Transcriptomic Profiling for the Autophagy Pathway in Colorectal Cancer



Abstract
1. Introduction
2. Results
2.1. Ensemble of Gene Set Enrichment Analysis (EGSEA)
2.2. Unsupervised Clustering of Colorectal Tumor Samples and Cell Lines
2.3. Molecular Characteristics of Autophagy Clusters (RNA-seq Data)
2.4. Molecular Characteristics of Autophagy Clusters (Affymetrix Data)
2.5. Selection of the Autophagy Genes that Best Define Autophagy Clusters
2.6. Differential Expression (DE) of Autophagy Genes
3. Discussion
4. Study Limitations
5. Materials and Methods
5.1. Data Acquisition
5.2. Consensus Molecular Subtyping
5.3. Data Pre-Processing
5.4. Selection of Genes of Interest
5.5. Ensemble of Gene Set Enrichment Analysis
5.6. Unsupervised Clustering of Autophagy Genes
5.7. Selection of the Autophagy Genes that Best Define Autophagy Clusters
5.8. Differential Expression (DE) Analysis
5.9. Comparison of Expression of Selected Autophagy-Related Genes in CRC Cell Lines
5.10. Molecular Characteristics of Clusters
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BH | Benjamini–Hochberg p-value correction for multiple testing |
| CMS | consensus molecular subtype |
| COMMUNAL | Combined Mapping of Multiple Clustering Algorithms |
| CRC | colorectal cancer |
| DE | differential expression |
| EGSEA | Ensemble of Gene Set Enrichment Analyses |
| FDR | False Discovery Rate |
| logFC | log fold change |
| MSI | microsatellite instability; microsatellite instable |
| MSS | microsatellite stability; microsatellite stable |
| NTP | nearest template prediction |
| RNA-seq | RNA sequencing |
| sPLS-DA | Sparse Partial Least Squares Discriminant Analysis |
| TCGA | The Cancer Genome Atlas |
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Hashim, D.; Boffetta, P.; La Vecchia, C.; Rota, M.; Bertuccio, P.; Malvezzi, M.; Negri, E. The global decrease in cancer mortality: Trends and disparities. Ann. Oncol. 2016, 27, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Welch, H.G.; Robertson, D.J. Colorectal Cancer on the Decline--Why Screening Can’t Explain It All. N. Engl. J. Med. 2016, 374, 1605–1607. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, J.; Davidson, C.; Hall, C.; Pearson, J.; Eglinton, T.; Wakeman, C.; Frizelle, F. Population-based study demonstrating an increase in colorectal cancer in young patients. Br. J. Surg. 2017, 104, 1063–1068. [Google Scholar] [CrossRef]
- Young, J.P.; Win, A.K.; Rosty, C.; Flight, I.; Roder, D.; Young, G.P.; Frank, O.; Suthers, G.K.; Hewett, P.J.; Ruszkiewicz, A.; et al. Rising incidence of early-onset colorectal cancer in Australia over two decades: Report and review. J. Gastroenterol. Hepatol. 2015, 30, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Stembalska, A.; Łaczmańska, I.; Sąsiadek, M. Sporadic colorectal cancer—Factors modulating individual susceptibility to cancer. Współczesna Onkol. 2010, 3, 123–128. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Linnekamp, J.F.; van Hooff, S.R.; Prasetyanti, P.R.; Kandimalla, R.; Buikhuisen, J.Y.; Fessler, E.; Ramesh, P.; Lee, K.A.S.T.; Bochove, G.G.W.; de Jong, J.H.; et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018, 25, 616–633. [Google Scholar] [CrossRef]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilertsen, I.A.; Ramirez, L.; Murumagi, A.; Arjama, M.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal cancer consensus molecular subtypes translated to preclinical models uncover potentially targetable cancer cell dependencies. Clin. Cancer Res. 2018, 24, 794–806. [Google Scholar] [CrossRef]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Pesz, K.A.; Sąsiadek, M.M. May autophagy be a novel biomarker and antitumor target in colorectal cancer? Biomark. Med. 2016, 10, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Devenport, S.N.; Shah, Y.M. Functions and Implications of Autophagy in Colon Cancer. Cells 2019, 8, 1349. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Yang, X.; Yu, D.D.; Yan, F.; Jing, Y.Y.; Han, Z.P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The Four Faces of Autophagy: Implications for Cancer Therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef]
- Weldon Gilcrease, G.; Stenehjem, D.D.; Wade, M.L.; Weis, J.; McGregor, K.; Whisenant, J.; Boucher, K.M.; Thorne, K.; Orgain, N.; Garrido-Laguna, I.; et al. Phase I/II study of everolimus combined with mFOLFOX-6 and bevacizumab for first–line treatment of metastatic colorectal cancer. Investig. New Drugs 2019, 37, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Ervin, T.J.; Senzer, N.N.; Richards, D.A.; Firdaus, I.; Lockhart, A.C.; Cohn, A.L.; Saleh, M.N.; Gardner, L.R.; Sportelli, P.; et al. Results of the X-PECT study: A phase III randomized double-blind, placebo-controlled study of perifosine plus capecitabine (P-CAP) versus placebo plus capecitabine (CAP) in patients (pts) with refractory metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2012, 30, LBA3501. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Vassilopoulou, L.; Fragkiadaki, P.; Sapsakos, T.M.; Papadakis, G.Z.; Spandidos, D.A.; Tsatsakis, A.M.; Tsiaoussis, J. Improving diagnosis, prognosis and prediction by using biomarkers in CRC patients (Review). Oncol. Rep. 2018, 39, 2455–2472. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Mieczkowski, J.; Swiatek-Machado, K.; Kaminska, B. Identification of pathway deregulation—Gene expression based analysis of consistent signal transduction. PLoS ONE 2012, 7, e41541. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Alessandrini, F.; Pezzè, L.; Ciribilli, Y. LAMPs: Shedding light on cancer biology. Semin. Oncol. 2017, 44, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Furuta, K.; Ikeda, M.; Nakayama, Y.; Nakamura, K.; Tanaka, M.; Hamasaki, N.; Himeno, M.; Hamilton, S.R.; August, J.T. Expression of lysosome-associated membrane proteins in human colorectal neoplasms and inflammatory diseases. Am. J. Pathol. 2001, 159, 449–455. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Yao, J.; Jin, Q.; Wang, X.; Zhu, H.; Huang, F.; Wang, W.; Qiang, J.; Ni, Q. LAMP1 expression is associated with poor prognosis in breast cancer. Oncol. Lett. 2017, 14, 4729–4735. [Google Scholar] [CrossRef]
- Sarafian, V.S.; Koev, I.; Mehterov, N.; Kazakova, M.; Dangalov, K. LAMP-1 gene is overexpressed in high grade glioma. APMIS 2018, 126, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Okato, A.; Goto, Y.; Kurozumi, A.; Kato, M.; Kojima, S.; Matsushita, R.; Yonemori, M.; Miyamoto, K.; Ichikawa, T.; Seki, N. Direct regulation of LAMP1 by tumor-suppressive microRNA-320a in prostate cancer. Int. J. Oncol. 2016, 49, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Koseki, J.; Takahashi, H.; Miyoshi, N.; Nishida, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Yamamoto, H.; et al. Disruption of endolysosomal Rab5/7 efficiently eliminates colorectal cancer stem cells. Cancer Res. 2019, 79, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.Y.; O’Prey, J.; Baudot, A.D.; Hoekstra, A.; Ryan, K.M. DRAM-1 encodes multiple isoforms that regulate autophagy. Autophagy 2012, 8, 18–28. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Guan, J.-J.; Zhang, X.-D.; Sun, W.; Qi, L.; Wu, J.-C.; Qin, Z.-H. DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis. 2015, 6, e1624. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, J.; Luo, L.; Sheng, P.; Wang, B.; Zhang, J.; Peng, S. Genome-Wide Identification of a Novel Autophagy-Related Signature for Colorectal Cancer. Dose Response 2019, 17, 155932581989417. [Google Scholar] [CrossRef]
- Zhou, Z.; Mo, S.; Dai, W.; Ying, Z.; Zhang, L.; Xiang, W.; Han, L.; Wang, Z.; Li, Q.; Wang, R.; et al. Development and Validation of an Autophagy Score Signature for the Prediction of Post-operative Survival in Colorectal Cancer. Front. Oncol. 2019, 9, 878. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Kyriakopoulou, G.; Papavassiliou, A.G.; Karamouzis, M. V The Interplay of Autophagy and Tumor Microenvironment in Colorectal Cancer-Ways of Enhancing Immunotherapy Action. Cancers 2019, 11, 533. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.W.; Loi, M.; Ligeon, L.-A.; Gannagé, M.; Lünemann, J.D.; Münz, C. Endocytosis regulation by autophagy proteins in MHC restricted antigen presentation. Curr. Opin. Immunol. 2018, 52, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Jackson, L.K.; Johnson, W.E.; Li, D.Y.; Bild, A.H.; Piccolo, S.R. Alternative preprocessing of RNA-Sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics 2015, 31, 3666–3672. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.J.; Smith, J.J.; Chen, X.; Washington, M.K.; Roland, J.T.; Means, A.L.; Eschrich, S.A.; Yeatman, T.J.; Deane, N.G.; Beauchamp, R.D. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of β-catenin. Gastroenterology 2012, 142, 562–571.e2. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Gibbs, P.; Christie, M.; Prakash, S.; Lipton, L.; Desai, J.; Kerr, D.; Aaltonen, L.A.; Arango, D.; Kruhøffer, M.; et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin. Cancer Res. 2009, 15, 7642–7651. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Lipton, L.; Gibbs, P.; Chapman, M.; Desai, J.; Jones, I.T.; Yeatman, T.J.; East, P.; Tomlinson, I.P.M.; Verspaget, H.; et al. DNA copy-number alterations underlie gene expression differences between microsatellite stable and unstable colorectal cancers. Clin. Cancer Res. 2008, 14, 8061–8069. [Google Scholar] [CrossRef]
- Kemper, K.; Versloot, M.; Cameron, K.; Colak, S.; de Sousa e Melo, F.; de Jong, J.H.; Bleackley, J.; Vermeulen, L.; Versteeg, R.; Koster, J.; et al. Mutations in the Ras-Raf Axis underlie the prognostic value of CD133 in colorectal cancer. Clin. Cancer Res. 2012, 18, 3132–3141. [Google Scholar] [CrossRef]
- LaPointe, L.C.; Dunne, R.; Brown, G.S.; Worthley, D.L.; Molloy, P.L.; Wattchow, D.; Young, G.P. Map of differential transcript expression in the normal human large intestine. Physiol. Genomics 2008, 33, 50–64. [Google Scholar] [CrossRef]
- Li, S.; Lu, X.; Chi, P.; Pan, J. Identification of HOXB8 and KLK11 expression levels as potential biomarkers to predict the effects of FOLFOX4 chemotherapy. Futur. Oncol. 2013, 9, 727–736. [Google Scholar] [CrossRef]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene expression classification of colon cancer into molecular subtypes: Characterization, validation, and prognostic value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef]
- Matsuyama, T.; Ishikawa, T.; Mogushi, K.; Yoshida, T.; Iida, S.; Uetake, H.; Mizushima, H.; Tanaka, H.; Sugihara, K. MUC12 mRNA expression is an independent marker of prognosis in stage II and stage III colorectal cancer. Int. J. Cancer 2010, 127, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Molnár, B.; Galamb, O.; Péterfia, B.; Wichmann, B.; Csabai, I.; Bodor, A.; Kalmár, A.; Szigeti, K.A.; Barták, B.K.; Nagy, Z.B.; et al. Gene promoter and exon DNA methylation changes in colon cancer development—mRNA expression and tumor mutation alterations. BMC Cancer 2018, 18, 695. [Google Scholar] [CrossRef] [PubMed]
- Sabates-Bellver, J.; van der Flier, L.G.; De Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, A.; Beran, G.; Chresta, C.M.; McWalter, G.; Pritchard, A.; Weston, S.; Runswick, S.; Davenport, S.; Heathcote, K.; Castro, D.A.; et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med. Genomics 2012, 5, 66. [Google Scholar] [CrossRef]
- Tsuji, S.; Midorikawa, Y.; Takahashi, T.; Yagi, K.; Takayama, T.; Yoshida, K.; Sugiyama, Y.; Aburatani, H. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br. J. Cancer 2012, 106, 126–132. [Google Scholar] [CrossRef]
- Valcz, G.; Patai, A.V.; Kalmár, A.; Péterfia, B.; Fűri, I.; Wichmann, B.; Műzes, G.; Sipos, F.; Krenács, T.; Mihály, E.; et al. Myofibroblast-derived SFRP1 as potential inhibitor of colorectal carcinoma field effect. PLoS ONE 2014, 9, e106143. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]
- Eide, P.W.; Bruun, J.; Lothe, R.A.; Sveen, A. CMScaller: An R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 2017, 7, 16618. [Google Scholar] [CrossRef]
- Filzmoser, P.; Todorov, V. Robust tools for the imperfect world. Inf. Sci. 2013, 245, 4–20. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R.; Larsson, O. Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinform. 2007, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Alhamdoosh, M.; Ng, M.; Wilson, N.J.; Sheridan, J.M.; Huynh, H.; Wilson, M.J.; Ritchie, M.E. Combining multiple tools outperforms individual methods in gene set enrichment analyses. Bioinformatics 2017, 33, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Rodchenkov, I.; Babur, O.; Luna, A.; Aksoy, B.A.; Wong, J.V.; Fong, D.; Franz, M.; Siper, M.C.; Cheung, M.; Wrana, M.; et al. Pathway Commons 2019 Update: Integration, analysis and exploration of pathway data. Nucleic Acids Res. 2020, 48, D489–D497. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.E.; Chen, A.C.; Gevaert, O. Combined Mapping of Multiple clUsteriNg ALgorithms (COMMUNAL): A Robust Method for Selection of Cluster Number, K. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
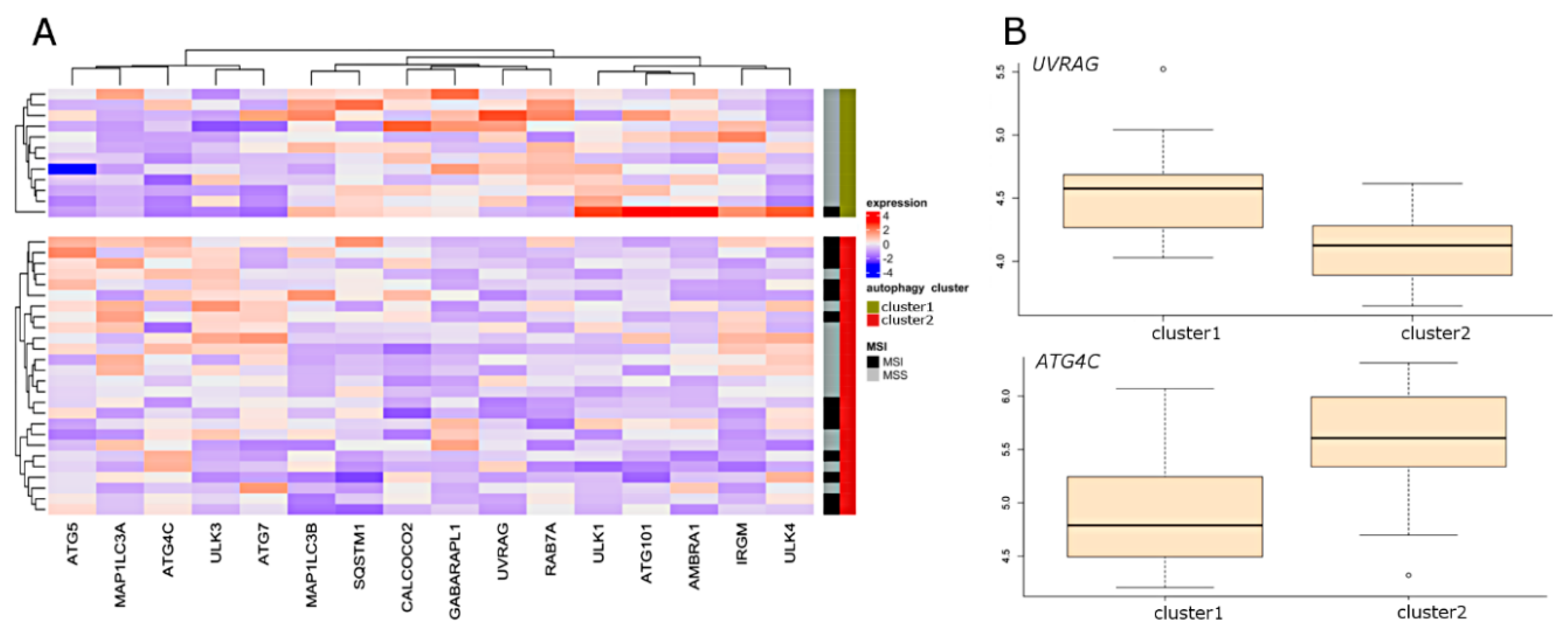{kind=link}
{kind=link}
| Clinicopathological Characteristics | [ALL] | Cluster 1 | Cluster 2 | adj. p-Value * | N ** |
|---|---|---|---|---|---|
| N = 502 | N = 232 | N = 270 | |||
| MSI: | <0.001 | 497 | |||
| MSI | 71 (14.3%) | 11 (4.8%) | 60 (22.2%) | ||
| MSS | 426 (85.7%) | 216 (95.2%) | 210 (77.8%) | ||
| CMScaller: | <0.001 | 457 | |||
| CMS1 | 83 (18.2%) | 18 (9.0%) | 65 (25.4%) | ||
| CMS2 | 141 (30.9%) | 104 (51.7%) | 37 (14.5%) | ||
| CMS3 | 82 (17.9%) | 21 (10.4%) | 61 (23.8%) | ||
| CMS4 | 151 (33.0%) | 58 (28.9%) | 93 (36.3%) | ||
| age | 68.0 [58.0; 76.0] | 68.0 [59.0; 75.0] | 68.0 [57.0; 77.0] | 0.828 | 502 |
| location: | <0.001 | 487 | |||
| distal | 273 (56.1%) | 149 (66.2%) | 124 (47.3%) | ||
| proximal | 214 (43.9%) | 76 (33.8%) | 138 (52.7%) | ||
| stage: | 0.003 | 486 | |||
| I | 84 (17.3%) | 27 (12.1%) | 57 (21.8%) | ||
| II | 189 (38.9%) | 84 (37.5%) | 105 (40.1%) | ||
| III | 138 (28.4%) | 67 (29.9%) | 71 (27.1%) | ||
| IV | 75 (15.4%) | 46 (20.5%) | 29 (11.1%) | ||
| gender: | 0.941 | 502 | |||
| female | 227 (45.2%) | 104 (44.8%) | 123 (45.6%) | ||
| male | 275 (54.8%) | 128 (55.2%) | 147 (54.4%) | ||
| Single nucleotide variants | 54.0 [41.0; 80.0] | 51.0 [37.0; 66.0] | 60.0 [41.0; 303.5] | <0.001 | 389 |
| Non-silent mutation rate | 2.7 [2.1; 3.8] | 2.6 [1.9; 3.3] | 2.9 [2.2; 5.0] | <0.001 | 382 |
| Aneuploidy score | 10.0 [4.0; 16.0] | 11.0 [7.0; 16.0] | 8.0 [2.0; 16.0] | <0.001 | 469 |
| APCmut: | <0.001 | 422 | |||
| 0 | 96 (22.7%) | 24 (13.0%) | 72 (30.4%) | ||
| 1 | 326 (77.3%) | 161 (87.0%) | 165 (69.6%) | ||
| TP53mut: | <0.001 | 422 | |||
| 0 | 172 (40.8%) | 45 (24.3%) | 127 (53.6%) | ||
| 1 | 250 (59.2%) | 140 (75.7%) | 110 (46.4%) | ||
| KRASmut: | 0.837 | 422 | |||
| 0 | 252 (59.7%) | 112 (60.5%) | 140 (59.1%) | ||
| 1 | 170 (40.3%) | 73 (39.5%) | 97 (40.9%) | ||
| BRAFmut: | <0.001 | 422 | |||
| 0 | 365 (86.5%) | 175 (94.6%) | 190 (80.2%) | ||
| 1 | 57 (13.5%) | 10 (5.4%) | 47 (19.8%) |
| Clinicopathological Characteristics | [ALL] | Cluster 1 | Cluster 2 | adj. p-Value * | N ** |
|---|---|---|---|---|---|
| N = 1229 | N = 505 | N = 724 | |||
| age | 69.0 [60.0; 77.0] | 68.0 [59.0; 75.0] | 70.0 [60.0; 78.0] | 0.022 | 566 |
| gender: | 0.302 | 703 | |||
| female | 304 (43.2%) | 135 (41.0%) | 169 (45.2%) | ||
| male | 399 (56.8%) | 194 (59.0%) | 205 (54.8%) | ||
| stage: | 0.081 | 603 | |||
| I | 70 (11.6%) | 31 (10.9%) | 39 (12.3%) | ||
| II | 265 (43.9%) | 119 (41.8%) | 146 (45.9%) | ||
| III | 189 (31.3%) | 87 (30.5%) | 102 (32.1%) | ||
| IV | 79 (13.1%) | 48 (16.8%) | 31 (9.7%) | ||
| MSI_native: | <0.001 | 685 | |||
| MSI | 162 (23.6%) | 32 (10.4%) | 130 (34.6%) | ||
| MSS | 523 (76.4%) | 277 (89.6%) | 246 (65.4%) | ||
| CMScaller: | <0.001 | 1229 | |||
| CMS1 | 218 (17.7%) | 37 (7.3%) | 181 (25.0%) | ||
| CMS2 | 373 (30.3%) | 254 (50.3%) | 119 (16.4%) | ||
| CMS3 | 215 (17.5%) | 87 (17.2%) | 128 (17.7%) | ||
| CMS4 | 423 (34.4%) | 127 (25.1%) | 296 (40.9%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, J.; Karpiński, P.; Sąsiadek, M.M. Transcriptomic Profiling for the Autophagy Pathway in Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 7101. https://doi.org/10.3390/ijms21197101
Gil J, Karpiński P, Sąsiadek MM. Transcriptomic Profiling for the Autophagy Pathway in Colorectal Cancer. International Journal of Molecular Sciences. 2020; 21(19):7101. https://doi.org/10.3390/ijms21197101
Chicago/Turabian StyleGil, Justyna, Paweł Karpiński, and Maria M. Sąsiadek. 2020. "Transcriptomic Profiling for the Autophagy Pathway in Colorectal Cancer" International Journal of Molecular Sciences 21, no. 19: 7101. https://doi.org/10.3390/ijms21197101
APA StyleGil, J., Karpiński, P., & Sąsiadek, M. M. (2020). Transcriptomic Profiling for the Autophagy Pathway in Colorectal Cancer. International Journal of Molecular Sciences, 21(19), 7101. https://doi.org/10.3390/ijms21197101