Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
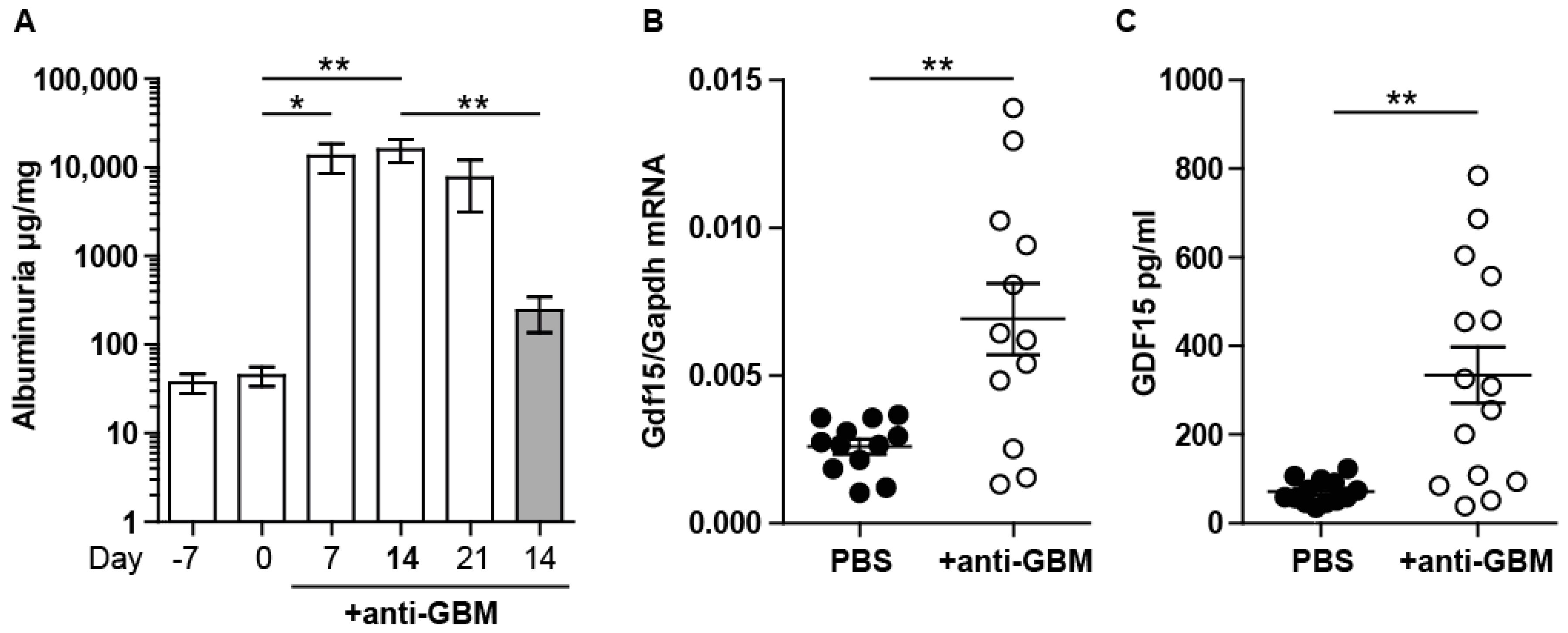
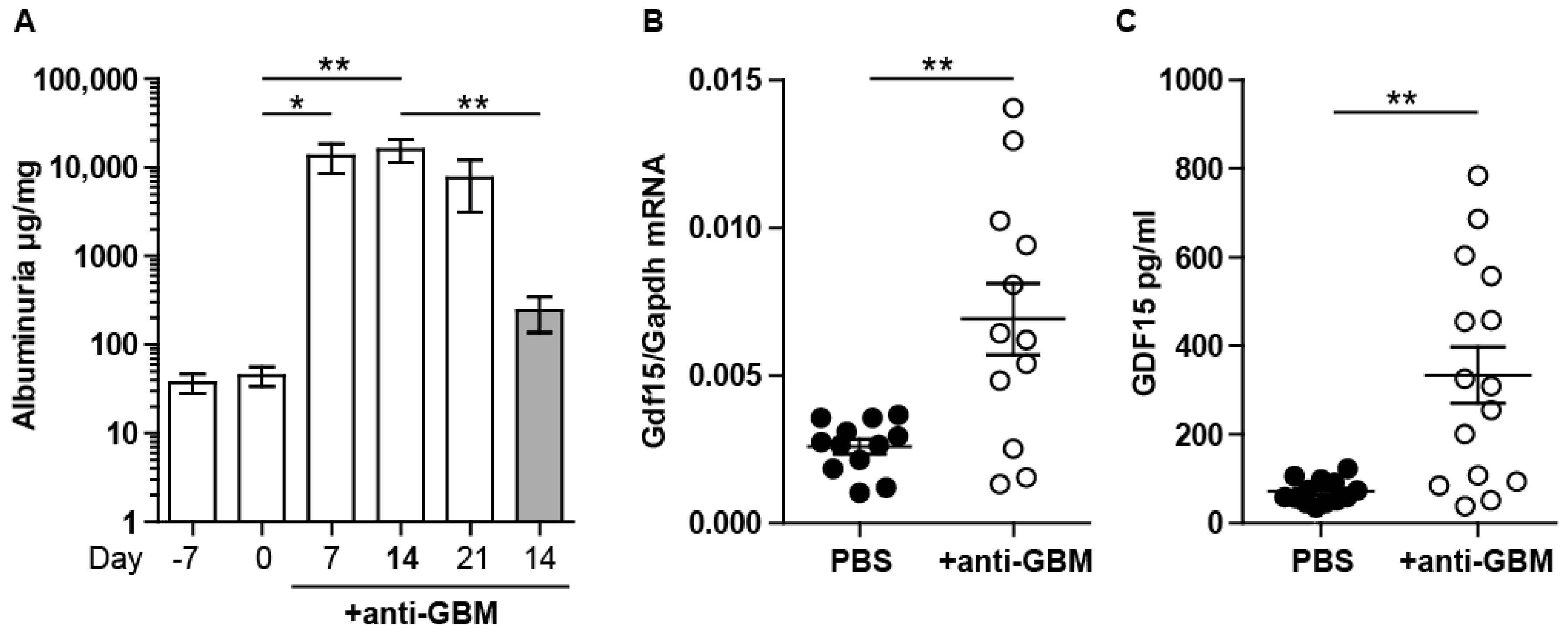
2.1. Experimental Anti-GBM Nephritis Is Associated with Higher Systemic and Intrarenal Expression of GDF15
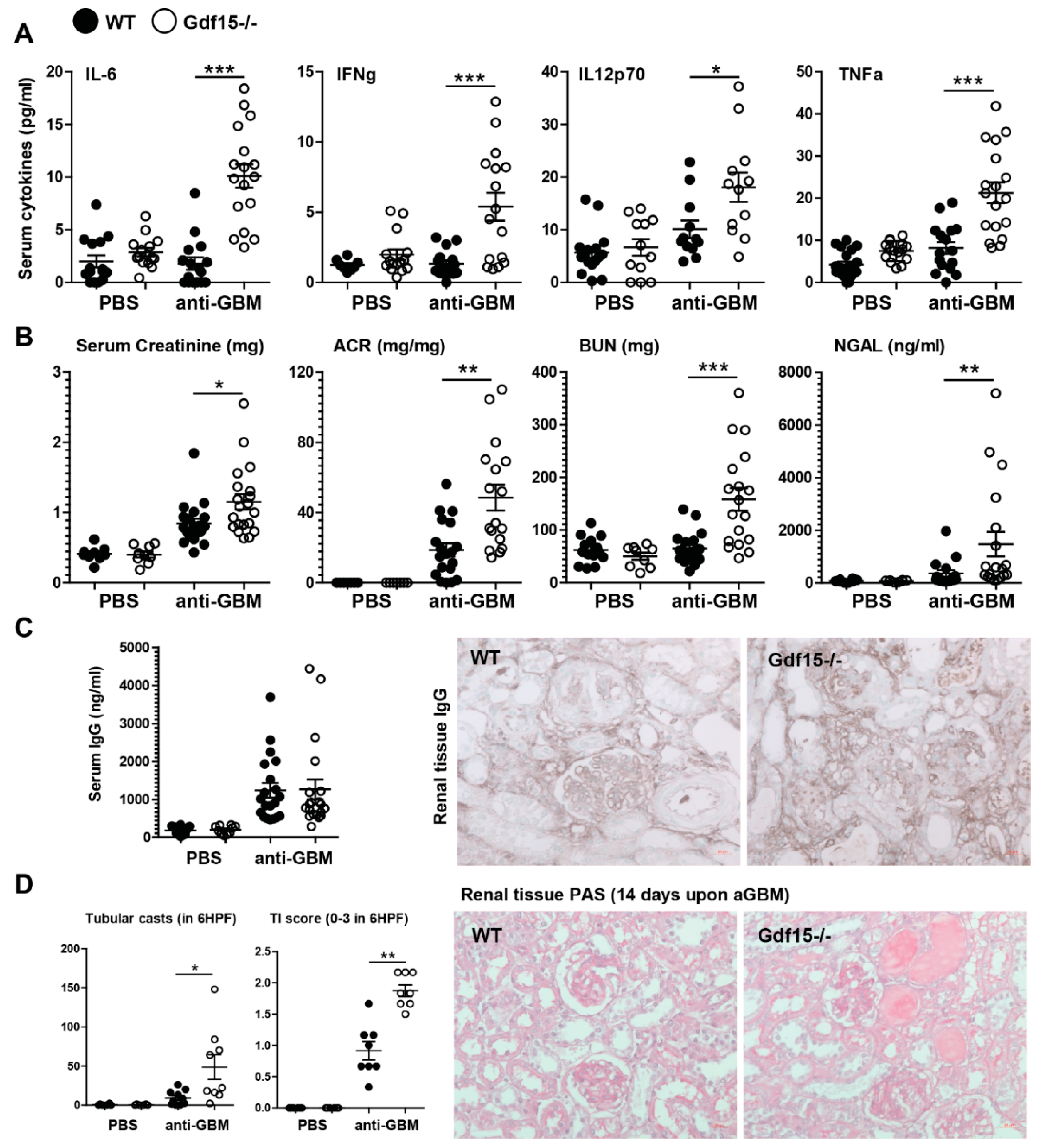
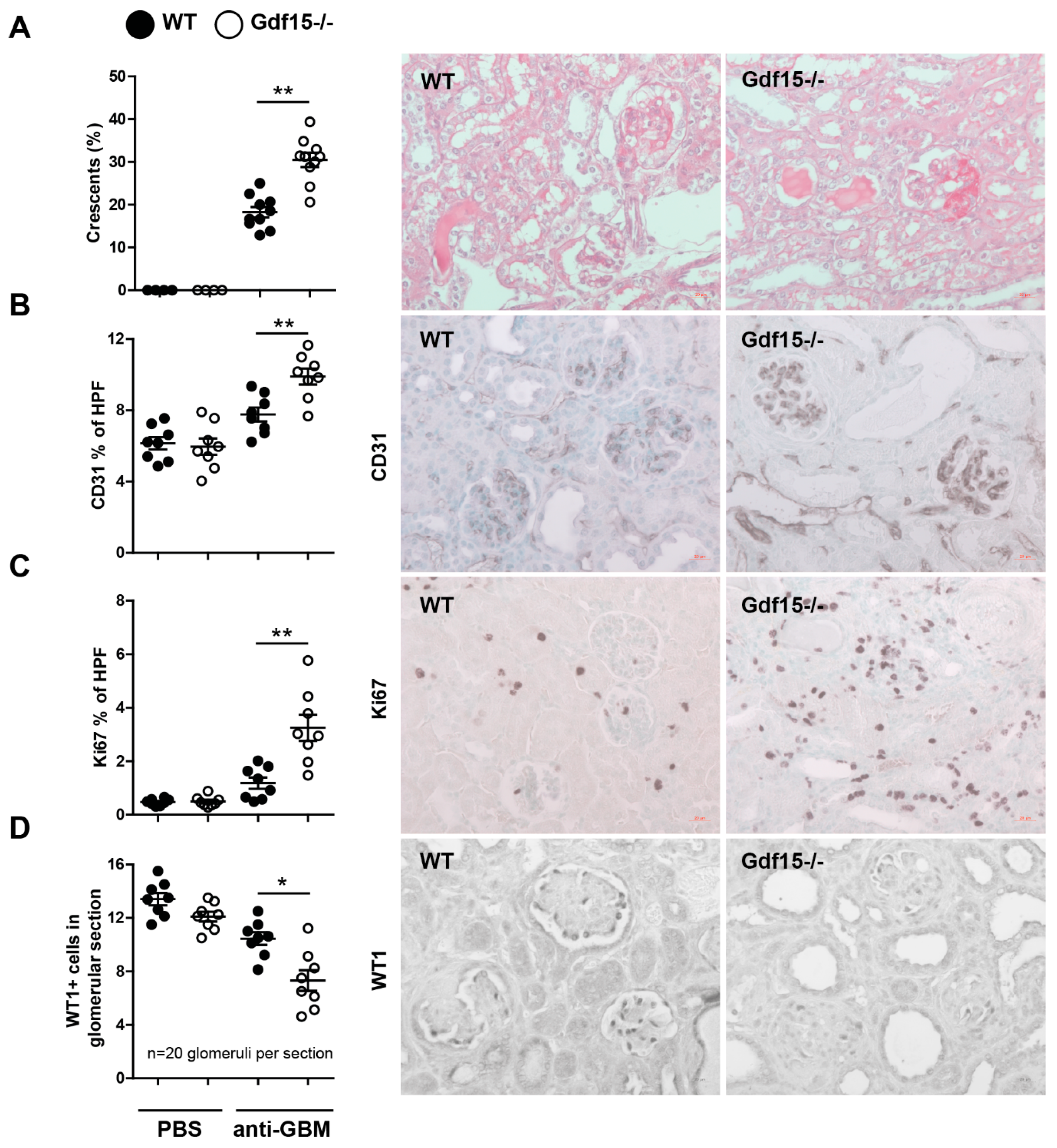
2.2. GDF15 Deficiency Aggravates Albuminuria, Kidney Function Loss, and More Severe Tubular and Glomerular Injury in Anti-GBM Nephritis
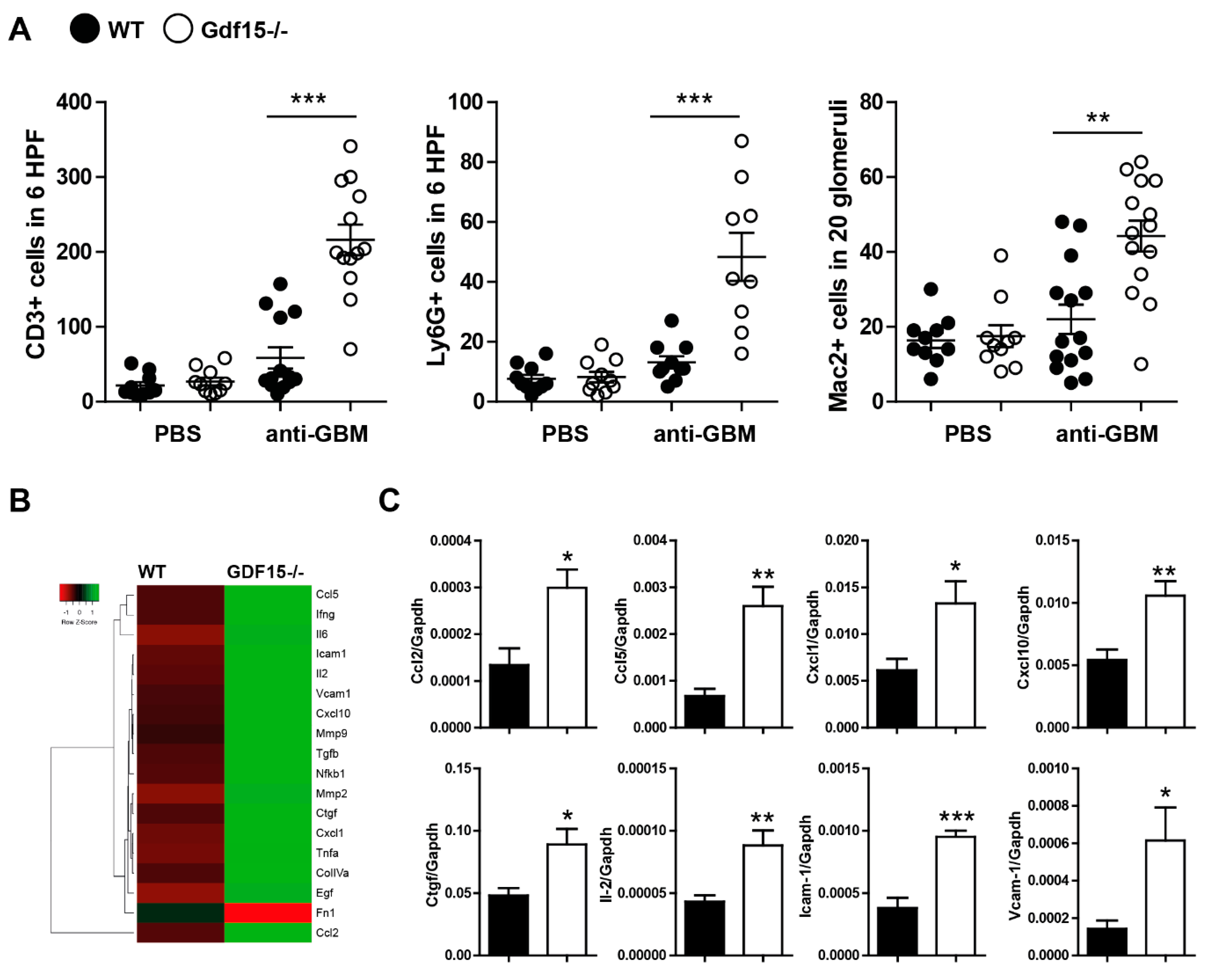
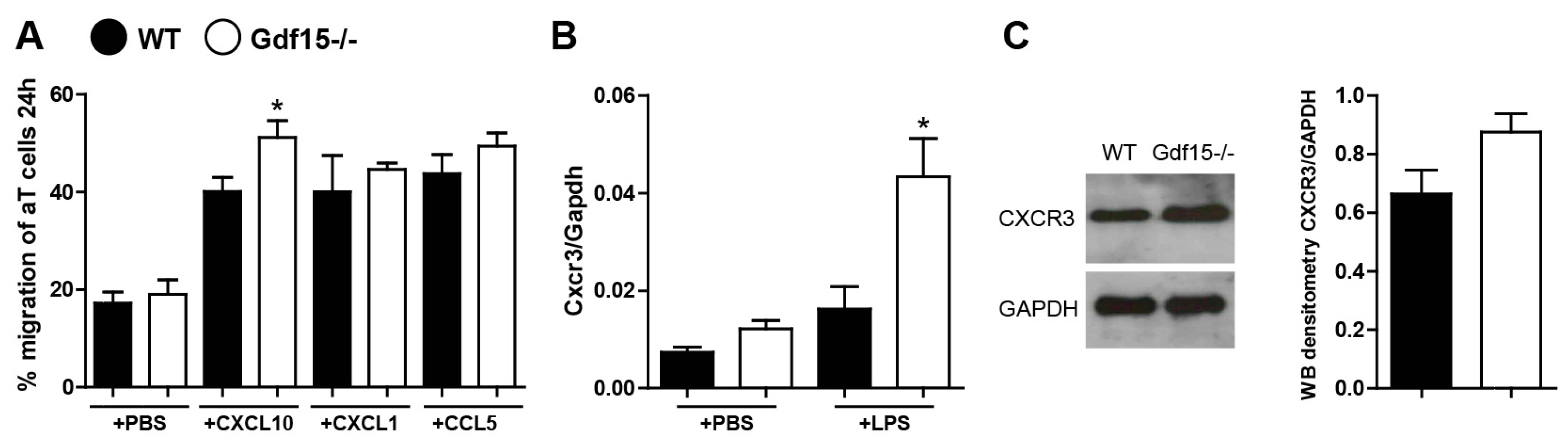
2.3. Gdf15-Deficient Mice Exhibit Increased Renal Inflammation in Anti-GBM Nephritis Model
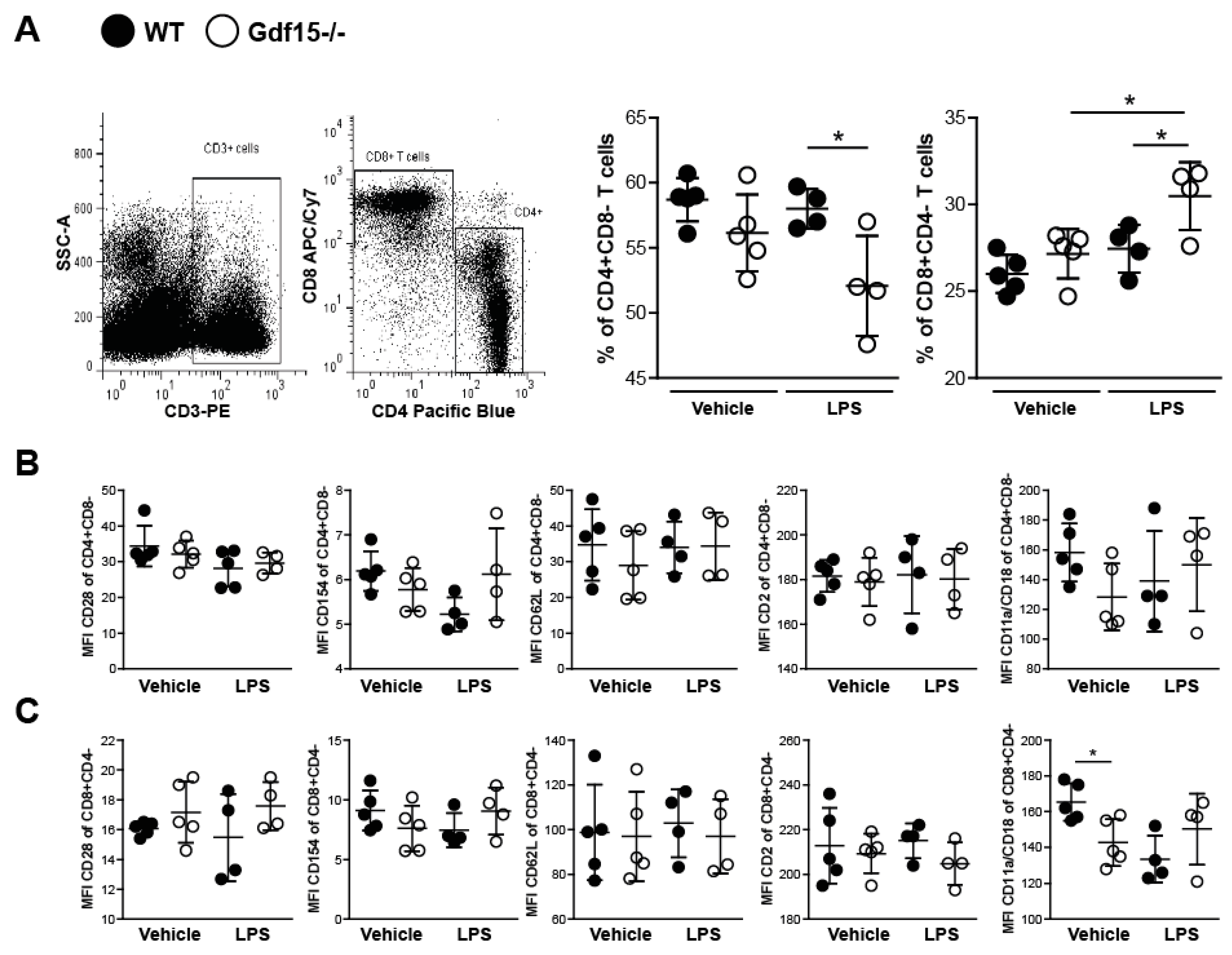
2.4. T Cells from Wild Type and Knockout Mice Display No Differences in Expression of Adhesions Markers
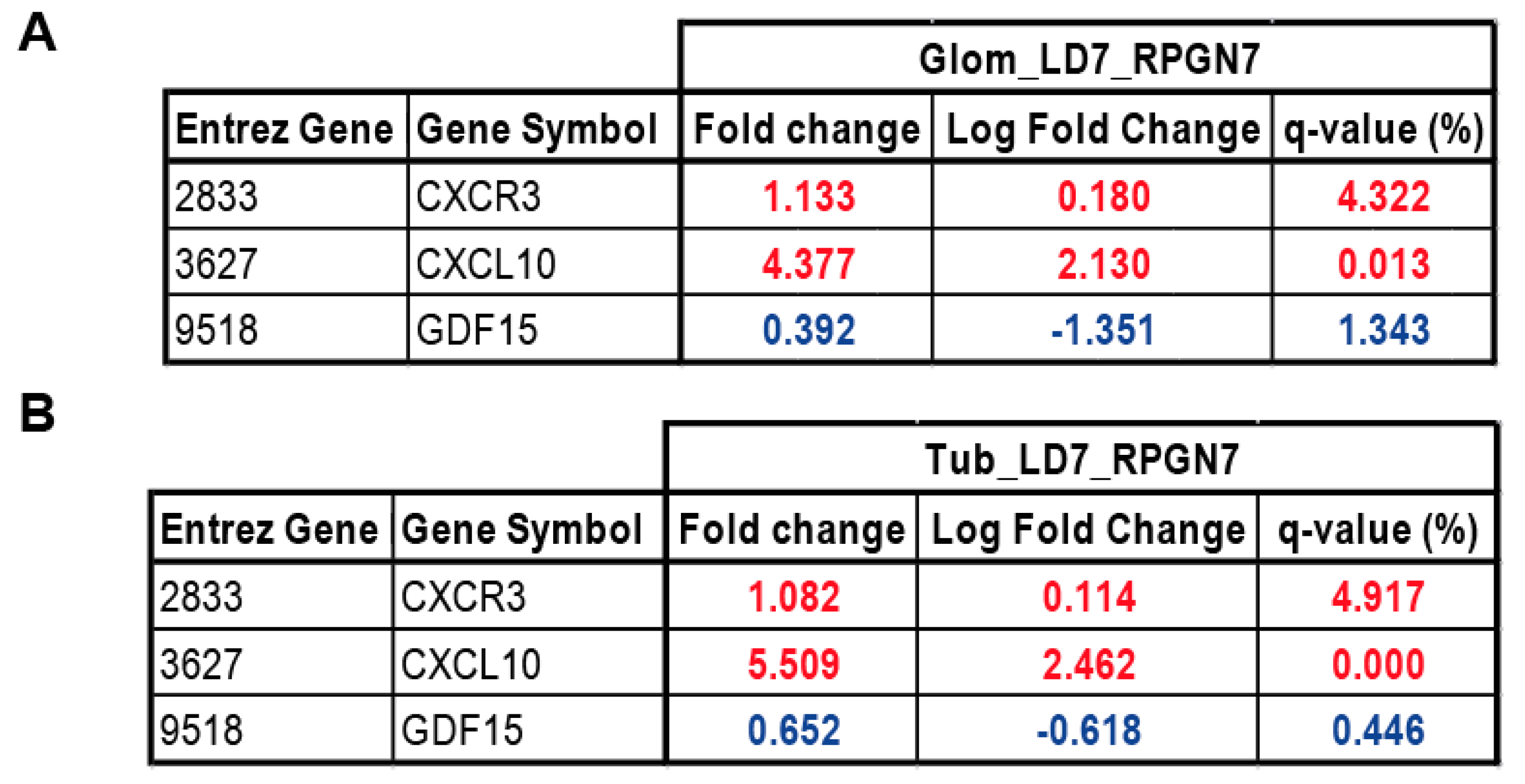
2.5. Gene Expression Analysis of GDF15, CXCL10 and CXCR3 in Patients with RPGN
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
References
- McAdoo, S.P.; Pusey, C.D. Anti-Glomerular Basement Membrane Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1162–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canney, M.; O’Hara, P.V.; McEvoy, C.M.; Medani, S.; Connaughton, D.M.; Abdalla, A.A.; Doyle, R.; Stack, A.G.; O’Seaghdha, C.M.; Clarkson, M.R.; et al. Spatial and Temporal Clustering of Anti-Glomerular Basement Membrane Disease. Clin. J. Am. Soc. Nephrol. 2016, 11, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Muller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artinger, K.; Kirsch, A.H.; Aringer, I.; Moschovaki-Filippidou, F.; Eller, P.; Rosenkranz, A.R.; Eller, K. Innate and adaptive immunity in experimental glomerulonephritis: A pathfinder tale. Pediatr. Nephrol. 2017, 32, 943–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Fueyo, A.; Bradley, S.J.; Klatzmann, D.; Tsokos, G.C. T cells and autoimmune kidney disease. Nat. Rev. Nephrol. 2017, 13, 329–343. [Google Scholar] [CrossRef]
- Krebs, C.F.; Steinmetz, O.M. CD4(+) T Cell Fate in Glomerulonephritis: A Tale of Th1, Th17, and Novel Treg Subtypes. Mediat. Inflamm. 2016, 2016, 5393894. [Google Scholar] [CrossRef] [Green Version]
- Summers, S.A.; Steinmetz, O.M.; Li, M.; Kausman, J.Y.; Semple, T.; Edgtton, K.L.; Borza, D.B.; Braley, H.; Holdsworth, S.R.; Kitching, A.R. Th1 and Th17 cells induce proliferative glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 2518–2524. [Google Scholar] [CrossRef] [Green Version]
- Hopfer, H.; Holzer, J.; Hunemorder, S.; Paust, H.J.; Sachs, M.; Meyer-Schwesinger, C.; Turner, J.E.; Panzer, U.; Mittrucker, H.W. Characterization of the renal CD4+ T-cell response in experimental autoimmune glomerulonephritis. Kidney Int. 2012, 82, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Odobasic, D.; Gan, P.Y.; Summers, S.A.; Semple, T.J.; Muljadi, R.C.; Iwakura, Y.; Kitching, A.R.; Holdsworth, S.R. Interleukin-17A promotes early but attenuates established disease in crescentic glomerulonephritis in mice. Am. J. Pathol. 2011, 179, 1188–1198. [Google Scholar] [CrossRef]
- Paust, H.J.; Turner, J.E.; Riedel, J.H.; Disteldorf, E.; Peters, A.; Schmidt, T.; Krebs, C.; Velden, J.; Mittrucker, H.W.; Steinmetz, O.M.; et al. Chemokines play a critical role in the cross-regulation of Th1 and Th17 immune responses in murine crescentic glomerulonephritis. Kidney Int. 2012, 82, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, A.; Ueno, H.; Shimomura, M.; Tanaka, R.; Shirakawa, T.; Nakamura, H.; Matsuo, M.; Iijima, K. Blockade of TGF-beta action ameliorates renal dysfunction and histologic progression in anti-GBM nephritis. Kidney Int. 2003, 64, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesnard, L.; Keller, A.C.; Michel, M.L.; Vandermeersch, S.; Rafat, C.; Letavernier, E.; Tillet, Y.; Rondeau, E.; Leite-de-Moraes, M.C. Invariant natural killer T cells and TGF-beta attenuate anti-GBM glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 1282–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamaru, Y.; Nakao, A.; Mamura, M.; Suzuki, Y.; Shirato, I.; Okumura, K.; Tomino, Y.; Ra, C. Blockade of TGF-beta signaling in T cells prevents the development of experimental glomerulonephritis. J. Immunol. 2001, 166, 2818–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Li, W.; Song, Y.; Wang, L.; Zhang, K.; Yang, J.; Zhang, W.; Su, H.; Zhang, Y. Growth differentiation factor-15 suppresses maturation and function of dendritic cells and inhibits tumor-specific immune response. PLoS ONE 2013, 8, e78618. [Google Scholar] [CrossRef] [PubMed]
- Soucek, K.; Slabakova, E.; Ovesna, P.; Malenovska, A.; Kozubik, A.; Hampl, A. Growth/differentiation factor-15 is an abundant cytokine in human seminal plasma. Hum. Reprod 2010, 25, 2962–2971. [Google Scholar] [CrossRef] [Green Version]
- Rochette, L.; Meloux, A.; Zeller, M.; Cottin, Y.; Vergely, C. Functional roles of GDF15 in modulating microenvironment to promote carcinogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165798. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Baek, S.J.; Eling, T. Growth differentiation factor 15 (GDF15): A survival protein with therapeutic potential in metabolic diseases. Pharmacol. Ther. 2019, 198, 46–58. [Google Scholar] [CrossRef]
- Tsai, V.W.W.; Husaini, Y.; Sainsbury, A.; Brown, D.A.; Breit, S.N. The MIC-1/GDF15-GFRAL Pathway in Energy Homeostasis: Implications for Obesity, Cachexia, and Other Associated Diseases. Cell Metab. 2018, 28, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yang, X.; Dai, J.; Lu, Y.; Zhang, J.; Keller, E.T. Prostate cancer promotes a vicious cycle of bone metastasis progression through inducing osteocytes to secrete GDF15 that stimulates prostate cancer growth and invasion. Oncogene 2019, 38, 4540–4559. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, Y.M.; Zheng, P.S.; Zhang, P. GDF15 promotes the proliferation of cervical cancer cells by phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J. Exp. Clin. Cancer Res. 2018, 37, 80. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, P.J.; Duffin, K.L.; Chintharlapalli, S.; Wu, X. GDF15 and Growth Control. Front. Physiol. 2018, 9, 1712. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Danoff, T.M.; Okada, H.; Neilson, E.G. Susceptibility to anti-glomerular basement membrane disease and Goodpasture syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in mice. J. Clin. Investig. 1997, 100, 2263–2275. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.R.; Holdsworth, S.R.; Tipping, P.G. Evidence for delayed-type hypersensitivity mechanisms in glomerular crescent formation. Kidney Int. 1994, 46, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Tipping, P.G.; Huang, X.R.; Qi, M.; Van, G.Y.; Tang, W.W. Crescentic glomerulonephritis in CD4- and CD8-deficient mice. Requirement for CD4 but not CD8 cells. Am. J. Pathol. 1998, 152, 1541–1548. [Google Scholar]
- Huang, X.R.; Tipping, P.G.; Apostolopoulos, J.; Oettinger, C.; D’Souza, M.; Milton, G.; Holdsworth, S.R. Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin. Exp. Immunol. 1997, 109, 134–142. [Google Scholar] [CrossRef]
- Reynolds, J.; Norgan, V.A.; Bhambra, U.; Smith, J.; Cook, H.T.; Pusey, C.D. Anti-CD8 monoclonal antibody therapy is effective in the prevention and treatment of experimental autoimmune glomerulonephritis. J. Am. Soc. Nephrol. 2002, 13, 359–369. [Google Scholar]
- Fujinaka, H.; Yamamoto, T.; Feng, L.; Kawasaki, K.; Yaoita, E.; Hirose, S.; Goto, S.; Wilson, C.B.; Uchiyama, M.; Kihara, I. Crucial role of CD8-positive lymphocytes in glomerular expression of ICAM-1 and cytokines in crescentic glomerulonephritis of WKY rats. J. Immunol. 1997, 158, 4978–4983. [Google Scholar]
- Kim, Y.I.; Shin, H.W.; Chun, Y.S.; Park, J.W. CST3 and GDF15 ameliorate renal fibrosis by inhibiting fibroblast growth and activation. Biochem. Biophys. Res. Commun. 2018, 500, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.; Liu, Y.; Chen, R.; Zhao, D.; McAlister, V.; Mele, T.; Liu, K.; Zheng, X. GDF15 Regulates Malat-1 Circular RNA and Inactivates NFkappaB Signaling Leading to Immune Tolerogenic DCs for Preventing Alloimmune Rejection in Heart Transplantation. Front. Immunol. 2018, 9, 2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jiang, M.; Nouraie, M.; Roth, M.G.; Tabib, T.; Winters, S.; Chen, X.; Sembrat, J.; Chu, Y.; Cardenes, N.; et al. GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2019, 317, L510–L521. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.H.; Wang, A.; Hilliard, B.K.; Carvalho, F.; Rosen, C.E.; Ahasic, A.M.; Herzog, E.L.; Kang, I.; Pisani, M.A.; Yu, S.; et al. GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 2019, 178, 1231–1244. [Google Scholar] [CrossRef]
- Lichtnekert, J.; Kulkarni, O.P.; Mulay, S.R.; Rupanagudi, K.V.; Ryu, M.; Allam, R.; Vielhauer, V.; Muruve, D.; Lindenmeyer, M.T.; Cohen, C.D.; et al. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PLoS ONE 2011, 6, e26778. [Google Scholar] [CrossRef] [Green Version]
- Tipping, P.G.; Holdsworth, S.R. T cells in glomerulonephritis. Springer Semin. Immunopathol. 2003, 24, 377–393. [Google Scholar] [CrossRef]
- Zhang, Y.; Thai, K.; Kepecs, D.M.; Winer, D.; Gilbert, R.E. Reversing CXCL10 Deficiency Ameliorates Kidney Disease in Diabetic Mice. Am. J. Pathol. 2018, 188, 2763–2773. [Google Scholar] [CrossRef] [Green Version]
- Gniewkiewicz, M.S.; Czerwinska, M.; Gozdowska, J.; Czerwinska, K.; Sadowska, A.; Deborska-Materkowska, D.; Perkowska-Ptasinska, A.; Kosieradzki, M.; Durlik, M. Urinary levels of CCL2 and CXCL10 chemokines as potential biomarkers of ongoing pathological processes in kidney allograft: An association with BK virus nephropathy. Pol. Arch. Intern. Med. 2019, 129, 592–597. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, I.; Wada, T.; Furuichi, K.; Sakai, N.; Kitagawa, K.; Yokoyama, H.; Ishida, Y.; Kondo, T.; Sugaya, T.; Kawachi, H.; et al. Blockade of IP-10/CXCR3 promotes progressive renal fibrosis. Nephron Exp. Nephrol. 2007, 107, e12–e21. [Google Scholar] [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011, 89, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, T.; Zarbock, A.; Widera, C.; Butz, S.; Stadtmann, A.; Rossaint, J.; Bolomini-Vittori, M.; Korf-Klingebiel, M.; Napp, L.C.; Hansen, B.; et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat. Med. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Eggenhuizen, P.; O’Sullivan, K.M.; Alikhan, M.A.; Holdsworth, S.R.; Ooi, J.D.; Kitching, A.R. CD8+ T Cells Effect Glomerular Injury in Experimental Anti-Myeloperoxidase GN. J. Am. Soc. Nephrol. 2017, 28, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.S.; Lin, E.; Zhang, B.; Luster, A.D.; Tollett, J.; Samuel, M.A.; Engle, M.; Diamond, M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005, 79, 11457–11466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, J.A.; Jans, D.A.; Jans, P.J.; Smyth, M.J.; Browne, K.A.; Sutton, V.R. Efficient nuclear targeting of granzyme B and the nuclear consequences of apoptosis induced by granzyme B and perforin are caspase-dependent, but cell death is caspase-independent. J. Biol. Chem. 1998, 273, 27934–27938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef]
- Nagata, S.; Golstein, P. The Fas death factor. Science 1995, 267, 1449–1456. [Google Scholar] [CrossRef]
- Fung-Leung, W.P.; Schilham, M.W.; Rahemtulla, A.; Kundig, T.M.; Vollenweider, M.; Potter, J.; van Ewijk, W.; Mak, T.W. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell 1991, 65, 443–449. [Google Scholar] [CrossRef]
- Brown, H.J.; Lock, H.R.; Wolfs, T.G.; Buurman, W.A.; Sacks, S.H.; Robson, M.G. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J. Am. Soc. Nephrol. 2007, 18, 1732–1739. [Google Scholar] [CrossRef] [Green Version]
- Allam, R.; Anders, H.J. The role of innate immunity in autoimmune tissue injury. Curr. Opin. Rheumatol. 2008, 20, 538–544. [Google Scholar] [CrossRef]
- Greenhall, G.H.; Salama, A.D. What is new in the management of rapidly progressive glomerulonephritis? Clin. Kidney J. 2015, 8, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Xing, L.; Cerise, J.; Wang, E.H.; Jabbari, A.; de Jong, A.; Petukhova, L.; Christiano, A.M.; Clynes, R. CXCR3 Blockade Inhibits T Cell Migration into the Skin and Prevents Development of Alopecia Areata. J. Immunol. 2016, 197, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.; Toksoy, A.; Spandau, U.; Engelhardt, E.; Brocker, E.B.; Gillitzer, R. The C-X-C chemokine Mig is highly expressed in the papillae of psoriatic lesions. J. Pathol. 1998, 184, 89–95. [Google Scholar] [CrossRef]
- Yellin, M.; Paliienko, I.; Balanescu, A.; Ter-Vartanian, S.; Tseluyko, V.; Xu, L.A.; Tao, X.; Cardarelli, P.M.; Leblanc, H.; Nichol, G.; et al. A phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Robinson-Cohen, C.; Smith, M.R.; Bellovich, K.A.; Bhat, Z.Y.; Bobadilla, M.; Brosius, F.; de Boer, I.H.; Essioux, L.; Formentini, I.; et al. Growth Differentiation Factor-15 and Risk of CKD Progression. J. Am. Soc. Nephrol. 2017, 28, 2233–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breit, S.N.; Carrero, J.J.; Tsai, V.W.; Yagoutifam, N.; Luo, W.; Kuffner, T.; Bauskin, A.R.; Wu, L.; Jiang, L.; Barany, P.; et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15) and mortality in end-stage renal disease. Nephrol. Dial. Transplant. 2012, 27, 70–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajer, M.; Jorsal, A.; Tarnow, L.; Parving, H.H.; Rossing, P. Plasma growth differentiation factor-15 independently predicts all-cause and cardiovascular mortality as well as deterioration of kidney function in type 1 diabetic patients with nephropathy. Diabetes Care 2010, 33, 1567–1572. [Google Scholar] [CrossRef] [Green Version]
- Anand, I.S.; Kempf, T.; Rector, T.S.; Tapken, H.; Allhoff, T.; Jantzen, F.; Kuskowski, M.; Cohn, J.N.; Drexler, H.; Wollert, K.C. Serial measurement of growth-differentiation factor-15 in heart failure: Relation to disease severity and prognosis in the Valsartan Heart Failure Trial. Circulation 2010, 122, 1387–1395. [Google Scholar] [CrossRef] [Green Version]
- Eggers, K.M.; Kempf, T.; Wallentin, L.; Wollert, K.C.; Lind, L. Change in growth differentiation factor 15 concentrations over time independently predicts mortality in community-dwelling elderly individuals. Clin. Chem. 2013, 59, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Na, K.R.; Kim, Y.H.; Chung, H.K.; Yeo, M.K.; Ham, Y.R.; Jeong, J.Y.; Kim, K.S.; Lee, K.W.; Choi, D.E. Growth differentiation factor 15 as a predictor of adverse renal outcomes in patients with immunoglobulin A nephropathy. Intern. Med. J. 2017, 47, 1393–1399. [Google Scholar] [CrossRef]
- Ham, Y.R.; Song, C.H.; Bae, H.J.; Jeong, J.Y.; Yeo, M.K.; Choi, D.E.; Na, K.R.; Lee, K.W. Growth Differentiation Factor-15 as a Predictor of Idiopathic Membranous Nephropathy Progression: A Retrospective Study. Dis. Markers 2018, 2018, 1463940. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.C.; Nowak, C.; Lind, L.; Ostgren, C.J.; Nystrom, F.H.; Sundstrom, J.; Carrero, J.J.; Riserus, U.; Ingelsson, E.; Fall, T.; et al. Growth differentiation factor 15 (GDF-15) is a potential biomarker of both diabetic kidney disease and future cardiovascular events in cohorts of individuals with type 2 diabetes: A proteomics approach. Upsala J. Med. Sci 2020, 125, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonson, M.S.; Tiktin, M.; Debanne, S.M.; Rahman, M.; Berger, B.; Hricik, D.; Ismail-Beigi, F. The renal transcriptome of db/db mice identifies putative urinary biomarker proteins in patients with type 2 diabetes: A pilot study. Am. J. Physiol. Renal Physiol. 2012, 302, F820–F829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, O.E.; Skjaervik, A.; Stordal, B.F.; Sundan, A.; Holien, T. TGF-beta contamination of purified recombinant GDF15. PLoS ONE 2017, 12, e0187349. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.D.; Frach, K.; Schlondorff, D.; Kretzler, M. Quantitative gene expression analysis in renal biopsies: A novel protocol for a high-throughput multicenter application. Kidney Int. 2002, 61, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.D.; Klingenhoff, A.; Boucherot, A.; Nitsche, A.; Henger, A.; Brunner, B.; Schmid, H.; Merkle, M.; Saleem, M.A.; Koller, K.P.; et al. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 5682–5687. [Google Scholar] [CrossRef] [Green Version]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Shved, N.; Warsow, G.; Eichinger, F.; Hoogewijs, D.; Brandt, S.; Wild, P.; Kretzler, M.; Cohen, C.D.; Lindenmeyer, M.T. Transcriptome-based network analysis reveals renal cell type-specific dysregulation of hypoxia-associated transcripts. Sci. Rep. 2017, 7, 8576. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moschovaki-Filippidou, F.; Steiger, S.; Lorenz, G.; Schmaderer, C.; Ribeiro, A.; von Rauchhaupt, E.; Cohen, C.D.; Anders, H.-J.; Lindenmeyer, M.; Lech, M. Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice. Int. J. Mol. Sci. 2020, 21, 6978. https://doi.org/10.3390/ijms21196978
Moschovaki-Filippidou F, Steiger S, Lorenz G, Schmaderer C, Ribeiro A, von Rauchhaupt E, Cohen CD, Anders H-J, Lindenmeyer M, Lech M. Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice. International Journal of Molecular Sciences. 2020; 21(19):6978. https://doi.org/10.3390/ijms21196978
Chicago/Turabian StyleMoschovaki-Filippidou, Foteini, Stefanie Steiger, Georg Lorenz, Christoph Schmaderer, Andrea Ribeiro, Ekaterina von Rauchhaupt, Clemens D. Cohen, Hans-Joachim Anders, Maja Lindenmeyer, and Maciej Lech. 2020. "Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice" International Journal of Molecular Sciences 21, no. 19: 6978. https://doi.org/10.3390/ijms21196978
APA StyleMoschovaki-Filippidou, F., Steiger, S., Lorenz, G., Schmaderer, C., Ribeiro, A., von Rauchhaupt, E., Cohen, C. D., Anders, H.-J., Lindenmeyer, M., & Lech, M. (2020). Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice. International Journal of Molecular Sciences, 21(19), 6978. https://doi.org/10.3390/ijms21196978