Inositol Polyphosphate-Based Compounds as Inhibitors of Phosphoinositide 3-Kinase-Dependent Signaling



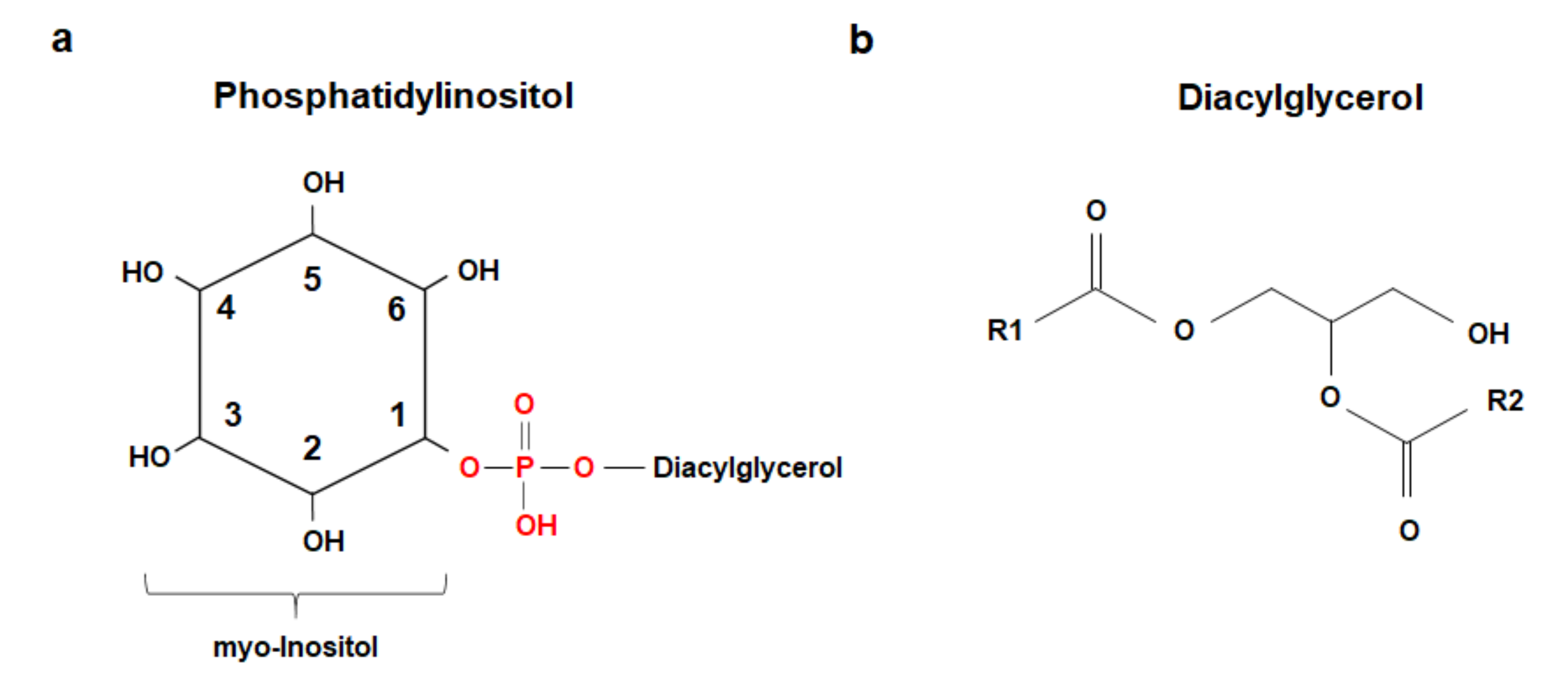{kind=link}
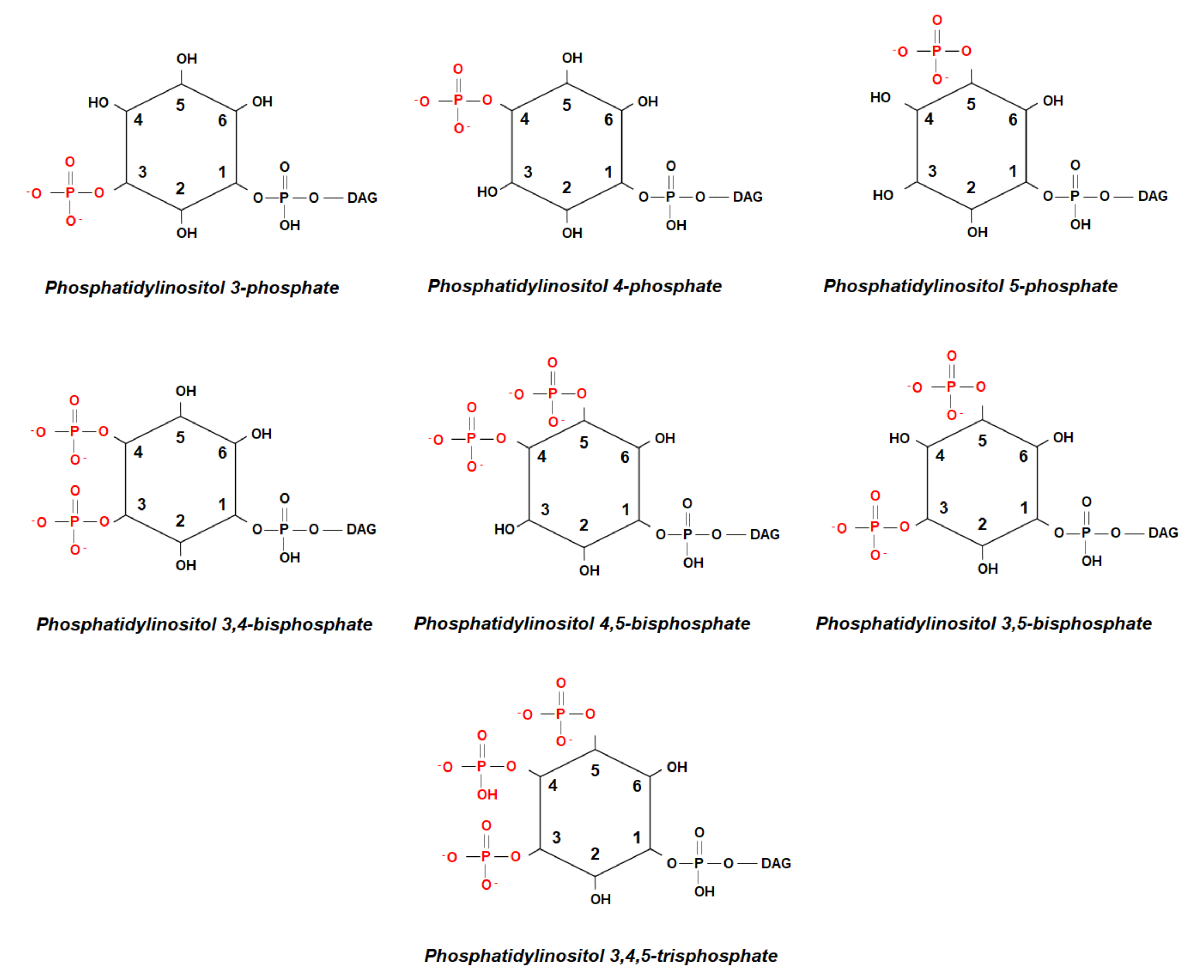{kind=link}
{kind=link}
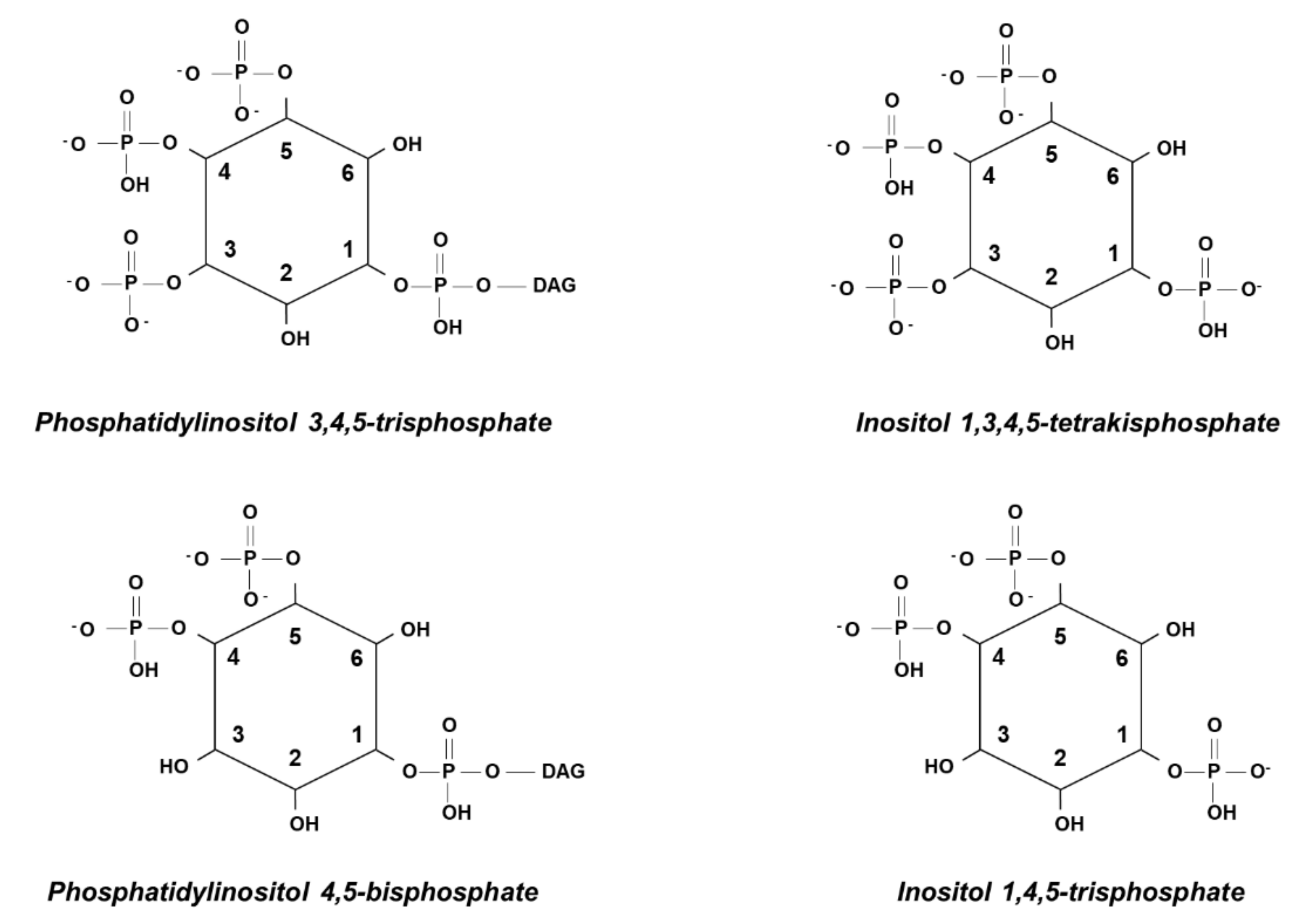{kind=link}
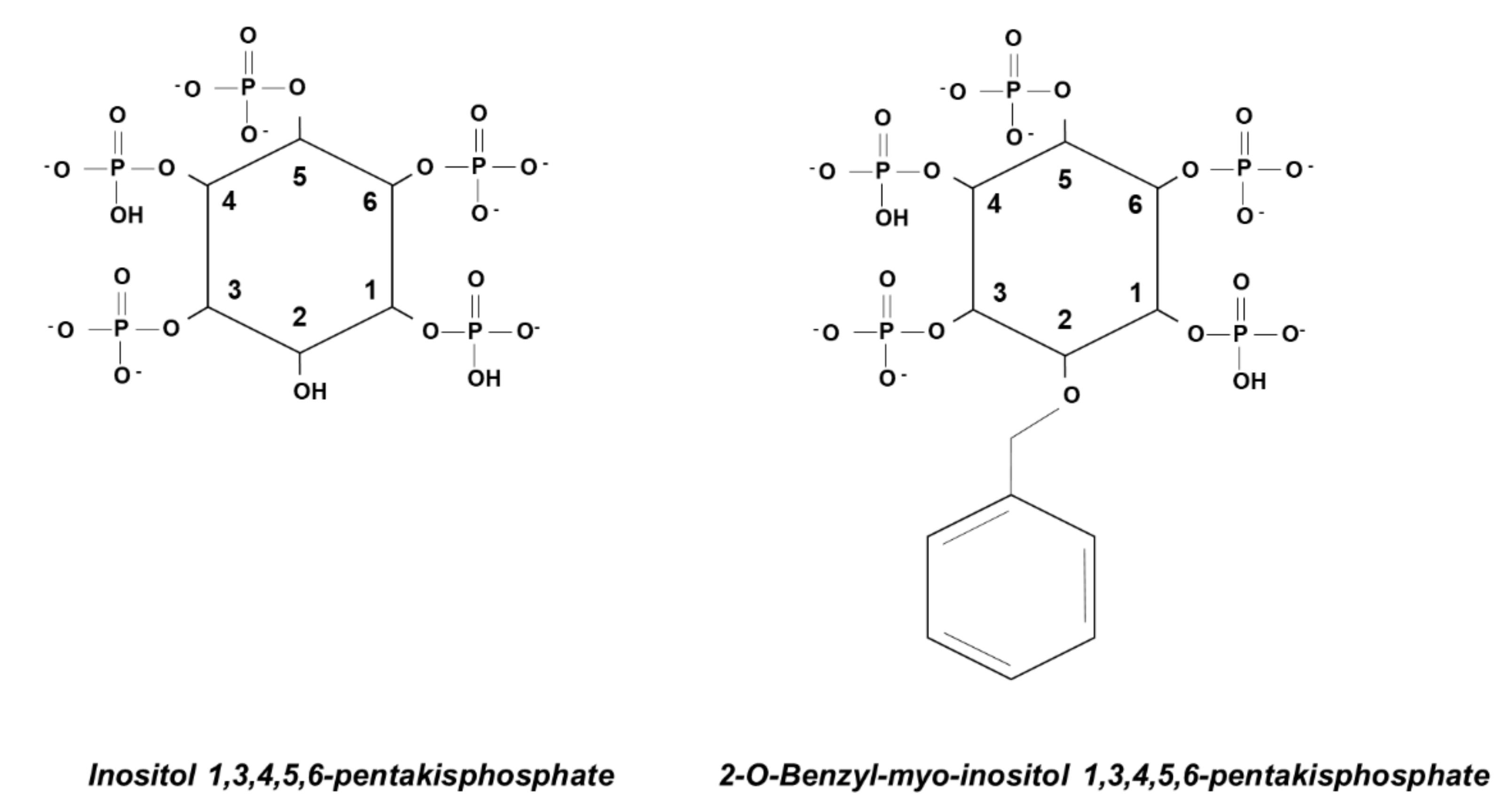{kind=link}
Abstract
1. Phosphoinositides and Protein Binding Domains
2. Akt PH Domain and PtdIns(3,4,5)P3: A “Textbook” Example of Phosphoinositide-Dependent Regulation of Signaling Pathways and Cellular Functions
3. PI3Ks, PtdIns(3,4,5)P3, and Akt in Cancer
4. Inhibiting Akt by Targeting Its Mechanism of Activation
4.1. Preventing Akt Translocation to the Plasma Membrane
4.2. Targeting Akt PH/Kinase Domain Interaction
4.2.1. Allosteric Inhibitors
4.2.2. Phosphatidylinositol Ether Lipid Analogues
4.2.3. Inositol Polyphosphates
5. Improving the Activity of InsP5—Chemical Modifications
5.1. Chemical Modifications that Resulted in Inhibition of Additional and Selective Targets
5.2. Chemical Modifications That Increase Intracellular Delivery
5.3. Nanodelivery
6. Targeting PH Domains of Signaling Proteins: Beyond Akt
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALPs | Alkyl-lysophospholipids |
| Btk | Bruton’s tyrosine kinase |
| Cnk1 | Connector enhancer of kinase suppressor of Ras 1 |
| FGF-2 | Basic fibroblast growth factor |
| FYVE | Fab1/YOTB/Vac1/EEA1 |
| GFP | Green Fluorescent Protein |
| Ins(1,3,4,5)P4 | Inositol 1,3,4,5-tetrakisphosphate |
| InsP5 | Inositol 1,3,4,5,6-pentakisphosphate |
| InsP6 | Inositol 1,2,3,4,5,6-hexakisphosphate |
| Itk | Interleukin-2–inducible T cell kinase |
| mTOR | Mechanistic target of rapamycin |
| PDK1 | 3-Phosphoinositide-dependent kinase 1 |
| PH | Pleckstrin homology |
| PI3K | Phosphoinositide 3-kinase |
| PLC | Phospholipase C |
| PtdIns3P | Phosphatidylinositol 3-phosphate |
| PtdIns(3,4)P2 | Phosphatidylinositol 3,4-bisphosphate |
| PtdIns(4,5)P2 | Phosphatidylinositol 4,5-bisphosphate |
| PtdIns(3,4,5)P3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PTEN | Phosphatase and tensin homolog |
| PX | Phox homology |
| 2-O-Bn-InsP5 | 2-O-benzyl-myo-inositol 1,3,4,5,6-pentakisphosphate |
| 5-InsP7 | 5-diphosphoinositolpentakisphosphate |
References
- Irvine, R.F. A short history of inositol lipids. J. Lipid Res. 2016, 57, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Michell, R.H. Inositol derivatives: Evolution and functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Barneda, D.; Cosulich, S.; Stephens, L.; Hawkins, P. How is the acyl chain composition of phosphoinositides created and does it matter? Biochem. Soc. Trans. 2019, 47, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, T. An Introduction to Phosphoinositides; Springer Science and Business Media LLC: Berlin, Germany, 2012; Volume 362, pp. 1–42. [Google Scholar]
- Gillooly, D.J.; Simonsen, A.; Stenmark, H. Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins. Biochem. J. 2001, 355, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, T.G. Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2006, 1761, 868–877. [Google Scholar] [CrossRef]
- Haslam, R.J.; Koide, H.B.; Hemmings, B.A. Pleckstrin domain homology. Nature 1993, 363, 309–310. [Google Scholar] [CrossRef]
- Mayer, B.J.; Ren, R.; Clark, K.L.; Baltimore, D. A putative modular domain present in diverse signaling proteins. Cell 1993, 73, 629–630. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Falasca, M.; Schlessinger, J.; Ferguson, K. Regulatory recruitment of signalling molecules to the cell membrane by pleckstrinhomology domains. Trends Cell Biol. 1997, 7, 237–242. [Google Scholar] [CrossRef]
- Maffucci, T.; Falasca, M. Specificity in pleckstrin homology (PH) domain membrane targeting: A role for a phosphoinositide-protein co-operative mechanism. FEBS Lett. 2001, 506, 173–179. [Google Scholar] [CrossRef]
- Vonkova, I.; Saliba, A.-E.; Deghou, S.; Anand, K.; Ceschia, S.; Doerks, T.; Galih, A.; Kugler, K.G.; Maeda, K.; Rybin, V.; et al. Lipid Cooperativity as a General Membrane-Recruitment Principle for PH Domains. Cell Rep. 2015, 12, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Kalli, A.C.; Yasuoka, K.; Sansom, M.S.P. Interactions of Pleckstrin Homology Domains with Membranes: Adding Back the Bilayer via High-Throughput Molecular Dynamics. Structure 2016, 24, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Domański, J.; Naughton, F.B.; Best, R.B.; Kalli, A.C.; Stansfeld, P.J.; Sansom, M.S.P. Multiple lipid binding sites determine the affinity of PH domains for phosphoinositide-containing membranes. Sci. Adv. 2020, 6, eaay5736. [Google Scholar] [CrossRef] [PubMed]
- Picas, L.; Gaits, F.; Goud, B. The emerging role of phosphoinositide clustering in intracellular trafficking and signal transduction. F1000Research 2016, 5, 422. [Google Scholar] [CrossRef]
- Cantley, L.C.; Wajant, H. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.F.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and Function of 3-Phosphorylated Inositol Lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef]
- Falasca, M.; Maffucci, T. Role of class II phosphoinositide 3-kinase in cell signalling. Biochem. Soc. Trans. 2007, 35, 211–214. [Google Scholar] [CrossRef]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Whitehead, M.A.; Piñeiro, R. Molecules in medicine mini-review: Isoforms of PI3K in biology and disease. J. Mol. Med. 2015, 94, 5–11. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P.T. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Ghigo, A.; Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-Kinases in Health and Disease. In Plant-Microbe Interactions; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2012; Volume 58, pp. 183–213. [Google Scholar]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Laguerre, M.; Larijani, B. 3-D structure and dynamics of protein kinase B—New mechanism for the allosteric regulation of an AGC kinase. J. Chem. Biol. 2009, 2, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Laguerre, M.; Parker, P.J.; Larijani, B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 2009, 7, e17. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Alcor, D.; Laguerre, M.; Park, J.; Vojnovic, B.; A Hemmings, B.; Downward, J.; Parker, P.J.; Larijani, B. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007, 5, e95. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Maehama, T. PTEN: A tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999, 9, 125–128. [Google Scholar] [CrossRef]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef]
- Siess, K.M.; Leonard, T. Lipid-dependent Akt-ivity: Where, when, and how. Biochem. Soc. Trans. 2019, 47, 897–908. [Google Scholar] [CrossRef]
- Kang, J.K.; Kim, O.-H.; Hur, J.; Yu, S.H.; Lamichhane, S.; Lee, J.W.; Ojha, U.; Hong, J.H.; Lee, C.S.; Cha, J.-Y.; et al. Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 11926–11931. [Google Scholar] [CrossRef]
- Razzini, G.; Ingrosso, A.; Brancaccio, A.; Sciacchitano, S.; Esposito, D.L.; Falasca, M. Different subcellular localization and phosphoinositides binding of insulin receptor substrate protein pleckstrin homology domains. Mol. Endocrinol. 2000, 14, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2014, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Pandolfi, P.P. The PTEN⁻PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Williams, R.L.; Hawkins, P.T. Phosphoinositide 3-kinases as drug targets in cancer. Curr. Opin. Pharmacol. 2005, 5, 357–365. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Paddock, M.N.; Field, S.J.; Cantley, L.C. Treating cancer with phosphatidylinositol-3-kinase inhibitors: Increasing efficacy and overcoming resistance. J. Lipid Res. 2019, 60, 747–752. [Google Scholar] [CrossRef]
- Kienle, D.L.; Stilgenbauer, S. Approved and emerging PI3K inhibitors for the treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma. Expert Opin. Pharmacother. 2020, 21, 917–929. [Google Scholar] [CrossRef]
- Verret, B.; Cortes, J.; Bachelot, T.; Andre, F.; Arnedos, M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann. Oncol. 2019, 30, x12–x20. [Google Scholar] [CrossRef]
- Kondapaka, S.B.; Singh, S.S.; DasMahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003, 2, 1093–1103. [Google Scholar]
- Ruiter, G.A.; Zerp, S.F.; Bartelink, H.; Van Blitterswijk, W.J.; Verheij, M. Anti-cancer alkyl-lysophospholipids inhibit the phosphatidylinositol 3-kinase-Akt/PKB survival pathway. Anti Cancer Drugs 2003, 14, 167–173. [Google Scholar] [CrossRef]
- Meuillet, E.J.; Zuohe, S.; Lemos, R.; Ihle, N.; Kingston, J.; Watkins, R.; Moses, S.A.; Zhang, S.; Du Cuny, L.; Herbst, R.; et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol. Cancer Ther. 2010, 9, 706–717. [Google Scholar] [CrossRef]
- Meuillet, E.J. Novel inhibitors of AKT: Assessment of a different approach targeting the pleckstrin homology domain. Curr. Med. Chem. 2011, 18, 2727–2742. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Marco, P.; Marco, C.; Gálvez, X.; Jiménez-López, J.M.; Carrasco, M.P. Alkylphospholipids: An update on molecular mechanisms and clinical relevance. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2016, 172, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, J.-M.; Eathiraj, S.; Vensel, D.; Liu, Y.; Bull, C.O.; Cornell-Kennon, S.; Iimura, S.; Kelleher, E.W.; Kizer, D.E.; Koerner, S.; et al. Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): An Orally Bioavailable, Selective, and Potent Allosteric AKT Inhibitor. J. Med. Chem. 2016, 59, 6455–6469. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Savage, R.E.; Eathiraj, S.; Meade, J.; Wick, M.J.; Hall, T.; Abbadessa, G.; Schwartz, B. Targeting AKT1-E17K and the PI3K/AKT Pathway with an Allosteric AKT Inhibitor, ARQ 092. PLoS ONE 2015, 10, e0140479. [Google Scholar] [CrossRef]
- Politz, O.; Siegel, F.; Bärfacker, L.; Bömer, U.; Hägebarth, A.; Scott, W.J.; Michels, M.; Ince, S.; Neuhaus, R.; Meyer, K.; et al. BAY 1125976, a selective allosteric AKT1/2 inhibitor, exhibits high efficacy on AKT signaling-dependent tumor growth in mouse models. Int. J. Cancer 2016, 140, 449–459. [Google Scholar] [CrossRef]
- Yan, L. Abstract #DDT01-1: MK-2206: A potent oral allosteric AKT inhibitor. Cancer Res. 2009, 69 (Suppl. S9: DDT01-1-DDT-1). Available online: https://cancerres.aacrjournals.org/content/69/9_Supplement/DDT01-1 (accessed on 8 August 2020).
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- Powis, G.; Aksoy, I.A.; Melder, D.C.; Aksoy, S.; Eichinger, H.; Fauq, A.H.; Kozikowski, A.P. D-3-deoxy-3-substituted myo-inositol analogues as inhibitors of cell growth. Cancer Chemother. Pharmacol. 1991, 29, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Meuillet, E.J.; Berggren, M.; Powis, G.; Kozikowski, A.P. 3-Deoxy-3-substituted-D-myo-inositol imidazolyl ether lipid phosphates and carbonate as inhibitors of the phosphatidylinositol 3-kinase pathway and cancer cell growth. Bioorgan. Med. Chem. Lett. 2001, 11, 173–176. [Google Scholar] [CrossRef]
- Hu, Y.; Qiao, L.; Wang, S.; Rong, S.-B.; Meuillet, E.J.; Berggren, M.; Gallegos, A.; Powis, G.; Kozikowski, A.P. 3-(Hydroxymethyl)-bearing phosphatidylinositol ether lipid analogues and carbonate surrogates block PI3-K, Akt, and cancer cell growth. J. Med. Chem. 2000, 43, 3045–3051. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Nan, F.; Kunkel, M.; Gallegos, A.; Powis, G.; Kozikowski, A.P. 3-Deoxy-d-myo-inositol 1-Phosphate, 1-Phosphonate, and Ether Lipid Analogues as Inhibitors of Phosphatidylinositol-3-kinase Signaling and Cancer Cell Growth. J. Med. Chem. 1998, 41, 3303–3306. [Google Scholar] [CrossRef] [PubMed]
- Meuillet, E.J.; Mahadevan, D.; Vankayalapati, H.; Berggren, M.; Williams, R.; Coon, A.; Kozikowski, A.P.; Powis, G. Specific inhibition of the Akt1 pleckstrin homology domain by D-3-deoxy-phosphatidyl-myo-inositol analogues. Mol. Cancer Ther. 2003, 2, 389–399. [Google Scholar]
- Takeuchi, H.; Kanematsu, T.; Misumi, Y.; Sakane, F.; Konishi, H.; Kikkawa, U.; Watanabe, Y.; Katan, M.; Hirata, M. Distinct specificity in the binding of inositol phosphates by pleckstrin homology domains of pleckstrin, RAC-protein kinase, diacylglycerol kinase and a new 130kDa protein. Biochim. Biophys. Acta BBA Bioenerg. 1997, 1359, 275–285. [Google Scholar] [CrossRef]
- Venkateswarlu, K.; Gunn-Moore, F.; Oatey, P.B.; Tavaré, J.M.; Cullen, P.J. Nerve growth factor- and epidermal growth factor-stimulated translocation of the ADP-ribosylation factor-exchange factor GRP1 to the plasma membrane of PC12 cells requires activation of phosphatidylinositol 3-kinase and the GRP1 pleckstrin homology domain. Biochem. J. 1998, 335, 139–146. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Ferguson, K.M.; O’Brien, R.; Sigler, P.B.; Schlessinger, J. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. USA 1995, 92, 10472–10476. [Google Scholar] [CrossRef]
- Berrie, C.P.; Falasca, M. Patterns within protein/polyphosphoinositide interactions provide specific targets for therapeutic intervention. FASEB J. 2000, 14, 2618–2622. [Google Scholar] [CrossRef]
- Razzini, G.; Berrie, C.; Vignati, S.; Broggini, M.; Mascetta, G.; Brancaccio, A.; Falasca, M. Novel functional PI 3-kinase antagonists inhibit cell growth and tumorigenicity in human cancer cell lines. FASEB J. 2000, 14, 1179–1187. [Google Scholar] [CrossRef]
- Piccolo, E.; Vignati, S.; Maffucci, T.; Innominato, P.F.; Riley, A.M.; Potter, B.V.; Pandolfi, P.P.; Broggini, M.; Iacobelli, S.; Innocenti, P.; et al. Inositol pentakisphosphate promotes apoptosis through the PI 3-K/Akt pathway. Oncogene 2004, 23, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, T. Inhibition of the Phosphatidylinositol 3-Kinase/Akt Pathway by Inositol Pentakisphosphate Results in Antiangiogenic and Antitumor Effects. Cancer Res. 2005, 65, 8339–8349. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Koldobskiy, M.A.; Bello, N.T.; Maxwell, M.; Potter, J.J.; Juluri, K.R.; Maag, D.; Kim, S.; Huang, A.S.; Dailey, M.J.; et al. Inositol Pyrophosphates Inhibit Akt Signaling, Thereby Regulating Insulin Sensitivity and Weight Gain. Cell 2010, 143, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Chiozzotto, D.; Godage, H.Y.; Mazzoletti, M.; Riley, A.M.; Previdi, S.; Potter, B.V.L.; Broggini, M.; Maffucci, T. A novel inhibitor of the PI3K/Akt pathway based on the structure of inositol 1,3,4,5,6-pentakisphosphate. Br. J. Cancer 2010, 102, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Calleja, V.; Ferro, R.; Fantin, A.; Riley, A.M.; Potter, B.V. A Small Molecule Inhibitor of PDK1/PLCγ1 Interaction Blocks Breast and Melanoma Cancer Cell Invasion. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Chikh, A.; Wheeler, A.P.; Maffucci, T.; Falasca, M. A novel regulatory mechanism links PLCγ1 to PDK1. J. Cell Sci. 2012, 125, 3153–3163. [Google Scholar] [CrossRef]
- Ferry, S.; Matsuda, M.; Yoshida, H.; Hirata, M. Inositol hexakisphosphate blocks tumor cell growth by activating apoptotic machinery as well as by inhibiting the Akt/NFκB-mediated cell survival pathway. Carcinogenesis 2002, 23, 2031–2041. [Google Scholar] [CrossRef]
- Nagy, R.; Grob, H.; Weder, B.; Green, P.; Klein, M.; Frelet-Barrand, A.; Schjoerring, J.K.; Brearley, C.; Martinoia, E. The Arabidopsis ATP-binding cassette protein AtMRP5/AtABCC5 is a high affinity inositol hexakisphosphate transporter involved in guard cell signaling and phytate storage. J. Biol. Chem. 2009, 284, 33614–33622. [Google Scholar] [CrossRef]
- Kobes, J.E.; Daryaei, I.; Howison, C.M.; Bontrager, J.G.; Sirianni, R.W.; Meuillet, E.J.; Pagel, M.D. Improved Treatment of Pancreatic Cancer With Drug Delivery Nanoparticles Loaded With a Novel AKT/PDK1 Inhibitor. Pancreas 2016, 45, 1158–1166. [Google Scholar] [CrossRef]
- Pagel, M.D.; Jeffery, J.J.; Abril, E.R.; Nagle, R.B.; Guzmán, R.; Meuillet, E.J.; Lucero-Acuña, A. Nanoparticle delivery of an AKT/PDK1 inhibitor improves the therapeutic effect in pancreatic cancer. Int. J. Nanomed. 2014, 9, 5653–5665. [Google Scholar] [CrossRef][Green Version]
- Emmanouilidi, A.; Fyffe, C.A.; Ferro, R.; Edling, C.E.; Capone, E.; Sestito, S.; Rapposelli, S.; Lattanzio, R.; Iacobelli, S.; Sala, G.; et al. Preclinical validation of 3-phosphoinositide-dependent protein kinase 1 inhibition in pancreatic cancer. J. Exp. Clin. Cancer Res. 2019, 38, 191. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Lertpiriyapong, K.; Yang, L.V.; Martelli, A.M.; Cocco, L.; Ratti, S.; Falasca, M.; Murata, R.M.; Rosalen, P.L.; Lombardi, P.; et al. Introduction of WT-TP53 into pancreatic cancer cells alters sensitivity to chemotherapeutic drugs, targeted therapeutics and nutraceuticals. Adv. Biol. Regul. 2018, 69, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Candido, S.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Cocco, L.; Ramazzotti, G.; Ratti, S.; Follo, M.Y.; Martelli, A.M.; et al. Abilities of β-Estradiol to interact with chemotherapeutic drugs, signal transduction inhibitors and nutraceuticals and alter the proliferation of pancreatic cancer cells. Adv. Biol. Regul. 2020, 75, 100672. [Google Scholar] [CrossRef]
- Emmanouilidi, A.; Falasca, M. Targeting PDK1 for Chemosensitization of Cancer Cells. Cancers 2017, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hills, L.B.; Huang, Y.H. Lipid and Protein Co-Regulation of PI3K Effectors Akt and Itk in Lymphocytes. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Amatya, N.; Wales, T.E.; Kwon, A.; Yeung, W.; Joseph, R.E.; Fulton, D.B.; Kannan, N.; Engen, J.R.; Andreotti, A.H. Lipid-targeting pleckstrin homology domain turns its autoinhibitory face toward the TEC kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 21539–21544. [Google Scholar] [CrossRef]
- Yoon, Y. Small chemicals with inhibitory effects on PtdIns(3,4,5)P3 binding of Btk PH domain. Bioorgan. Med. Chem. Lett. 2014, 24, 2334–2339. [Google Scholar] [CrossRef]
- Kim, W.; Kim, E.; Min, H.; Kim, M.G.; Eisenbeis, V.B.; Dutta, A.K.; Pavlovic, I.; Jessen, H.J.; Kim, S.; Seong, R.H. Inositol polyphosphates promote T cell-independent humoral immunity via the regulation of Bruton’s tyrosine kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 12952–12957. [Google Scholar] [CrossRef]
- Wang, Q.; Vogan, E.M.; Nocka, L.M.; Rosen, C.E.; Zorn, J.A.; Harrison, S.C.; Kuriyan, J. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. eLife 2015, 4. [Google Scholar] [CrossRef]
- Cash, J.N.; Chandan, N.R.; Hsu, A.Y.; Sharma, P.V.; Deng, Q.; Smrcka, A.V.; Tesmer, J.J. Discovery of Small Molecules That Target the Phosphatidylinositol (3,4,5) Trisphosphate (PIP3)-Dependent Rac Exchanger 1 (P-Rex1) PIP3-Binding Site and Inhibit P-Rex1-Dependent Functions in Neutrophils. Mol. Pharmacol. 2020, 97, 226–236. [Google Scholar] [CrossRef]
- Indarte, M.; Puentes, R.; Maruggi, M.; Ihle, N.T.; Grandjean, G.; Scott, M.; Ahmed, Z.; Meuillet, E.J.; Zhang, S.; Lemos, M., Jr.; et al. An Inhibitor of the Pleckstrin Homology Domain of CNK1 Selectively Blocks the Growth of Mutant KRAS Cells and Tumors. Cancer Res. 2019, 79, 3100–3111. [Google Scholar] [CrossRef] [PubMed]
- Blind, R. Structural analyses of inositol phosphate second messengers bound to signaling effector proteins. Adv. Biol. Regul. 2020, 75, 100667. [Google Scholar] [CrossRef] [PubMed]
- Nawrotek, A.; Benabdi, S.; Niyomchon, S.; Kryszke, M.-H.; Ginestier, C.; Cañeque, T.; Tepshi, L.; Mariani, A.; Onge, R.P.S.; Giaever, G.; et al. PH-domain-binding inhibitors of nucleotide exchange factor BRAG2 disrupt Arf GTPase signaling. Nat. Methods 2019, 15, 358–366. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maffucci, T.; Falasca, M. Inositol Polyphosphate-Based Compounds as Inhibitors of Phosphoinositide 3-Kinase-Dependent Signaling. Int. J. Mol. Sci. 2020, 21, 7198. https://doi.org/10.3390/ijms21197198
Maffucci T, Falasca M. Inositol Polyphosphate-Based Compounds as Inhibitors of Phosphoinositide 3-Kinase-Dependent Signaling. International Journal of Molecular Sciences. 2020; 21(19):7198. https://doi.org/10.3390/ijms21197198
Chicago/Turabian StyleMaffucci, Tania, and Marco Falasca. 2020. "Inositol Polyphosphate-Based Compounds as Inhibitors of Phosphoinositide 3-Kinase-Dependent Signaling" International Journal of Molecular Sciences 21, no. 19: 7198. https://doi.org/10.3390/ijms21197198
APA StyleMaffucci, T., & Falasca, M. (2020). Inositol Polyphosphate-Based Compounds as Inhibitors of Phosphoinositide 3-Kinase-Dependent Signaling. International Journal of Molecular Sciences, 21(19), 7198. https://doi.org/10.3390/ijms21197198