosis in oral squamous cell carcinoma. Arch. Oral Biol. 2016, 69, 25–32. [Google Scholar] [CrossRef]
- Tarsitano, A.; Asioli, S.; Morandi, L.; Monti, V.; Righi, A.; Labate, A.M.M.; Nardi, E.; Foschini, M.P.; Marchetti, C. Laminin-5 and insulin-like growth factor-II mRNA binding protein-3 (IMP3) expression in preoperative biopsy specimens from oral cancer patients: Their role in neural spread risk and survival stratification. J. Cranio-Maxillofac. Surg. 2016, 44, 1896–1902. [Google Scholar] [CrossRef]
- Zhang, X.; Jung, I.H.; Hwang, Y.S. EGF enhances low-invasive cancer cell invasion by promoting IMP-3 expression. Tumour Biol. 2016, 37, 2555–2563. [Google Scholar] [CrossRef]
- Moss, E.G.; Lee, R.C.; Ambros, V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell 1997, 88, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, H.; Wu, T. LIN28: A cancer stem cell promoter for immunotherapy in head and neck squamous cell carcinoma. Oral Oncol. 2019, 98, 92–95. [Google Scholar] [CrossRef]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; LaPierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Jia, J.; Xiong, X.; He, H.; Bu, L.; Zhao, Z.; Huang, C.; Zhang, W. Increased expression of Lin28B associates with poor prognosis in patients with oral squamous cell carcinoma. PLoS ONE 2013, 8, e83869. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhu, Y.; Wang, Y.; Li, Z.; Yuan, C.; Zhang, W.; Yuan, H.; Ye, J.; Yang, J.; Jiang, H.; et al. The pluripotency factor LIN28B is involved in oral carcinogenesis and associates with tumor aggressiveness and unfavorable prognosis. Cancer Cell Int. 2015, 15, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.T.; Shieh, T.M.; Yang, L.C.; Wang, T.Y.; Chou, M.Y.; Yu, C.C. Elevated Lin28B expression is correlated with lymph node metastasis in oral squamous cell carcinomas. J. Oral Pathol. Med. 2015, 44, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.S.; Wang, M.L.; Chu, P.Y.; Chang, Y.L.; Liu, W.H.; Yu, C.C.; Lan, Y.T.; Huang, P.I.; Lee, Y.Y.; Chen, Y.W.; et al. Lin28B/Let-7 Regulates Expression of Oct4 and Sox2 and Reprograms Oral Squamous Cell Carcinoma Cells to a Stem-like State. Cancer Res. 2015, 75, 2553–2565. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Weng, H.; Chen, J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Jacob, R.; Zander, S.; Gutschner, T. The Dark Side of the Epitranscriptome: Chemical Modifications in Long Non-Coding RNAs. Int. J. Mol. Sci. 2017, 18, 2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; He, C. Dynamic RNA modifications in posttranscriptional regulation. Mol. Cell 2014, 56, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Chen, J.; Fang, X.; Zhong, P.; Song, Z.; Hu, X. N6-methyladenosine modifications: Interactions with novel RNA-binding proteins and roles in signal transduction. RNA Biol. 2019, 16, 991–1000. [Google Scholar] [CrossRef]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Sledz, P.; Jinek, M. Structural insights into the molecular mechanism of the m(6)A writer complex. eLife 2016, 5, e18434. [Google Scholar] [CrossRef]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C.; et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.Z.; Wu, Q.Q.; Zheng, Z.N.; Shao, T.R.; Chen, Y.C.; Zeng, W.S.; Lv, X.Z. M6A-related bioinformatics analysis reveals that HNRNPC facilitates progression of OSCC via EMT. Aging Albany N. Y. 2020, 12, 11667–11684. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Y.; Li, Q.; Liang, J.; He, Q.; Zhao, L.; Chen, J.; Cheng, M.; Huang, Z.; Ren, H.; et al. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 m(6)A Methylation in Oral Squamous Cell Carcinoma. Mol. Ther. 2020, in press. [Google Scholar] [CrossRef]
- Zhao, W.; Cui, Y.; Liu, L.; Ma, X.; Qi, X.; Wang, Y.; Liu, Z.; Ma, S.; Liu, J.; Wu, J. METTL3 Facilitates Oral Squamous Cell Carcinoma Tumorigenesis by Enhancing c-Myc Stability via YTHDF1-Mediated m(6)A Modification. Mol. Ther. Nucleic Acids 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Chen, D.; Wu, M.; Li, Y.; Chang, I.; Yuan, Q.; Ekimyan-Salvo, M.; Deng, P.; Yu, B.; Yu, Y.; Dong, J.; et al. Targeting BMI1(+) Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell 2017, 20, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Sasahira, T.; Kurihara, M.; Yamamoto, K.; Ueda, N.; Nakashima, C.; Matsushima, S.; Bhawal, U.K.; Kirita, T.; Kuniyasu, H. HuD promotes progression of oral squamous cell carcinoma. Pathobiology 2014, 81, 206–214. [Google Scholar] [CrossRef]
- Jia, R.; Zhang, S.; Liu, M.; Zhang, Y.; Liu, Y.; Fan, M.; Guo, J. HnRNP L is important for the expression of oncogene SRSF3 and oncogenic potential of oral squamous cell carcinoma cells. Sci. Rep. 2016, 6, 35976. [Google Scholar] [CrossRef]
- Ravindran, G.; Devaraj, H. Prognostic significance of neural stem cell markers, Nestin and Musashi-1, in oral squamous cell carcinoma: Expression pattern of Nestin in the precancerous stages of oral squamous epithelium. Clin. Oral Investig. 2015, 19, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.F.; Zhang, H.C.; Feng, X.M.; Song, X.M.; Wu, Y.N. Knockdown of MSI1 inhibits the proliferation of human oral squamous cell carcinoma by inactivating STAT3 signaling. Int. J. Mol. Med. 2019, 44, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Feng, F.; Xu, J.; Lu, X.; Wang, S.; Wang, L.; Lu, H.; Wei, M.; Yang, G.; Wang, L.; et al. QKI impairs self-renewal and tumorigenicity of oral cancer cells via repression of SOX2. Cancer Biol. Ther. 2014, 15, 1174–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.J.; Kim, J.S.; Lee, S.; Lee, H.; Yoon, J.S.; Hong, J.H.; Chun, S.H.; Sun, S.; Won, H.S.; Hong, S.A.; et al. QKI, a miR-200 target gene, suppresses epithelial-to-mesenchymal transition and tumor growth. Int. J. Cancer 2019, 145, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Guo, Z.; Yin, Y.; Jia, J.; Guo, J.; Jia, R. Expression of SRSF3 is Correlated with Carcinogenesis and Progression of Oral Squamous Cell Carcinoma. Int. J. Med. Sci. 2016, 13, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Sommer, G.; Rossa, C.; Chi, A.C.; Neville, B.W.; Heise, T. Implication of RNA-binding protein La in proliferation, migration and invasion of lymph node-metastasized hypopharyngeal SCC cells. PLoS ONE 2011, 6, e25402. [Google Scholar] [CrossRef]
- Zhu, S.; Rooney, S.; Michlewski, G. RNA-Targeted Therapies and High-Throughput Screening Methods. Int. J. Mol. Sci. 2020, 21, 2996. [Google Scholar] [CrossRef]
- Mohibi, S.; Chen, X.; Zhang, J. Cancer the ‘RBP’eutics-RNA-binding proteins as therapeutic targets for cancer. Pharmacol. Ther. 2019, 203, 107390. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Veliz, E.A.; Easterwood, L.M.; Beal, P.A. Substrate analogues for an RNA-editing adenosine deaminase: Mechanistic investigation and inhibitor design. J. Am. Chem. Soc. 2003, 125, 10867–10876. [Google Scholar] [CrossRef]
- Bol, G.M.; Vesuna, F.; Xie, M.; Zeng, J.; Aziz, K.; Gandhi, N.; Levine, A.; Irving, A.; Korz, D.; Tantravedi, S.; et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 2015, 7, 648–669. [Google Scholar] [CrossRef] [PubMed]
- Kondaskar, A.; Kondaskar, S.; Kumar, R.; Fishbein, J.C.; Muvarak, N.; Lapidus, R.G.; Sadowska, M.; Edelman, M.J.; Bol, G.M.; Vesuna, F.; et al. Novel, Broad Spectrum Anti-Cancer Agents Containing the Tricyclic 5:7:5-Fused Diimidazodiazepine Ring System. ACS Med. Chem. Lett. 2010, 2, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Vesuna, F.; Tantravedi, S.; Bol, G.M.; van Voss, M.R.H.; Nugent, K.; Malek, R.; Gabrielson, K.; van Diest, P.J.; Tran, P.T.; et al. RK-33 Radiosensitizes Prostate Cancer Cells by Blocking the RNA Helicase DDX3. Cancer Res. 2016, 76, 6340–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Vesuna, F.; Botlagunta, M.; Bol, G.M.; Irving, A.; Bergman, Y.; Hosmane, R.S.; Kato, Y.; Winnard, P.T., Jr.; Raman, V. NZ51, a ring-expanded nucleoside analog, inhibits motility and viability of breast cancer cells by targeting the RNA helicase DDX3. Oncotarget 2015, 6, 29901–29913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yedavalli, V.S.; Zhang, N.; Cai, H.; Zhang, P.; Starost, M.F.; Hosmane, R.S.; Jeang, K.T. Ring expanded nucleoside analogues inhibit RNA helicase and intracellular human immunodeficiency virus type 1 replication. J. Med. Chem. 2008, 51, 5043–5051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maga, G.; Falchi, F.; Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Irannejad, H.; Manetti, F.; Garbelli, A.; Samuele, A.; et al. Toward the discovery of novel anti-HIV drugs. Second-generation inhibitors of the cellular ATPase DDX3 with improved anti-HIV activity: Synthesis, structure-activity relationship analysis, cytotoxicity studies, and target validation. ChemMedChem 2011, 6, 1371–1389. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Falchi, F.; Garbelli, A.; Belfiore, A.; Witvrouw, M.; Manetti, F.; Botta, M. Pharmacophore modeling and molecular docking led to the discovery of inhibitors of human immunodeficiency virus-1 replication targeting the human cellular aspartic acid-glutamic acid-alanine-aspartic acid box polypeptide 3. J. Med. Chem. 2008, 51, 6635–6638. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Falchi, F.; Garbelli, A.; Samuele, A.; Bernardo, V.; Paolucci, S.; Baldanti, F.; Schenone, S.; Manetti, F.; Maga, G.; et al. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: Towards the next generation HIV-1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2094–2098. [Google Scholar] [CrossRef]
- Samal, S.K.; Routray, S.; Veeramachaneni, G.K.; Dash, R.; Botlagunta, M. Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci. Rep. 2015, 5, 9982. [Google Scholar] [CrossRef]
- Meisner, N.C.; Hintersteiner, M.; Mueller, K.; Bauer, R.; Seifert, J.M.; Naegeli, H.U.; Ottl, J.; Oberer, L.; Guenat, C.; Moss, S.; et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat. Chem. Biol. 2007, 3, 508–515. [Google Scholar] [CrossRef]
- Chae, M.J.; Sung, H.Y.; Kim, E.H.; Lee, M.; Kwak, H.; Chae, C.H.; Kim, S.; Park, W.Y. Chemical inhibitors destabilize HuR binding to the AU-rich element of TNF-alpha mRNA. Exp. Mol. Med. 2009, 41, 824–831. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, V.G.; Adami, V.; Provenzani, A. A novel high throughput biochemical assay to evaluate the HuR protein-RNA complex formation. PLoS ONE 2013, 8, e72426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Agostino, V.G.; Lal, P.; Mantelli, B.; Tiedje, C.; Zucal, C.; Thongon, N.; Gaestel, M.; Latorre, E.; Marinelli, L.; Seneci, P.; et al. Dihydrotanshinone-I interferes with the RNA-binding activity of HuR affecting its post-transcriptional function. Sci. Rep. 2015, 5, 16478. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bhattacharya, A.; Ivanov, D.N. Identification of Small-Molecule Inhibitors of the HuR/RNA Interaction Using a Fluorescence Polarization Screening Assay Followed by NMR Validation. PLoS ONE 2015, 10, e0138780. [Google Scholar] [CrossRef]
- Wu, X.; Lan, L.; Wilson, D.M.; Marquez, R.T.; Tsao, W.C.; Gao, P.; Roy, A.; Turner, B.A.; McDonald, P.; Tunge, J.A.; et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem. Biol. 2015, 10, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Wu, X.; Fields, J.K.; Johnson, D.K.; Lan, L.; Pratt, M.; Somoza, A.D.; Wang, C.C.C.; Karanicolas, J.; Oakley, B.R.; et al. The fungal natural product azaphilone-9 binds to HuR and inhibits HuR-RNA interaction in vitro. PLoS ONE 2017, 12, e0175471. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, L.; Ge, C.; Chen, L.; Fang, T.; Li, H.; Tian, H.; Liu, J.; Chen, T.; Jiang, G.; et al. An isocorydine derivative (d-ICD) inhibits drug resistance by downregulating IGF2BP3 expression in hepatocellular carcinoma. Oncotarget 2015, 6, 25149–25160. [Google Scholar] [CrossRef] [Green Version]
- Elagib, K.E.; Lu, C.H.; Mosoyan, G.; Khalil, S.; Zasadzinska, E.; Foltz, D.R.; Balogh, P.; Gru, A.A.; Fuchs, D.A.; Rimsza, L.M.; et al. Neonatal expression of RNA-binding protein IGF2BP3 regulates the human fetal-adult megakaryocyte transition. J. Clin. Investig. 2017, 127, 2365–2377. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, C.; Pasello, M.; Ventura, S.; Grilli, A.; Calzolari, L.; Toracchio, L.; Lollini, P.L.; Donati, D.M.; Picci, P.; Ferrari, S.; et al. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 is a Novel Post-Transcriptional Regulator of Ewing Sarcoma Malignancy. Clin. Cancer Res. 2018, 24, 3704–3716. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.; Byun, W.G.; Koo, J.Y.; Park, H.; Park, S.B. Discovery of a Small-Molecule Inhibitor of Protein-MicroRNA Interaction Using Binding Assay with a Site-Specifically Labeled Lin28. J. Am. Chem. Soc. 2016, 138, 13630–13638. [Google Scholar] [CrossRef]
- Roos, M.; Pradere, U.; Ngondo, R.P.; Behera, A.; Allegrini, S.; Civenni, G.; Zagalak, J.A.; Marchand, J.R.; Menzi, M.; Towbin, H.; et al. A Small-Molecule Inhibitor of Lin28. ACS Chem. Biol. 2016, 11, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.L.; Miska, E.A.; Balasubramanian, S. Identification of small molecule inhibitors of the Lin28-mediated blockage of pre-let-7g processing. Org. Biomol. Chem. 2016, 14, 10208–10216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Rowe, R.G.; Jaimes, A.; Yu, C.; Nam, Y.; Pearson, D.S.; Zhang, J.; Xie, X.; Marion, W.; Heffron, G.J.; et al. Small-Molecule Inhibitors Disrupt let-7 Oligouridylation and Release the Selective Blockade of let-7 Processing by LIN28. Cell Rep. 2018, 23, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Byun, W.G.; Lim, D.; Park, S.B. Discovery of Small-Molecule Modulators of Protein-RNA Interactions by Fluorescence Intensity-Based Binding Assay. ChemBioChem 2020, 21, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, D.A.; Kaur, T.; Kerk, S.A.; Gallagher, E.E.; Sandoval, J.; Garner, A.L. Expansion of cat-ELCCA for the Discovery of Small Molecule Inhibitors of the Pre-let-7-Lin28 RNA-Protein Interaction. ACS Med. Chem. Lett. 2018, 9, 517–521. [Google Scholar] [CrossRef]
- Bedi, R.K.; Huang, D.; Eberle, S.A.; Wiedmer, L.; Sledz, P.; Caflisch, A. Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. ChemMedChem 2020, 15, 744–748. [Google Scholar] [CrossRef]
- Minuesa, G.; Antczak, C.; Shum, D.; Radu, C.; Bhinder, B.; Li, Y.; Djaballah, H.; Kharas, M.G. A 1536-well fluorescence polarization assay to screen for modulators of the MUSASHI family of RNA-binding proteins. Comb. Chem. High Throughput Screen. 2014, 17, 596–609. [Google Scholar] [CrossRef]
- Minuesa, G.; Albanese, S.K.; Xie, W.; Kazansky, Y.; Worroll, D.; Chow, A.; Schurer, A.; Park, S.M.; Rotsides, C.Z.; Taggart, J.; et al. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nat. Commun. 2019, 10, 2691. [Google Scholar] [CrossRef] [Green Version]
- Clingman, C.C.; Deveau, L.M.; Hay, S.A.; Genga, R.M.; Shandilya, S.M.; Massi, F.; Ryder, S.P. Allosteric inhibition of a stem cell RNA-binding protein by an intermediary metabolite. eLife 2014, 3, e02848. [Google Scholar] [CrossRef]
- Lan, L.; Appelman, C.; Smith, A.R.; Yu, J.; Larsen, S.; Marquez, R.T.; Liu, H.; Wu, X.; Gao, P.; Roy, A.; et al. Natural product (-)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol. Oncol. 2015, 9, 1406–1420. [Google Scholar] [CrossRef]
- Tang, J.; Huang, Z.M.; Chen, Y.Y.; Zhang, Z.H.; Liu, G.L.; Zhang, J. A novel inhibitor of human La protein with anti-HBV activity discovered by structure-based virtual screening and in vitro evaluation. PLoS ONE 2012, 7, e36363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Das, M.; Sauceda, C.; Ellies, L.G.; Kuo, K.; Parwal, P.; Kaur, M.; Jih, L.; Bandyopadhyay, G.K.; Burton, D.; et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J. Clin. Investig. 2019, 129, 4477–4491. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.D.; MacCoss, M.; Lawson, A.D. Rings in drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, H.; Liu, T.; Zhou, S.; Du, Y.; Xiong, J.; Yi, S.; Qu, C.K.; Fu, H.; Zhou, M. Discovery of Dual Inhibitors of MDM2 and XIAP for Cancer Treatment. Cancer Cell 2016, 30, 623–636. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lai, H.; Fan, X.; Luo, L.; Duan, F.; Jiang, Z.; Wang, Q.; Leung, E.L.H.; Liu, L.; Yao, X. Gossypol Inhibits Non-small Cell Lung Cancer Cells Proliferation by Targeting EGFR(L858R/T790M). Front. Pharmacol. 2018, 9, 728. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Li, J.; Yang, Q.; Wang, J.; Su, T.; Zhou, S. Gossypol has anti-cancer effects by dual-targeting MDM2 and VEGF in human breast cancer. Breast Cancer Res. 2017, 19, 27. [Google Scholar] [CrossRef] [Green Version]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Huang, H.; Zeqiraj, E.; Dong, B.; Jha, B.K.; Duffy, N.M.; Orlicky, S.; Thevakumaran, N.; Talukdar, M.; Pillon, M.C.; Ceccarelli, D.F.; et al. Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol. Cell 2014, 53, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Hudson, W.H.; Ortlund, E.A. The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 2014, 15, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moududee, S.A.; Jiang, Y.; Gilbert, N.; Xie, G.; Xu, Z.; Wu, J.; Gong, Q.; Tang, Y.; Shi, Y. Structural and functional characterization of hMEX-3C Ring finger domain as an E3 ubiquitin ligase. Protein Sci. 2018, 27, 1661–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popow, J.; Alleaume, A.M.; Curk, T.; Schwarzl, T.; Sauer, S.; Hentze, M.W. FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA 2015, 21, 1873–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caudron-Herger, M.; Rusin, S.F.; Adamo, M.E.; Seiler, J.; Schmid, V.K.; Barreau, E.; Kettenbach, A.N.; Diederichs, S. R-DeeP: Proteome-wide and Quantitative Identification of RNA-Dependent Proteins by Density Gradient Ultracentrifugation. Mol. Cell 2019, 75, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, M.; Lawson, A.; Ladenburger, V.; Fredebohm, J.; Wolf, J.; Hoheisel, J.D.; Frezza, C.; Shlomi, T. High throughput synthetic lethality screen reveals a tumorigenic role of adenylate cyclase in fumarate hydratase-deficient cancer cells. BMC Genom. 2014, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, M.; Tian, R.; Blau, J.A.; Markegard, E.; Wagner, R.T.; Wu, D.; Mo, X.; Biton, A.; Zaitlen, N.; Fu, H.; et al. Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat. Biotechnol. 2018, 36, 170–178. [Google Scholar] [CrossRef]
- Colic, M.; Wang, G.; Zimmermann, M.; Mascall, K.; McLaughlin, M.; Bertolet, L.; Lenoir, W.F.; Moffat, J.; Angers, S.; Durocher, D.; et al. Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med. 2019, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Boutros, M. Towards a compendium of essential genes—From model organisms to synthetic lethality in cancer cells. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 74–85. [Google Scholar] [CrossRef] [Green Version]
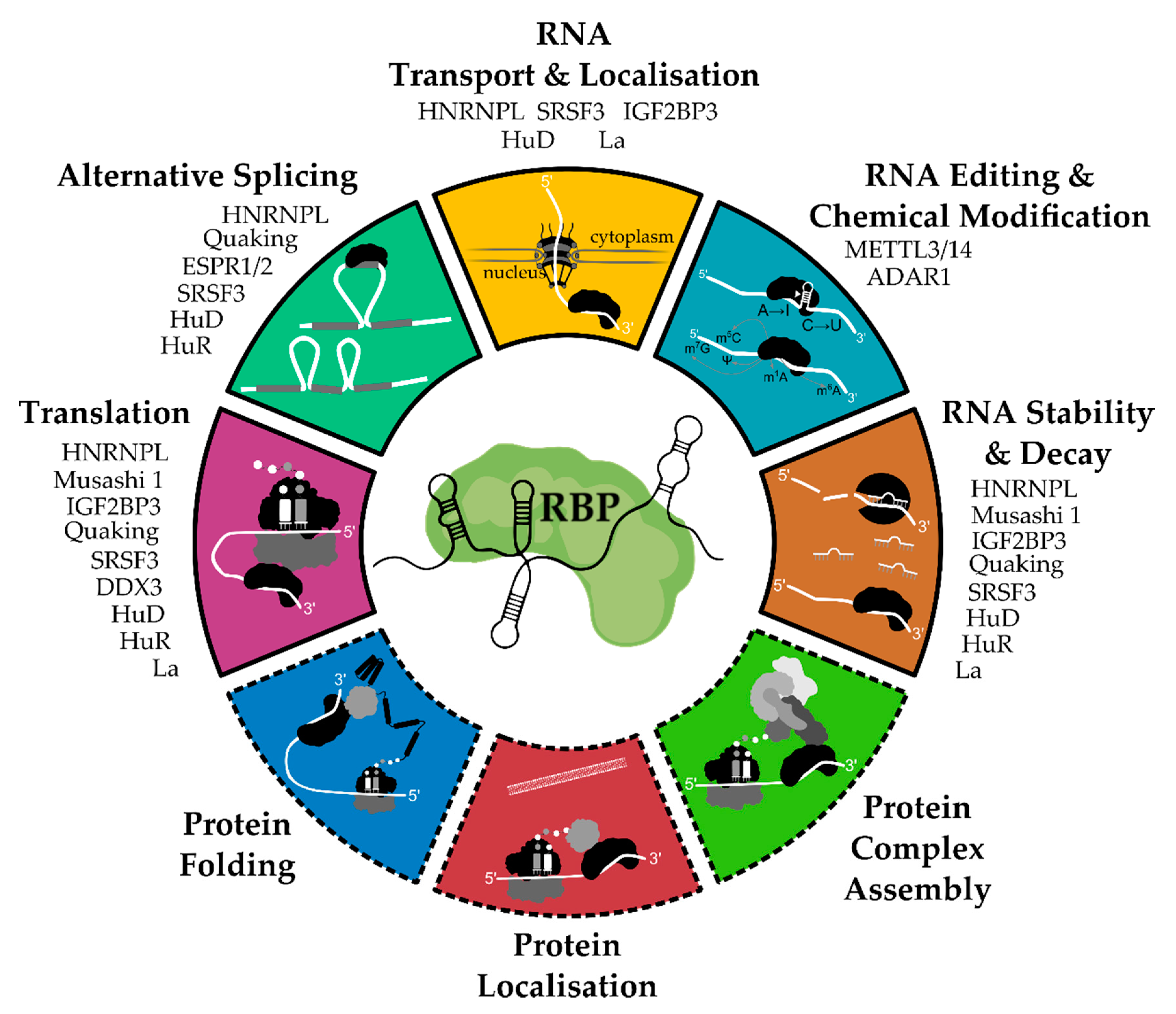

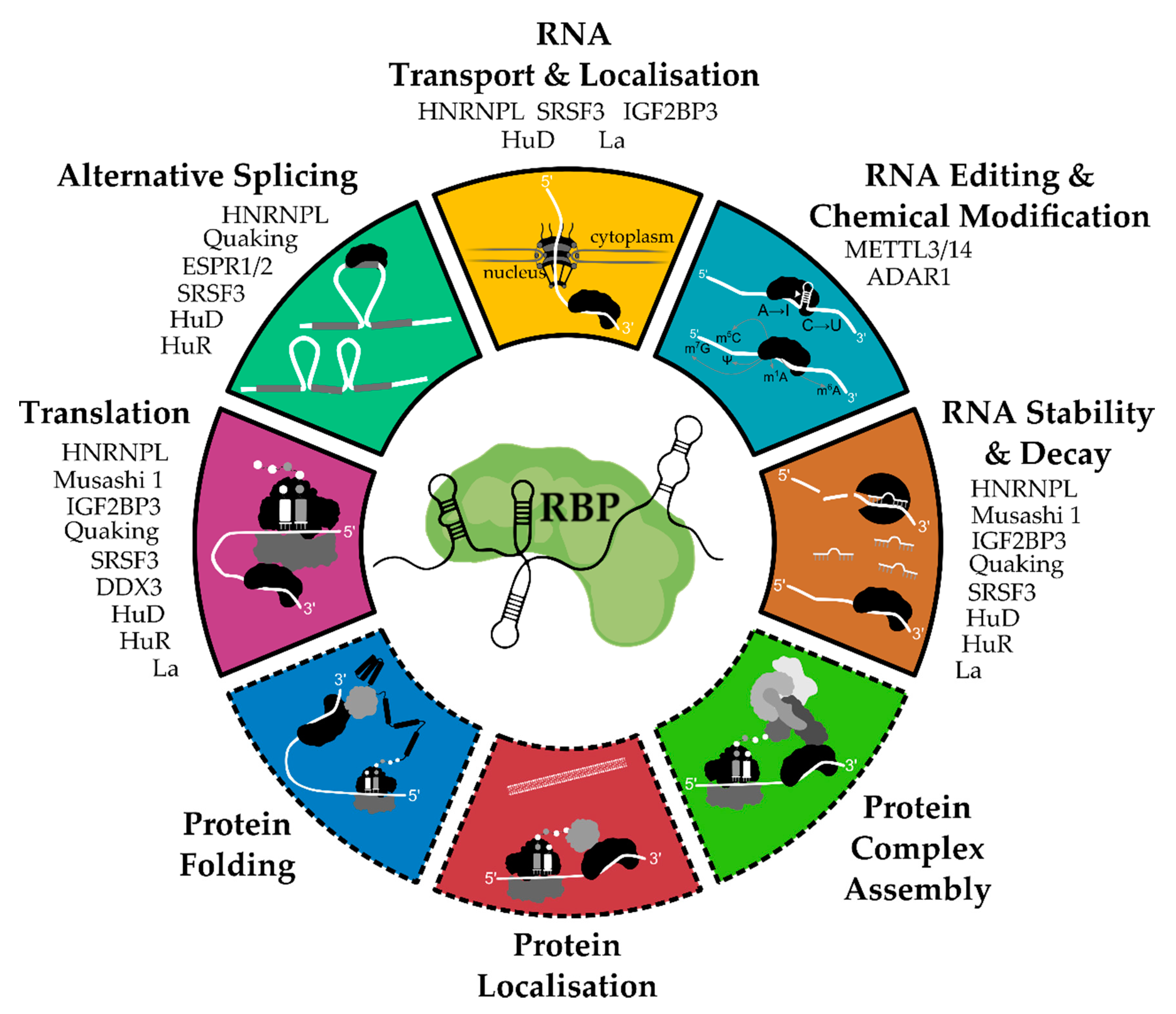{kind=link}
| RBP | Function & Mechanism | Reference |
|---|---|---|
| HuD | expression associated with lymph node metastasis and mode of invasion; depletion reduced invasiveness of cells and transcript levels of VEGF-A, VEGF-D, matrix metalloproteinase-2 (MMP-2) and MMP-9 | [180] |
| HNRNPL | overexpressed in OSCC compared to normal oral mucosa tissue; depletion reduced proliferation, viability, migration and in vivo growth; regulates SRSF3 RNA expression & alternative splicing of exon 4 | [181] |
| Musashi 1 (MSI1) | higher expression in higher stages and poorly differentiated tumors; independent prognostic marker of overall and disease-free survival | [182] |
| higher MSI1 mRNA level in OSCC compared to matched healthy tissue; depletion reduced proliferation, invasion, migration, and in vivo growth; might regulate c-MYC expression and STAT3 activation | [183] | |
| Quaking (QKI) | lower expression in OSCC compared to normal mucosal samples; overexpression reduced stemness, tumor growth and metastasis; depletion enhanced Sox2 mRNA stability and expression | [184] |
| low expression in HNSCC resulted in shorter overall survival; targeted by miR-200 family; knockdown induced EMT and enhanced migration and invasion in vitro and tumor growth and metastasis in vivo; | [185] | |
| SRSF3 | up-regulated in moderate or severe dysplasia tissues compared with normal oral mucosal tissues, and higher grade cancers express more SRSF3; expression correlated with lymph node metastasis and its depletion reduced EMT-related genes SNAI2 and N-cadherin | [186] |
| La | overexpressed in OSCC tissue compared to normal epithelial tissue; depletion inhibited proliferation, migration and invasion; knockdown reduced MMP-2 and β-catenin protein expression | [187] |
| Target RBP | Compound | Mode of Action | Reference |
|---|---|---|---|
| ADAR1 | 8-Azaadenosine | Inhibits RNA editing activity | [191] |
| DDX3 | RK-33 | Inhibits helicase or ATPase activity | [192,193,194] |
| NZ51 | Inhibits helicase activity | [195,196] | |
| FE15/FE87/FE98/FE109 | Inhibit the ATPase activity | [197,198] | |
| Compounds 1 & 3 | Target the RNA binding site | [199] | |
| Compounds 6 & 8 | Inhibit helicase and ATPase activity | [199] | |
| Ketorolac salt | Reduces DDX3 expression and inhibits ATPase activity | [200] | |
| HuR | MS-444, Okicenone, Dehydromutactin | Interfere with formation of HuR dimers and thereby RNA binding | [201] |
| Quercetin, b-40 | Inhibit HuR:ARE (TNF-α) complex formation | [202] | |
| Mitoxantrone | Inhibit HuR:ARE (TNF-α) complex formation | [203] | |
| dihydrotanshinone-I | Inhibits binding of HuR to several RNAs | [204] | |
| Compound 10 | Disrupts HuR oligomerization | [205] | |
| CMLD-2 | Inhibits binding of HuR to ARE-containing target RNAs (Bcl-2, MSI1 and XIAP) | [206] | |
| Azaphilone-9 | Inhibits HuR:ARE interaction by competitive binding in the RNA-binding cleft | [207] | |
| IGF2BP3 | d-ICD | Inhibits IGF2BP3 expression | [208] |
| JQ1, iBET | Inhibit IGF2BP3 expression | [209,210] | |
| LIN28 | Compound 1 | Inhibits LIN28–pre-let-7 interaction | [211] |
| Compound 1632 | Inhibits LIN28–pre-let-7 interaction | [212] | |
| 6-hydroxy-DL-DOPA, SB/ZW/0065 | Inhibit LIN28–pre-let-7 interaction | [213] | |
| LI38 (TPEN), LI71 | Inhibit LIN28-mediated oligouridylation of let-7 | [214] | |
| KCB170522, Luteolin | Inhibit LIN28–pre-let-7 interaction | [215] | |
| CCG-233094, CCG-234459 | Inhibit LIN28–pre-let-7 interaction | [216] | |
| METTL3 | ribofuranuronic acid analogues of adenosine, adenosine analogue with a tetrahydropyran ring | Competitors of S-adenosyl-L-methionine (SAM) for METTL3 binding | [217] |
| MSI1 | Inhibitor #1-3 | Inhibit RNA binding activity of MSI1/2 | [218] |
| Ro 08-2750 | Inhibits RNA binding activity of MSI1/2 | [219] | |
| Oleic acid | Induces a conformational change that prevents RNA association | [220] | |
| (-)-gossypol | Interacts with RNA binding pocket and blocks MSI1-RNA interaction | [221] | |
| La | HBSC-11 | Reduced La mRNA and protein levels | [222] |
| SRSF3 | Palmitic Acid | Increases neddylation and degradation of SRSF3 protein | [223] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiße, J.; Rosemann, J.; Krauspe, V.; Kappler, M.; Eckert, A.W.; Haemmerle, M.; Gutschner, T. RNA-Binding Proteins as Regulators of Migration, Invasion and Metastasis in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 6835. https://doi.org/10.3390/ijms21186835
Weiße J, Rosemann J, Krauspe V, Kappler M, Eckert AW, Haemmerle M, Gutschner T. RNA-Binding Proteins as Regulators of Migration, Invasion and Metastasis in Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2020; 21(18):6835. https://doi.org/10.3390/ijms21186835
Chicago/Turabian StyleWeiße, Jonas, Julia Rosemann, Vanessa Krauspe, Matthias Kappler, Alexander W. Eckert, Monika Haemmerle, and Tony Gutschner. 2020. "RNA-Binding Proteins as Regulators of Migration, Invasion and Metastasis in Oral Squamous Cell Carcinoma" International Journal of Molecular Sciences 21, no. 18: 6835. https://doi.org/10.3390/ijms21186835
APA StyleWeiße, J., Rosemann, J., Krauspe, V., Kappler, M., Eckert, A. W., Haemmerle, M., & Gutschner, T. (2020). RNA-Binding Proteins as Regulators of Migration, Invasion and Metastasis in Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences, 21(18), 6835. https://doi.org/10.3390/ijms21186835