Phenotypic Subtyping and Re-Analysis of Existing Methylation Data from Autistic Probands in Simplex Families Reveal ASD Subtype-Associated Differentially Methylated Genes and Biological Functions


Abstract
1. Introduction
2. Results and Discussion
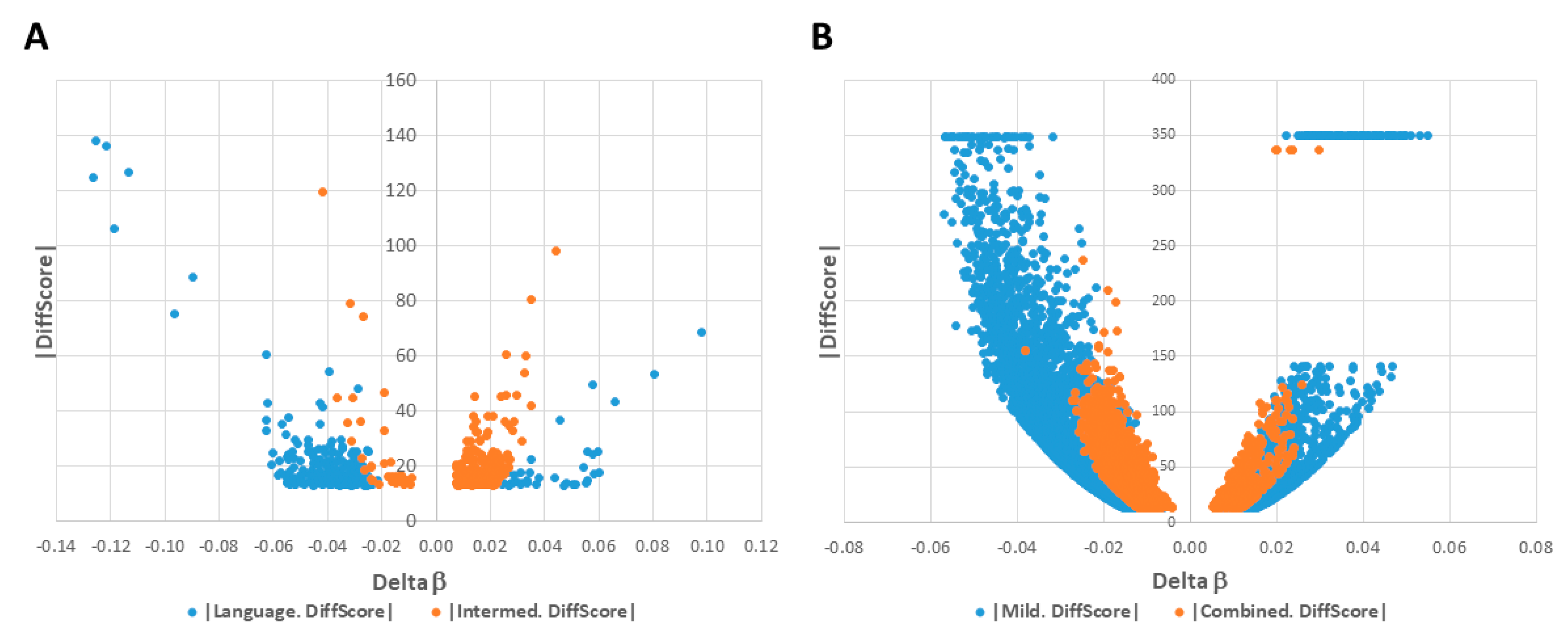
2.1. DAGs Associated with ASD Subphenotypes and the Combined Case Group
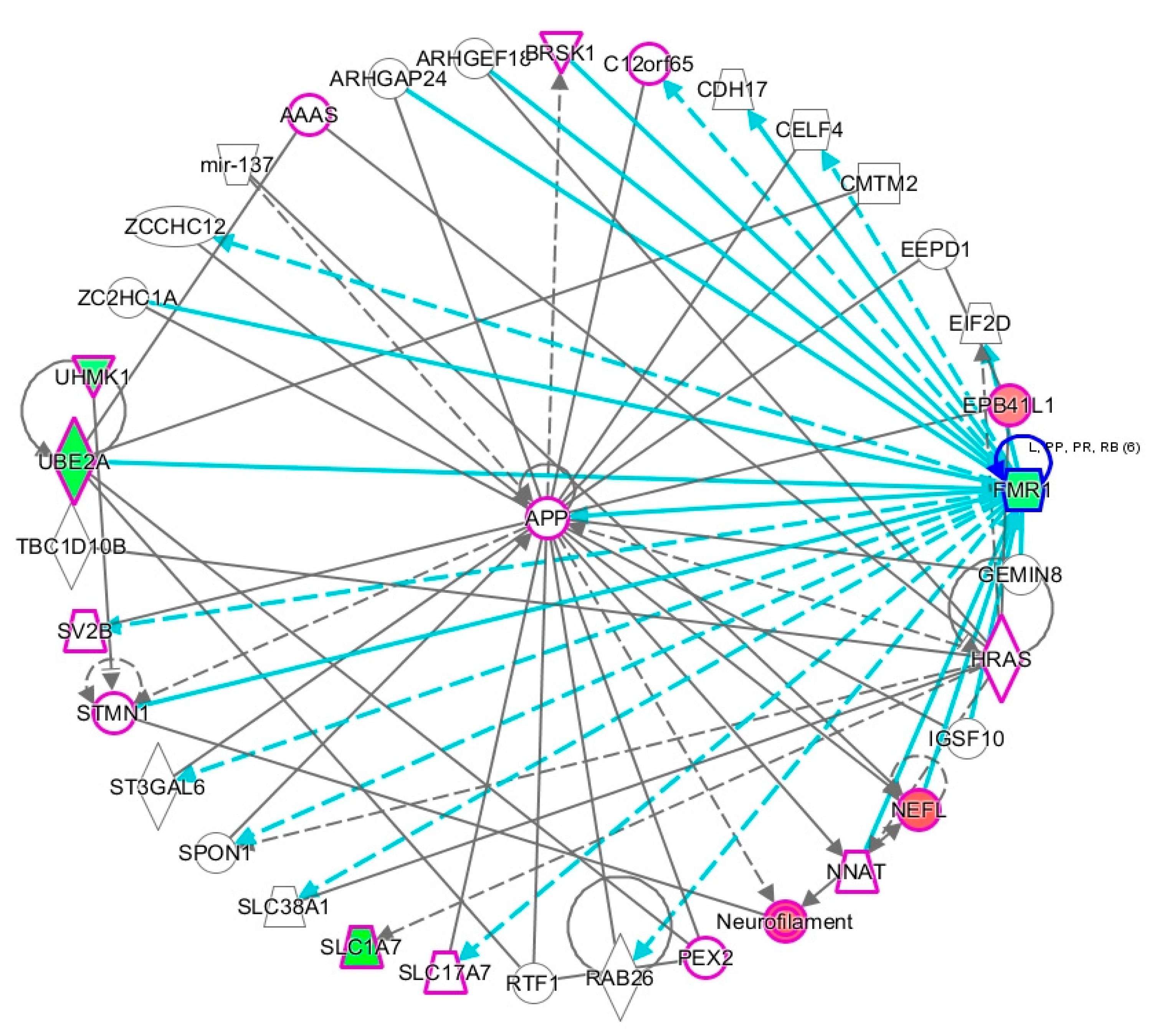
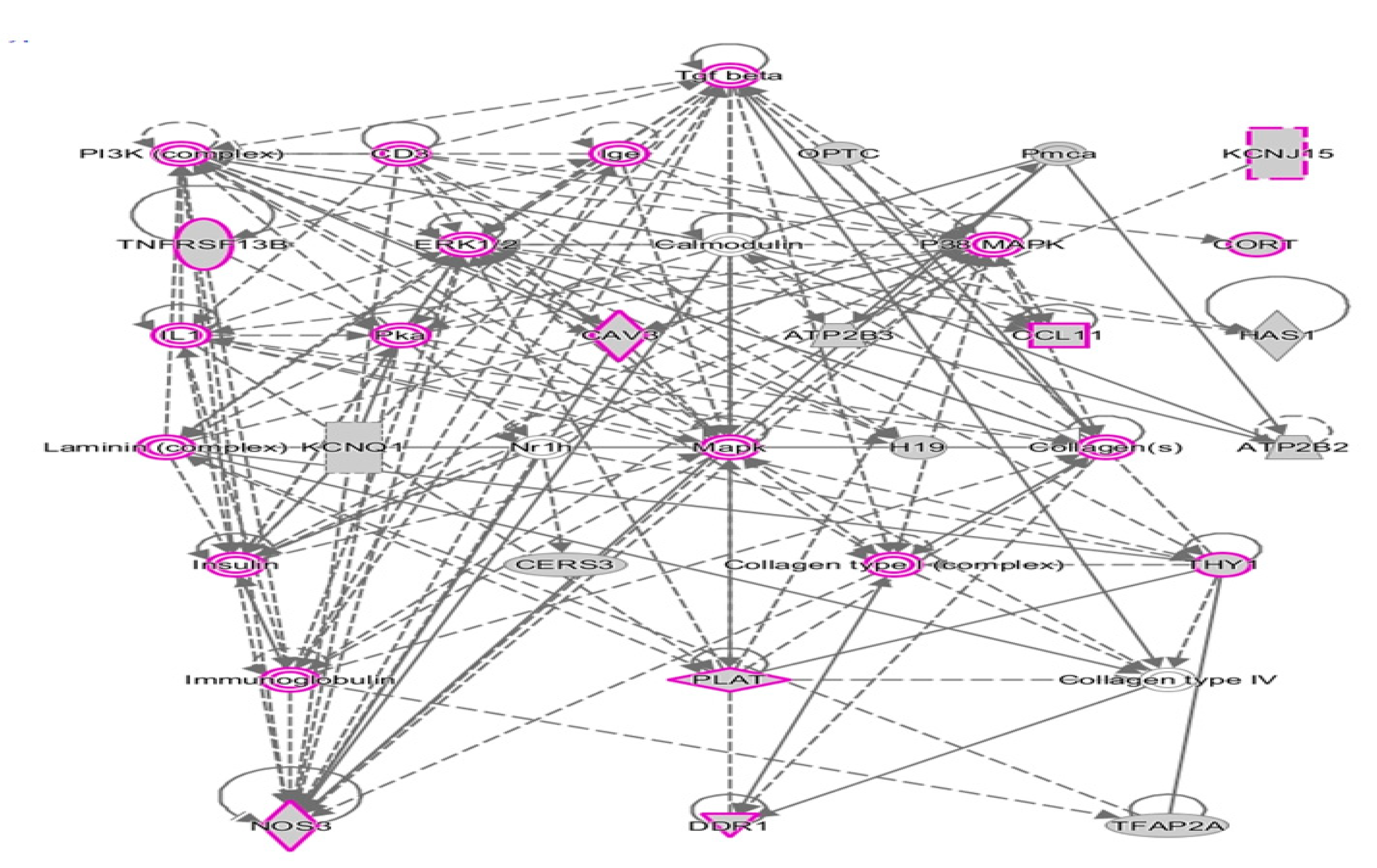
2.2. Network Prediction Analyses of Subgroup-Associated DAGs
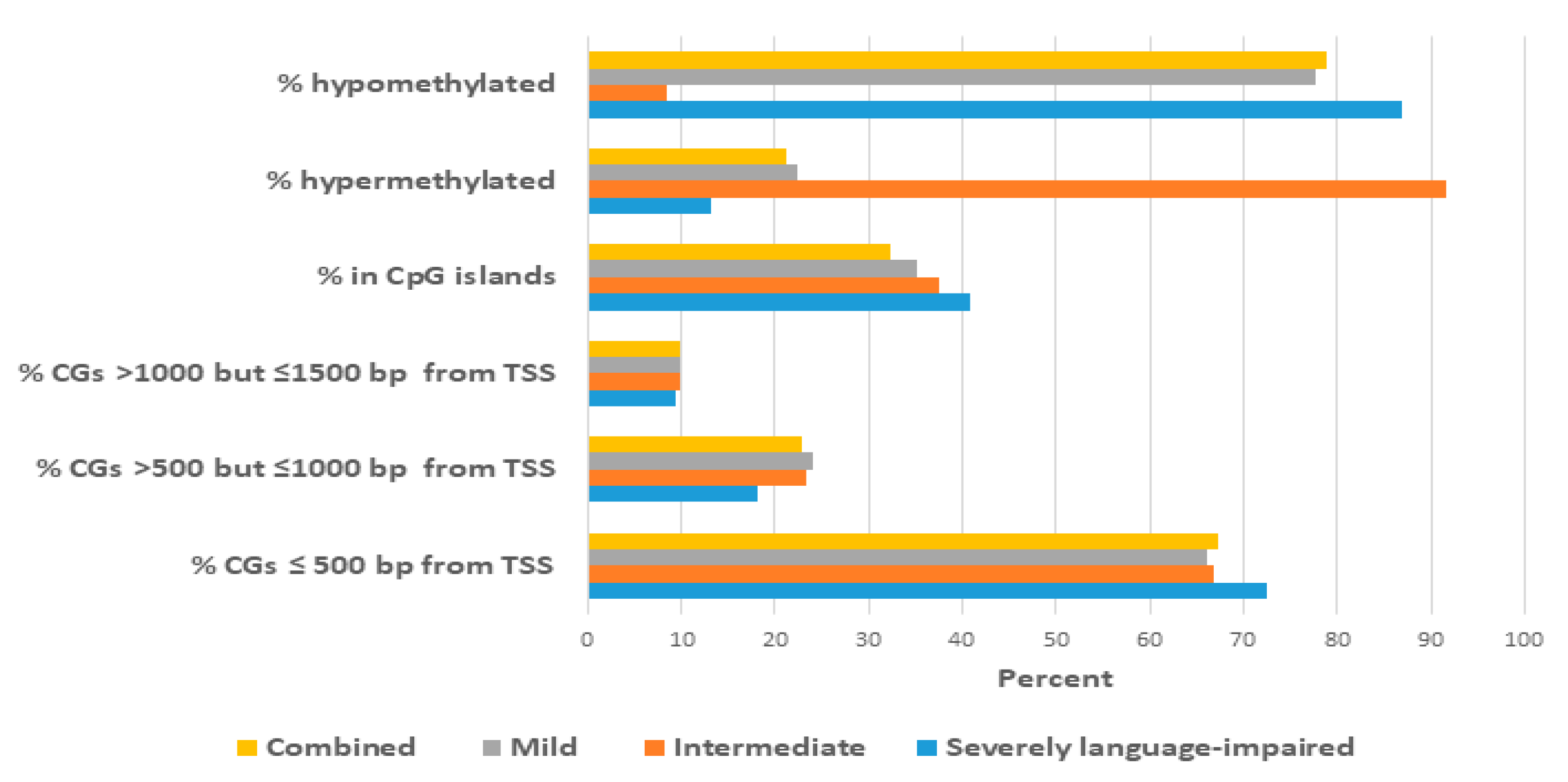
2.3. Proximity of Hypermethylated and Hypomethylated CpGs to the TSS of the DAGs
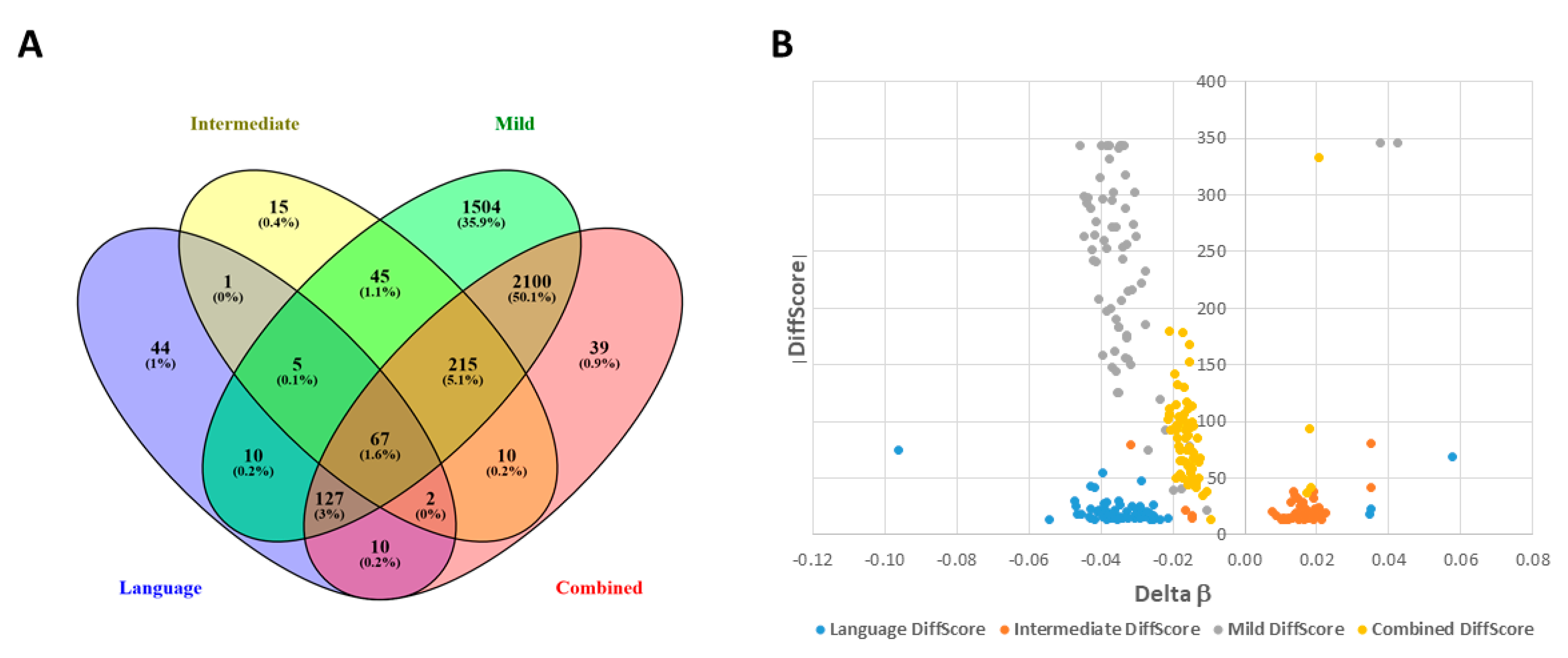
2.4. Shared DAGs among Case Groups Converge on Inflammatory Responses
2.5. Overlap of DAGs and Differentially Expressed Genes (DEGs) from Analogous Phenotypic Subgroups from the Simplex Population
2.6. Comparison with Other DNA Methylation Studies of ASD
2.7. Advantages and Limitations of Study Design and Future Considerations
3. Materials and Methods
3.1. Acquisition of Methylation Data for Individuals with ASD from the Simplex Population
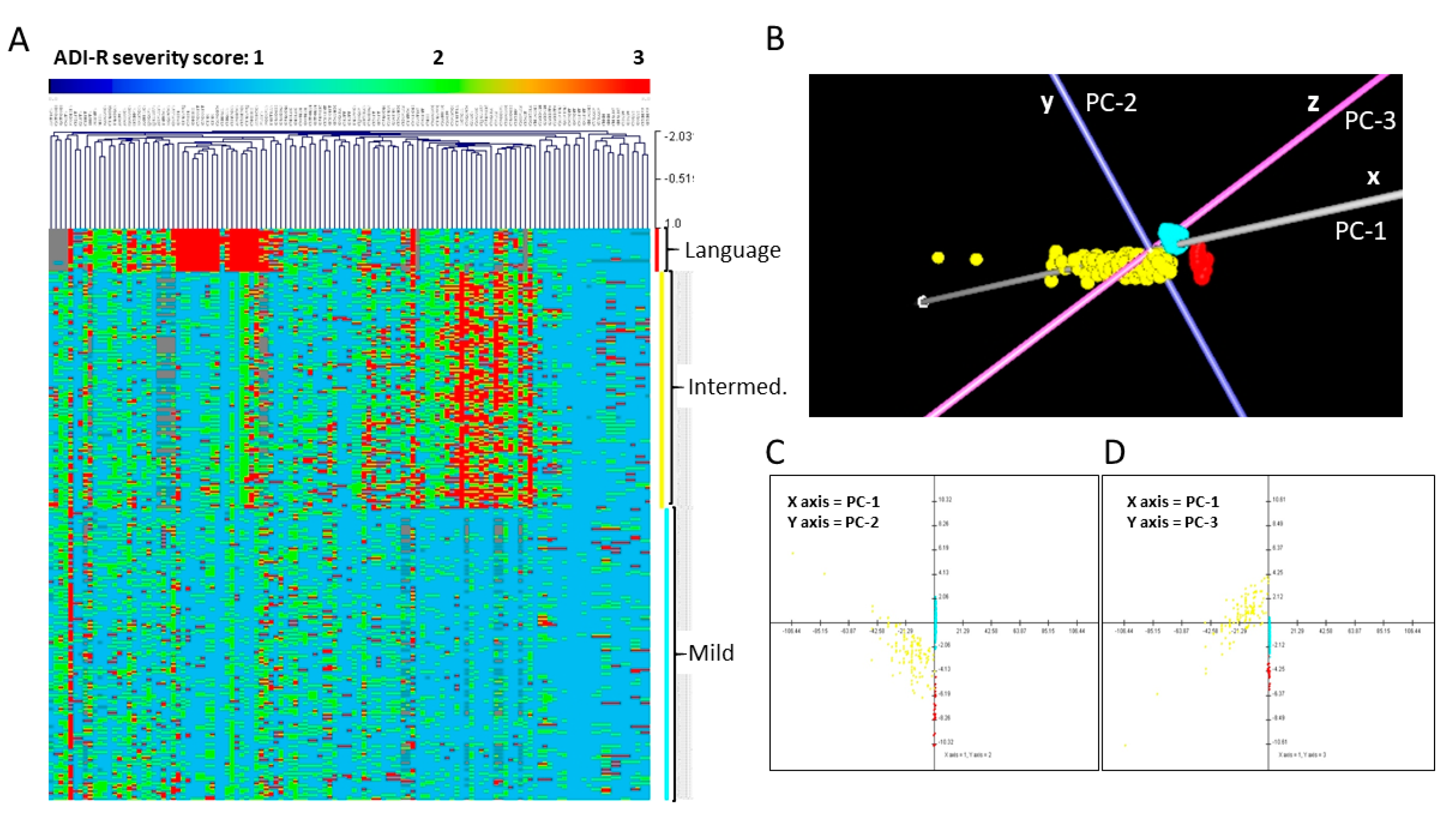
3.2. Phenotypic Subtyping for ASD Individuals from Simplex Families
3.3. Identification of Differentially Methylated Regions (DMR) and DMR-Associated Genes (DAG)
3.4. Network Prediction Analyses of DAGs
3.5. Hypergeometric Distribution Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Bakulski, K.M.; Singer, A.B.; Fallin, M.D. Genes and environment in autism spectrum disorders: An integrated perspective. In Frontiers in Autism Research: New Horizons for Diagnosis and Treatment; Hu, V.W., Ed.; World Scientific Publishing Co.: Singapore, 2014; pp. 335–374. [Google Scholar]
- Siu, M.T.; Weksberg, R. Epigenetics of Autism Spectrum Disorder. In Neuroepigenomics in Aging and Disease; Delgado-Morales, R., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 63–90. [Google Scholar]
- Moosa, A.; Shu, H.; Sarachana, T.; Hu, V.W. Are Endocrine Disrupting Compounds Environmental Risk Factors for Autism Spectrum Disorder? Horm. Behav. 2018, 101, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ciernia, A.V.; LaSalle, J. The Landscape of DNA Methylation Amid a Perfect Storm of Autism Aetiologies. Nat. Rev. Neurosci. 2016, 17, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Fallin, M.D. DNA Methylation Signatures as Biomarkers of Prior Environmental Exposures. Curr. Epidemiol. Rep. 2019, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global Methylation Profiling of Lymphoblastoid Cell Lines Reveals Epigenetic Contributions to Autism Spectrum Disorders and a Novel Autism Candidate Gene, RORA, Whose Protein Product is Reduced in Autistic Brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef]
- Ginsberg, M.R.; Rubin, R.A.; Falcone, T.; Ting, A.H.; Natowicz, M.R. Brain Transcriptional and Epigenetic Associations with Autism. PLoS ONE 2012, 7, e44736. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA Methylation Alterations in Multiple Brain Regions in Autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef]
- Nardone, S.; Sharan Sams, D.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA Methylation Analysis of the Autistic Brain Reveals Multiple Dysregulated Biological Pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef]
- Berko, E.R.; Suzuki, M.; Beren, F.; Lemetre, C.; Alaimo, C.M.; Calder, R.B.; Ballaban-Gil, K.; Gounder, B.; Kampf, K.; Kirschen, J.; et al. Mosaic Epigenetic Dysregulation of Ectodermal Cells in Autism Spectrum Disorder. PLoS Genet. 2014, 10, e1004402. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Meaburn, E.L.; Ronald, A.; Price, T.S.; Jeffries, A.R.; Schalkwyk, L.C.; Plomin, R.; Mill, J. Methylomic Analysis of Monozygotic Twins Discordant for Autism Spectrum Disorder and Related Behavioural Traits. Mol. Psychiatry 2014, 19, 495–503. [Google Scholar] [CrossRef]
- Feinberg, J.I.; Bakulski, K.M.; Jaffe, A.E.; Tryggvadottir, R.; Brown, S.C.; Goldman, L.R.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Fallin, M.D.; et al. Paternal Sperm DNA Methylation Associated with Early Signs of Autism Risk in an Autism-Enriched Cohort. Int. J. Epidemiol. 2015, 14, 1–12. [Google Scholar] [CrossRef]
- Massrali, A.; Brunel, H.; Hannon, E.; Wong, C.; Baron-Cohen, S.; Warrier, V. Integrated Genetic and Methylomic Analyses Identify Shared Biology between Autism and Autistic Traits. Mol. Autism 2019, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-Wide DNA Methylation Profiling Identifies Convergent Molecular Signatures Associated with Idiopathic and Syndromic Autism in Post-Mortem Human Brain Tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Steinberg, M.E. Novel Clustering of Items from the Autism Diagnostic Interview-Revised to Define Phenotypes within Autism Spectrum Disorders. Autism Res. 2009, 2, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Sarachana, T.; Kim, K.S.; Nguyen, A.; Kulkarni, S.; Steinberg, M.E.; Luu, T.; Lai, Y.; Lee, N.H. Gene Expression Profiling Differentiates Autism Case-Controls and Phenotypic Variants of Autism Spectrum Disorders: Evidence for Circadian Rhythm Dysfunction in Severe Autism. Autism Res. 2009, 2, 78–97. [Google Scholar] [CrossRef]
- Hu, V.W.; Addington, A.; Hyman, A. Novel Autism Subtype-Dependent Genetic Variants are Revealed by Quantitative Trait and Subphenotype Association Analyses of Published GWAS Data. PLoS ONE 2011, 6, e19067. [Google Scholar] [CrossRef]
- Talebizadeh, Z.; Arking, D.E.; Hu, V.W. A Novel Stratification Method in Linkage Studies to Address Inter and Intra Family Heterogeneity in Autism. PLoS ONE 2013, 8, e67569. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, V.W.; Devlin, C.A.; Debski, J.J. ASD Phenotype-genotype Associations in Concordant and Discordant Monozygotic and Dizygotic Twins Stratified by Severity of Autistic Traits. Int. J. Mol. Sci. 2019, 20, 3804. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Pramparo, T.; Gazestani, V.H.; Lombardo, M.V.; Pierce, K.; Lewis, N.E. The ASD Living Biology: From Cell Proliferation to Clinical Phenotype. Mol. Psychiatry 2019, 24, 88–107. [Google Scholar] [CrossRef]
- Bakos, J.; Bacova, Z.; Grant, S.G.; Castejon, A.M.; Ostatnikova, D. Are Molecules Involved in Neuritogenesis and Axon Guidance Related to Autism Pathogenesis? Neuromol. Med. 2015, 17, 297–304. [Google Scholar] [CrossRef]
- Nguyen, H.T.N.; Kato, H.; Masuda, K.; Yamaza, H.; Hirofuji, Y.; Sato, H.; Pham, T.T.M.; Takayama, F.; Sakai, Y.; Ohga, S.; et al. Impaired Neurite Development Associated with Mitochondrial Dysfunction in Dopaminergic Neurons Differentiated from Exfoliated Deciduous Tooth-Derived Pulp Stem Cells of Children with Autism Spectrum Disorder. Biochem. Biophys. Rep. 2018, 16, 24–31. [Google Scholar] [CrossRef]
- Phillips, M.; Pozzo-Miller, L. Dendritic Spine Dysgenesis in Autism Related Disorders. Neurosci. Lett. 2014, 601, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial Activation and Neuroinflammation in the Brain of Patients with Autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Prata, J.; Machado, A.S.; von Doellinger, O.; Almeida, M.I.; Barbosa, M.A.; Coelho, R.; Santos, S.G. The Contribution of Inflammation to Autism Spectrum Disorders: Recent Clinical Evidence. Methods Mol. Biol. 2019, 2011, 493–510. [Google Scholar] [PubMed]
- Chang, M.C.; Parham, L.D.; Blanche, E.I.; Schell, A.; Chou, C.-P.; Dawson, M.; Clark, F. Autonomic and Behavioral Responses of Children with Autism to Auditory Stimuli. Am. J. Occup. Ther. 2012, 66, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Baum, S.H.; Stevenson, R.A.; Wallace, M.T. Behavioral, Perceptual, and Neural Alterations in Sensory and Multisensory Function in Autism Spectrum Disorder. Prog. Neurobiol. 2015, 134, 140–160. [Google Scholar] [CrossRef]
- Wang, G.; Li, W.; Han, Y.; Gao, L.; Dai, W.; Su, Y.; Zhang, X. Sensory Processing Problems and Comorbidities in Chinese Preschool Children with Autism Spectrum Disorders. J. Autism Dev. Disord. 2019, 49, 4097–4108. [Google Scholar] [CrossRef]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- McKeague, I.W.; Brown, A.S.; Bao, Y.; Hinkka-Yli-Salomäki, S.; Huttunen, J.; Sourander, A. Autism with Intellectual Disability Related to Dynamics of Head Circumference Growth during Early Infancy. Biol. Psychiatry 2015, 77, 833–840. [Google Scholar] [CrossRef]
- Bryson, S.E.; Bradley, E.A.; Thompson, A.; Wainwright, A. Prevalence of Autism among Adolescents with Intellectual Disabilities. Can. J. Psychiatry 2008, 53, 449–459. [Google Scholar] [CrossRef]
- McCarthy, J. Children with Autism Spectrum Disorders and Intellectual Disability. Curr. Opin. Psychiatry 2007, 20, 472–476. [Google Scholar] [CrossRef] [PubMed]
- McCleery, J.P.; Elliott, N.A.; Sampanis, D.S.; Stefanidou, C.A. Motor Development and Motor Resonance Difficulties in Autism: Relevance to Early Intervention for Language and Communication Skills. Front. Integr. Neurosci. 2013, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Whyatt, C.; Craig, C. Sensory-Motor Problems in Autism. Front. Integr. Neurosci. 2013, 7, 51. [Google Scholar]
- Kikkawa, T.; Casingal, C.R.; Chun, S.H.; Shinohara, H.; Hiraoka, K.; Osumi, N. The Role of Pax6 in Brain Development and its Impact on Pathogenesis of Autism Spectrum Disorder. Brain Res. 2019, 1705, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.M.; Liang, J.; Vlasac, I.; Anderson, S.G.; Bechtold, D.A.; Bowden, J.; Emsley, R.; Gill, S.; Little, M.A.; Luik, A.I.; et al. Genome-Wide Association Analyses of Sleep Disturbance Traits Identify New Loci and Highlight Shared Genetics with Neuropsychiatric and Metabolic Traits. Nat. Genet. 2017, 49, 274–281. [Google Scholar] [CrossRef]
- Jones, S.E.; Tyrrell, J.; Wood, A.R.; Beaumont, R.N.; Ruth, K.S.; Tuke, M.A.; Yaghootkar, H.; Hu, Y.; Teder-Laving, M.; Hayward, C.; et al. Genome-Wide Association Analyses in 128,266 Individuals Identifies New Morningness and Sleep Duration Loci. PLoS Genet. 2016, 12, e1006125. [Google Scholar] [CrossRef]
- Gong, X.H.; Wang, H.Y. SHANK1 and Autism Spectrum Disorders. Sci. China Life Sci. 2015, 58, 985–990. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Minami, A.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Neuron Membrane Trafficking and Protein Kinases Involved in Autism and ADHD. Int. J. Mol. Sci. 2015, 16, 3095–3115. [Google Scholar] [CrossRef]
- Campos, V.E.; Du, M.; Li, Y. Increased Seizure Susceptibility and Cortical Malformation in Beta-Catenin Mutant Mice. Biochem. Biophys. Res. Commun. 2004, 320, 606–614. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Reutiman, T.J.; Folsom, T.D.; Thuras, P.D. GABAA Receptor Downregulation in Brains of Subjects with Autism. J. Autism Dev. Disord. 2009, 39, 223–230. [Google Scholar] [CrossRef]
- Sherr, E.H. The ARX Story (Epilepsy, Mental Retardation, Autism, and Cerebral Malformations): One Gene Leads to Many Phenotypes. Curr. Opin. Pediatr. 2003, 15, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; Rea, A.; Guy, M.; Lin, S.; Cook, E.H.; et al. A Common Genetic Variant in the Neurexin Superfamily Member CNTNAP2 Increases Familial Risk of Autism. Am. J. Hum. Genet. 2008, 82, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, M.; Abrahams, B.S.; Stone, J.L.; Duvall, J.A.; Perederiy, J.V.; Bomar, J.M.; Sebat, J.; Wigler, M.; Martin, C.L.; Ledbetter, D.H.; et al. Linkage, Association, and Gene-Expression Analyses Identify CNTNAP2 as an Autism-Susceptibility Gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Bakkaloglu, B.; O’Roak, B.J.; Louvi, A.; Gupta, A.R.; Abelson, J.F.; Morgan, T.M.; Chawarska, K.; Klin, A.; Ercan-Sencicek, A.G.; Stillman, A.A.; et al. Molecular Cytogenetic Analysis and Resequencing of Contactin Associated Protein-Like 2 in Autism Spectrum Disorders. Am. J. Hum. Genet. 2008, 82, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 Leads to Epilepsy, Neuronal Migration Abnormalities, and Core Autism-Related Deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Canali, G.; Garcia, M.; Hivert, B.; Pinatel, D.; Goullancourt, A.; Oguievetskaia, K.; Saint-Martin, M.; Girault, J.-A.; Faivre-Sarrailh, C.; Goutebroze, L. Genetic Variants in Autism-Related CNTNAP2 Impair Axonal Growth of Cortical Neurons. Hum. Mol. Genet. 2018, 27, 1941–1954. [Google Scholar] [CrossRef]
- Poot, M. Connecting the CNTNAP2 Networks with Neurodevelopmental Disorders. Mol. Syndr. 2015, 6, 7–22. [Google Scholar] [CrossRef]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, Neuroglia and Neuroinflammation in Autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The Influence of Neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of Fragile X Syndrome and Fragile X-Related Disorders. Dev. Med. Child Neurol. 2019, 61, 121–127. [Google Scholar] [CrossRef]
- Lavery, L.A.; Zoghbi, H.Y. The Distinct Methylation Landscape of Maturing Neurons and its Role in Rett Syndrome Pathogenesis. Curr. Opin. Neurobiol. 2019, 59, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.W.; Jiang, Y. DNA Methylation and Susceptibility to Autism Spectrum Disorder. Annu. Rev. Med. 2019, 70, 151. [Google Scholar] [CrossRef] [PubMed]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-Methylation-Dependent Long Gene Repression in Rett Syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef]
- Hu, V.W.; Frank, B.C.; Heine, S.; Lee, N.H.; Quackenbush, J. Gene Expression Profiling of Lymphoblastoid Cell Lines from Monozygotic Twins Discordant in Severity of Autism Reveals Differential Regulation of Neurologically Relevant Genes. BMC Genom. 2006, 7, 118. [Google Scholar] [CrossRef]
- Andrews, S.V.; Sheppard, B.; Windham, G.C.; Schieve, L.A.; Schendel, D.E.; Croen, L.A.; Chopra, P.; Alisch, R.S.; Newschaffer, C.J.; Warren, S.T.; et al. Case-Control Meta-Analysis of Blood DNA Methylation and Autism Spectrum Disorder. Mol. Autism 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Hannon, E.; Schendel, D.; Ladd-Acosta, C.; Grove, J.; Hansen, C.S.; Andrews, S.V.; Hougaard, D.M.; Bresnahan, M.; Mors, O.; Hollegaard, M.V.; et al. Elevated Polygenic Burden for Autism is Associated with Differential DNA Methylation at Birth. Genome Med. 2018, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Siu, M.T.; Butcher, D.T.; Turinsky, A.L.; Cytrynbaum, C.; Stavropoulos, D.J.; Walker, S.; Caluseriu, O.; Carter, M.; Lou, Y.; Nicolson, R.; et al. Functional DNA Methylation Signatures for Autism Spectrum Disorder Genomic Risk Loci: 16p11.2 Deletions and CHD8 Variants. Clin. Epigenetics 2019, 11, 103. [Google Scholar] [CrossRef]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism Diagnostic Interview-Revised: A Revised Version of a Diagnostic Interview for Caregivers of Individuals with Possible Pervasive Developmental Disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A Free, Open-Source System for Microarray Data Management and Analysis. BioTechniques 2003, 34, 374–378. [Google Scholar] [CrossRef]
- Luo, R.; Sanders, S.J.; Tian, Y.; Voineagu, I.; Huang, N.; Chu, S.H.; Klei, L.; Cai, C.; Ou, J.; Lowe, J.K.; et al. Genome-Wide Transcriptome Profiling Reveals the Functional Impact of Rare De Novo and Recurrent CNVs in Autism Spectrum Disorders. Am. J. Hum. Genet. 2012, 91, 38–55. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/ (accessed on 18 September 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nervous System Development and Function Severely Language Impaired (n = 22) | p-Value * | Number of Genes |
|---|---|---|
| Abnormal morphology of neurons | 2.21 × 10−4 | 19 |
| Sensorimotor integration | 3.39 × 10−3 | 2 |
| Neuritogenesis | 3.52 × 10−3 | 19 |
| Maturation of synapse | 4.25 × 10−3 | 3 |
| Size of neurites | 6.09 × 10−3 | 2 |
| Branching of neurites | 6.31 × 10−3 | 11 |
| Maturation of dendritic spines | 7.15 × 10−3 | 2 |
| Development of neurons | 9.24 × 10−3 | 22 |
| Intermediate (n = 121) | ||
| Activation of neuroglia | 7.35 × 10−5 | 11 |
| Activation of astrocytes | 4.77 × 10−4 | 6 |
| Abnormal morphology of neurons | 1.22 × 10−3 | 21 |
| Abnormal morphology of axons | 1.27 × 10−3 | 7 |
| Abnormal morphology of neurites | 1.48 × 10−3 | 9 |
| Loss of neurites | 2.91 × 10−3 | 4 |
| Mild (n = 149) | ||
| Abnormal morphology of nervous system | 4.34 × 10−15 | 297 |
| Sensory system development | 7.77 × 10−15 | 194 |
| Combined (n = 292) | ||
| Abnormal morphology of nervous system | 4.37 × 10−11 | 194 |
| Sensory system development | 6.17 × 10−9 | 121 |
| Abnormal morphology of neurons | 2.26 × 10−8 | 121 |
| Activation of neuroglia | 4.27 × 10−8 | 45 |
| Neurological Diseases | p-Value * | Number of Genes |
| Severely language impaired | ||
| Cognitive impairment | 1.32 × 10−5 | 26 |
| Syndromic X⁻linked mental retardation | 2.99 × 10−4 | 6 |
| Mental retardation | 4.13 × 10−3 | 14 |
| Motor dysfunction or movement disorder | 7.75 × 10−3 | 29 |
| Autosomal dominant mental retardation type 11 | 9.94 × 10−3 | 1 |
| Intermediate | ||
| Schizophrenia | 7.73 × 10−4 | 20 |
| Demyelination of nerves | 4.59 × 10−3 | 4 |
| Mild | ||
| Schizophrenia | 1.18 × 10−16 | 184 |
| Motor dysfunction or movement disorder | 5.98 × 10−14 | 393 |
| Movement Disorders | 3.93 × 10−13 | 384 |
| Combined | ||
| Schizophrenia | 1.81 × 10−7 | 106 |
| Developmental Disorders (Severely Language Impaired Only) | p-Value * | Number of Genes |
| Autism or intellectual disability | 3.31 × 10−3 | 15 |
| Autism spectrum disorder or intellectual disability | 3.67 × 10−3 | 17 |
| Dystrophy of muscle | 5.51 × 10−3 | 10 |
| Nervous System Development & Function | p-Value * | DAGs (<500 bp from TSS) |
| Severely language-impaired subtype | Hypermethylated | |
| Cell-cell adhesion of neurons | 2.29 × 10−4 | CADM1, NINJ2 |
| Induction of neural crest | 2.37 × 10−3 | TFAP2A |
| Scaffolding of postsynaptic region | 3.55 × 10−3 | CADM1 |
| Corticogenesis | 3.55 × 10−3 | PAX6 |
| Quantity of neurons | 7.25 × 10−3 | CADM1, NPTX2, PAX6, TFAP2A |
| Abnormal morphology of brain | 7.37 × 10−3 | CRMP1, EPB41L1, PAX6, TFAP2A |
| Size of growth cone | 1.65 × 10−2 | PAX6 |
| Morphology of nervous system | 3.71 × 10−2 | CADM1, CRMP1, EPB41L1, PAX6, TFAP2A |
| Memory consolidation | 3.73 × 10−2 | PAX6 |
| Quantity of nerve ending | 3.73 × 10−2 | CADM1 |
| Development of sensory neurons | 3.95 × 10−2 | PAX6 |
| Abnormal morphology of forebrain | 4.58 × 10−2 | PAX6, TFAP2A |
| Severely language-impaired subtype | Hypomethylated | |
| Abnormal morphology of sensory neurons | 6.21 × 10−4 | ATP2B2, CETN2, KCNQ1, OPN4, PAX8 |
| Delay in initiation of maturation of interneurons | 5.93 × 10−3 | FMR1 |
| Abnormal morphology of neurons | 6.49 × 10−3 | ATP2B2, CABP4, CCL11, CETN2, CORT, FMR1, KCNQ1, NOS3, OPN4, PAX8, SHANK1 |
| Long term synaptic depression of hippocampal cells | 9.57 × 10−3 | FMR1, INS |
| Circadian phase shifting | 1.18 × 10−2 | OPN4 |
| Remodeling of dendrites | 1.18 × 10−2 | FMR1 |
| Density of GABAergic synapse | 1.18 × 10−2 | FMR1 |
| Neurological Diseases | p-Value * | DAGs (<500 bp from TSS) |
| Severely language-impaired subtype | Hypermethylated | |
| Autosomal dominant mental retardation type 11 | 1.19 × 10−3 | EPB41L1 |
| Cognitive impairment | 1.33 × 10−3 | CRMP1, EPB41L1, GSTM1, NPTX2, PAX6, TFAP2A |
| Epileptic seizure | 3.70 × 10−2 | CCN1, NPTX2 |
| Mental retardation | 4.09 × 10−2 | EPB41L1, PAX6, TFAP2A |
| Abnormal morphology of forebrain | 4.58 × 10−2 | PAX6, TFAP2A |
| Severely language impaired subtype | Hypomethylated | |
| Syndromic X-linked mental retardation | 2.20 × 10−5 | BCAP31, FMR1, PHF8, SLC1A7, SLC9A6, UBE2A |
| Cognitive impairment | 1.65 × 10−3 | BCAP31, FMR1, GRM4, INS, KCNQ1, LRRN4, NOS3, PDZK1, PHF8, POLR3C, SHANK1, SLC1A7, SLC9A6, UBE2A, UHMK1 |
| Fragile X-associated tremor ataxia syndrome | 5.93 × 10−3 | FMR1 |
| Fragile X syndrome with Prader-Willi-like phenotype | 5.93 × 10−3 | FMR1 |
| Movement Disorders | 1.15 × 10−2 | ATP2B2, ATP2B3, BCAP31, C9, CCL11, CETN2, COL6A3, F8A1, FCGR3A/FCGR3B, FMR1, KCNQ1, MYH7, OPN4, SDC4, SGCG, SLC17A4, SLC1A7, SLC9A6, TGM6 |
| Nervous System Development & Function | p-Value * | DAGs (<500 bp from TSS) |
| Intermediate subtype | Hypermethylated | |
| Loss of neurites | 4.64 × 10−4 | CORT, GJB1, NTF3, SERPINA3 |
| Activation of neuroglia | 1.11 × 10−3 | C1QA, CCL11, CCL22, FGF1, GJB1, NOS3, SMPD3 |
| Abnormal morphology of neurons | 1.22 × 10−3 | ATP2B2, C1QA, CABP4, CCL11, CORT, GJB1, KCNQ1, NOS3, NTF3, OPN4, PAX8, PLP1, RHO, SERPINA3, SORBS2 |
| Abnormal morphology of nerve ending | 1.66 × 10−3 | C1QA, NTF3 |
| Abnormal morphology of sensory neurons | 2.16 × 10−3 | ATP2B2, KCNQ1, NTF3, OPN4, PAX8 |
| Loss of axons | 2.38 × 10−3 | CORT, GJB1, NTF3 |
| Activation of astrocytes | 3.04 × 10−3 | C1QA, CCL11, FGF1, SMPD3 |
| Formation of excitatory synapses | 3.22 × 10−3 | NTF3, SORBS2 |
| Abnormal morphology of neurites | 5.98 × 10−3 | CORT, GJB1, NTF3, PLP1, SERPINA3, SORBS2 |
| Evoked potential | 6.24 × 10−3 | ATP2B2, KCNQ1, NTF3, PAX8 |
| Abnormal morphology of nervous system | 6.49 × 10−3 | ATP2B2, C1QA, CABP4, CCL11, CNGA2, CORT, FGF1, GJB1, KCNQ1, NOS3, NR5A1, NTF3, OPN4, PAX8, PLP1, RHO, SERPINA3, SMPD3, SORBS2 |
| Intermediate subtype | Hypomethylated | |
| Morphology of brain | 8.83 × 10−4 | ARSA, CTNNB1, GBX2, GSX1, TFAP2A |
| Differentiation of sensory progenitor cells | 9.40 × 10−4 | CTNNB1 |
| Neurogenesis of dopaminergic neurons | 1.88 × 10−3 | CTNNB1 |
| Abnormal morphology of forebrain | 2.42 × 10−3 | ARSA, GSX1, TFAP2A |
| Abnormal morphology of brain | 3.12 × 10−3 | ARSA, GBX2, GSX1, TFAP2A |
| Auditory evoked potential | 3.15 × 10−3 | ARSA, KCNQ1 |
| Cell survival of dopaminergic neurons | 3.76 × 10−3 | CTNNB1 |
| Formation of forebrain | 3.77 × 10−3 | CTNNB1, GBX2, GSX1 |
| Accumulation of microglia | 7.50 × 10−3 | CTNNB1 |
| Lack of cerebellum | 8.43 × 10−3 | GBX2 |
| Neurological Diseases | p-Value * | DAGs (<500 bp from TSS) |
| Intermediate subtype | Hypermethylated | |
| Abnormal morphology of mechanosensory neurons | 8.56 × 10−4 | ATP2B2, KCNQ1, NTF3, PAX8 |
| Lack of muscle sensory neurons | 7.85 × 10−3 | NTF3 |
| Intermediate subtype | Hypomethylated | |
| Early-onset neurological disorder | 3.20 × 10−4 | ARSA, GABRA3, KCNQ1, PDZRN3 |
| Cognitive impairment | 3.54 × 10−4 | ARSA, CTNNB1, GABRA3, GSTM1, KCNQ1, TFAP2A |
| Autosomal dominant mental retardation type 19 | 9.40 × 10−4 | CTNNB1 |
| Early-onset schizophrenia | 1.13 × 10−3 | GABRA3, PDZRN3 |
| Lack of cerebellum | 8.43 × 10−3 | GBX2 |
| Nervous System Development & Function | p-Value * | DAGs (<500 bp from TSS) |
|---|---|---|
| Mild subtype | Hypermethylated | |
| Development of striatum | 1.26 × 10−6 | ARX, CNTNAP2, GSX1, PAX6 |
| Abnormal morphology of nervous system | 3.61 × 10−6 | ARX, ATP8A2, CDH11, CNTNAP2, EDN3, FOXB1, FST, GSX1, OSTM1, PAX6, RBP1, SYN2, TFAP2A, TLX3, ZIC1 |
| Abnormal morphology of glutamatergic neuron | 4.96 × 10−5 | PAX6,TLX3 |
| Migration of neurons | 5.84 × 10−5 | ARX, CNTNAP2, ERRFI1, FLRT2, LRP12, PAX6, TLX3 |
| Development of neurons | 9.20 × 10−5 | ARX, ATP8A2, CADM1, CCN1, CDH1, CNTNAP2, FLRT2, ITGA1, LRP12, PAX6, SYN2, TLX3, TPBG |
| Quantity of neurons | 1.19 × 10−4 | ARX, CADM1, CNTNAP2, ENTPD3, FST, PAX6, PTGER2, SYN2, TFAP2A |
| Abnormal morphology of brain | 1.23 × 10−4 | ARX, CNTNAP2, FOXB1, GSX1, OSTM1, PAX6, SYN2, TFAP2A, ZIC1 |
| Abnormal morphology of forebrain | 1.94 × 10−4 | ARX, CNTNAP2, FOXB1, GSX1, PAX6, TFAP2A |
| Growth of cerebellum | 2.30 × 10−4 | FOXB1, ZIC1 |
| Cell cycle progression of neurons | 9.71 × 10−4 | PAX6, ZIC1 |
| Growth of brain | 1.22 × 10−3 | ARX, FOXB1, PAX6, ZIC1 |
| Developmental process of synapse | 1.83 × 10−3 | CADM1, CDH1,FLRT2, SYN2, TPBG |
| Initiation of migration of neurons | 2.90 × 10−3 | ARX |
| Formation of forebrain | 3.02 × 10−3 | ARX, CNTNAP2, GSX1, PAX6, ZIC1 |
| Synaptic transmission of hippocampal CA1 region | 4.14 × 10−3 | CNTNAP2, SYN2 |
| Development of sensory neurons | 4.39 × 10−3 | PAX6, TLX3 |
| Association of synaptic vesicles | 5.80 × 10−3 | SYN2 |
| Mild subtype | Hypomethylated | |
| Sensory system development | 6.57 × 10−9 | ABCA4, ADCY1, AIPL1, AIRE, ALDH1A2, ALOX15, APOB, AQP1, ARSG, ASPA, ATP2B2, BBS1, BBS7, BCL9L, BFSP2, BMPR1B, BRD1, C5AR1, CABP4, CCL1, CDH5, CDK20, CNGA3, COL18A1, COL1A1, COL8A2, CPLX4, CRX, CRYAA/CRYAA2, CRYAB, CRYBA4, CRYBB2, CRYGA, CRYGB, CRYGC, CXCR3, DNMT3A, DPT, DSC1, EGFR, ELOVL4, EMX1, FABP7, FASLG, FEZF2, FGF1, FGF7, FGFR2, FYCO1, GDF3, GFRA1, GJA3, GNAT2, GRK1, GUCA1A, GUCY2F, HCN1, HGF, HK2, HRG, IL1R1, IRX3, KERA, KNG1, KRT12, KRT4, LCK, LCTL, LHX2, LRAT, LRP8, LYVE1, MARCKSL1, METRN, MFAP2, MFRP, MGAT5, MSX2, NEUROD2, NEUROD4, NOTCH3, NRTN, NTF3, NTF4, NXNL1, OLIG2, OPN4, PDCD1, PDE6B, PDE6C, PF4V1, PLA2G3, POR, POU4F2, POU4F3, PPT2, PRPH2, PTGS2, PTPRS, PYGO1, RB1, RBP3, RDH8, RHO, RPE65, RUNX3, S1PR3, SERPINF1, SIX3, SIX6, SLC17A8, SLC39A5, SOX10, SOX11, SPI1, TFB1M, TH, THBS1, TP63, TRPV4, TUB, TULP1, TYRP1, USH2A, VCAM1, VSX2, WNT2 |
| Activation of neuroglia | 1.35 × 10−8 | ABCA4, ADIPOQ, AGT, ALB, C1QA, C5AR1, CCL1, CCL11, CCL21, CCL22, CCL5, CCL7, CD40LG, CHGA, CNGA3, CSF3, DRD2, EGFR, F2, FASLG, FGA, FGF1, FGG, GFAP, GJB1, GJC2, GRK2, IL10, IL1R1, MC4R, MOG, MSTN, MYOD1, NOS3, NR4A1, NRG1, PDE6B, PDK4, PTGS2, RDH8, SERPINF2, SLC6A4, SST, TLR7, TLR9, TREM2, TRPM2, VTN |
| Neurological Diseases | p-Value * | DAGs (<500 bp from TSS) |
|---|---|---|
| Mild subtype | Hypermethylated | |
| Cerebellar ataxia with intellectual disability | 2.95 × 10−4 | ATP8A2, PAX6 |
| Movement Disorders | 6.67 × 10−4 | ARX, ATP8A2, CDH11, DKK3, ERRFI1, FGF12, FLRT2, GABRE, GYPC, NKX6-2, PAX6, PDE4B, SYN2, ZIC1 |
| Epilepsy | 7.73 × 10−4 | ARX, CCN1, CNTNAP2, ERRFI1, FGF12, GABRE, SYN2 |
| Epilepsy or neurodevelopmental disorder | 1.08 × 10−3 | ARX, CCN1, CNTNAP2, EDN3, ERRFI1, FGF12, GABRE, SYN2 |
| ARX-related X-linked mental retardation | 2.90 × 10−3 | ARX |
| Moderate to severe stage mental retardation | 2.90 × 10−3 | ARX |
| Severe hypotonia | 2.90 × 10−3 | ARX |
| Susceptibility to autism type 15 | 2.90 × 10−3 | CNTNAP2 |
| Cerebellar ataxia, mental retardation, and dysequilibrium syndrome type 4 | 2.90 × 10−3 | ATP8A2 |
| Autism | 3.95 × 10−3 | ARX, CNTNAP2, GABRE |
| Seizures | 4.52 × 10−3 | ARX, CCN1, CNTNAP2, ERRFI1, FGF12, GABRE, SYN2 |
| Familial pervasive developmental disorder | 4.64 × 10−3 | ARX, CNTNAP2 |
| Ataxia | 5.23 × 10−3 | ARX, ATP8A2, NKX6-2, PAX6, ZIC1 |
| Mild subtype | Hypomethylated | |
| Schizophrenia | 9.17 × 10−11 | ACSBG1, ADRA2B, APOA4, APOB, ATP1A4, ATP2B2, ATP4A, ATP4B, BPIFC, CA1, CA5A, CA7, CA9, CAD, CALY, CAP2, CCDC60, CCK, CCKAR, CHI3L1, CHRM1, CHRNA1, CHRNA2, CHRNA9, CHRNB4, CNR1, COL3A1, CPLX2, CRHBP, CYP2D6, CYP2E1, CYP3A5, DAB1, DAO, DDR1, DLG2, DRD1, DRD2, DRD5, DRP2, EGFR, ERBB4, FABP7, FAM3D, FCGR2A, FCGR3A/FCGR3B, GABRA3, GABRA5, GABRA6, GABRG3, GABRP, GABRR1, GFAP, GPR37, GRIK1, GRIK5, GRIN1, GRIN3A, GRM4, GRM7, HIPK3, HRH1, HTR2B, HTR3A, HTR3B, HTR3C, HTR3D, HTR3E, HTR5A, HTR6, INS, LAMA1, LGALS1, MAGEC1, MC4R, MEST, MT2A, MTNR1B, NEFM, NELL1, NOTCH4, NPAS3, NRG1, NRXN1, NTF3, NTNG2, OFCC1, OXTR, PDZRN3, PLP1, PMP22, POMC, PRL, PTGS2, RCAN2, RIT2, RPP21, SCG2, SCG5, SCN2B, SCN3B, SCN4A, SCN9A, SLC14A1, SLC18A1, SLC18A2, SLC31A2, SLC5A7, SLC6A4, SLC7A11, SOX10, SST, STON1, SYN3, SYT3, SYT4, TAC1, TF, THBS1, TRAK1, TTR, UGT1A3, XDH |
| Overlap between DAGs and DEGs in Language-Impaired Subgroup (hypergeom. q=0.30) | Overlap between DAGs and DEGs in Intermediate Subgroup (q = 0.35) | Overlap between DAGs and DEGs in Mild Subgroup (q = 7.14 × 10−4) | Overlap between DAGs and DEGs in Combined Case Group (q = 2.31 × 10−4) | ||
|---|---|---|---|---|---|
| ALOX15B | AIM2 | AADAC | LIMS2 | AIM2 | LCN2 |
| CARD14 | ARSA | AMPH | MATK | ASB10 | LHX3 |
| CAV3 | ATP8B1 | APOB | MEP1B | ASCL2 | LYST |
| CCL11 | KCNQ1 | BNC1 | MMP10 | BGN | MAGEA11 |
| COL6A3 | LDHC | C15orf32 | MS4A12 | BMP10 | MAGEA8 |
| CTRB1 | NEFL | C19orf18 | MS4A6A | BMX | MEPE |
| FAM83A | RGS3 | C8B | NKX6-2 | BTNL2 | MS4A12 |
| HIPK4 | SMPD3 | CADPS | NNAT | C14orf39 | MS4A6A |
| HSPB8 | CCDC54 | NR1I3 | C15orf32 | NR1I3 | |
| PSG4 | CDH5 | NR4A1 | CARD14 | NR5A1 | |
| SEMA3B | CEACAM3 | NRXN1 | CCR9 | OR10A4 | |
| WFDC8 | CHI3L1 | NRXN3 | CHRNA1 | OR10H1 | |
| COX4I2 | OR10H1 | CNKSR1 | OR1N1 | ||
| CPA6 | P4HA3 | CYP2C9 | OR3A3 | ||
| DCN | PCDHB7 | CYP4F3 | P4HA3 | ||
| DLK1 | PHYHIP | DEFB126 | PABPC5 | ||
| DNMT3L | RGS12 | EGFL7 | PLAT | ||
| DOCK1 | S100A3 | FBLN1 | PMCHL1 | ||
| EDNRB | SATB1 | FLRT2 | PNLIPRP1 | ||
| FMR1NB | SCARF1 | GALR1 | RASL12 | ||
| FRMPD2 | SCN3B | GATA5 | RGS13 | ||
| FXYD3 | SLC13A5 | GKN1 | RHOJ | ||
| GATA5 | SLC35E4 | GLIS1 | SCGB1D1 | ||
| GIPR | SLC6A11 | GRIK2 | SLC6A11 | ||
| GLIS1 | SPATA3 | GUCA2B | SPATA21 | ||
| GUCA2B | SPINK4 | GUCY2F | SPRR3 | ||
| HBE1 | SPOCK3 | HCRTR1 | SPTB | ||
| IFNA16 | SPTB | IFNA17 | TFAP2B | ||
| IQCF1 | ST6GAL2 | IGSF11 | TRPM5 | ||
| KLHDC7A | SYN3 | IQCF1 | TTLL2 | ||
| KLK3 | SYNE2 | KIF25 | WFDC5 | ||
| KLK9 | TMEM119 | KLK9 | WWP1 | ||
| KRTAP20-1 | TMEM40 | KRT1 | ZBED2 | ||
| LAD1 | TNNI1 | LCE3C | ZDHHC11 | ||
| LCE3C | TRIM16 | ||||
| LCN2 | USH2A | ||||
| LHX3 | ZDHHC11 | ||||
| LILRA5 | ZPLD1 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.C.; Hu, V.W. Phenotypic Subtyping and Re-Analysis of Existing Methylation Data from Autistic Probands in Simplex Families Reveal ASD Subtype-Associated Differentially Methylated Genes and Biological Functions. Int. J. Mol. Sci. 2020, 21, 6877. https://doi.org/10.3390/ijms21186877
Lee EC, Hu VW. Phenotypic Subtyping and Re-Analysis of Existing Methylation Data from Autistic Probands in Simplex Families Reveal ASD Subtype-Associated Differentially Methylated Genes and Biological Functions. International Journal of Molecular Sciences. 2020; 21(18):6877. https://doi.org/10.3390/ijms21186877
Chicago/Turabian StyleLee, Elizabeth C., and Valerie W. Hu. 2020. "Phenotypic Subtyping and Re-Analysis of Existing Methylation Data from Autistic Probands in Simplex Families Reveal ASD Subtype-Associated Differentially Methylated Genes and Biological Functions" International Journal of Molecular Sciences 21, no. 18: 6877. https://doi.org/10.3390/ijms21186877
APA StyleLee, E. C., & Hu, V. W. (2020). Phenotypic Subtyping and Re-Analysis of Existing Methylation Data from Autistic Probands in Simplex Families Reveal ASD Subtype-Associated Differentially Methylated Genes and Biological Functions. International Journal of Molecular Sciences, 21(18), 6877. https://doi.org/10.3390/ijms21186877