Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension

 ,
,  , and
, and
Abstract
:
1. Introduction
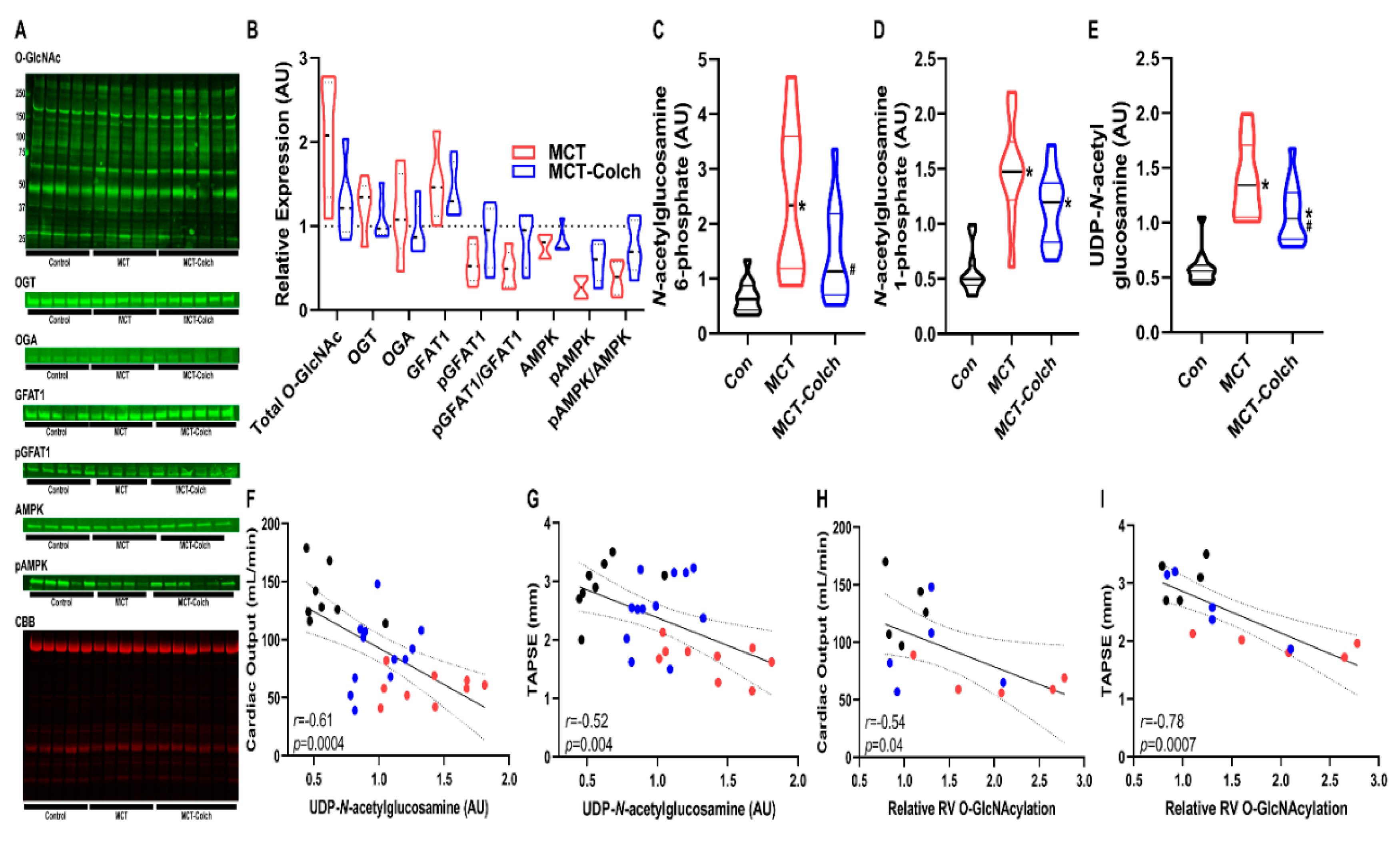
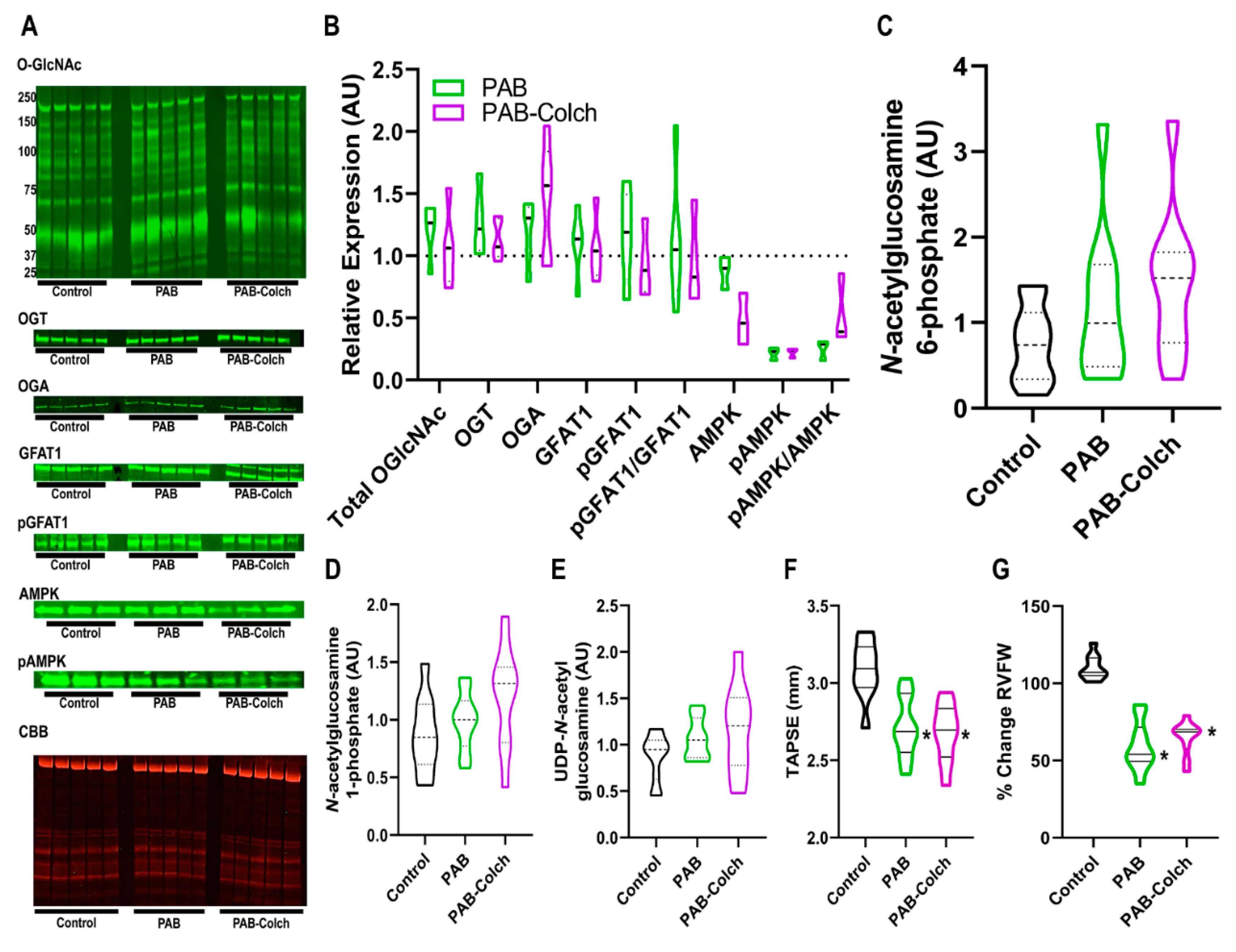
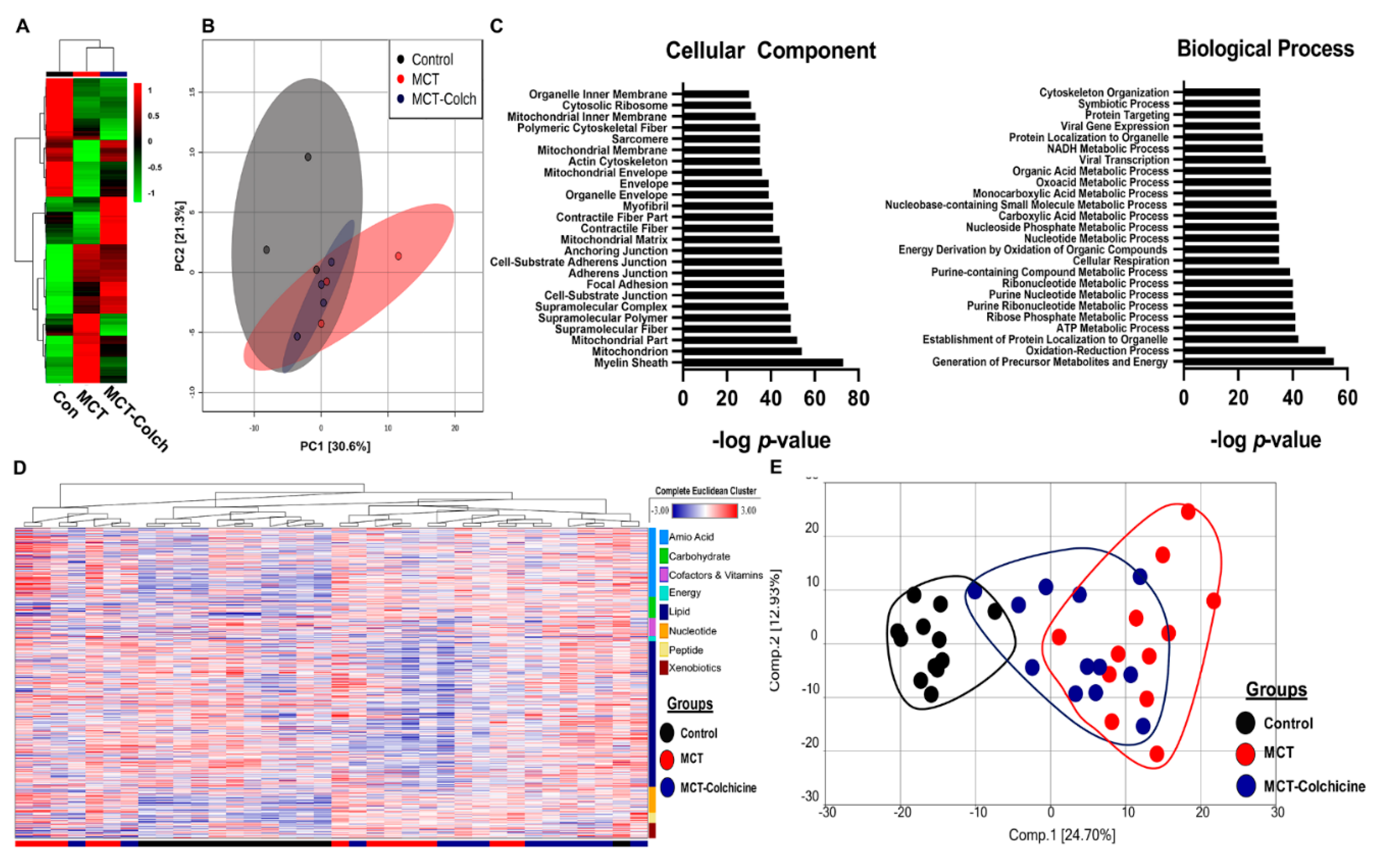
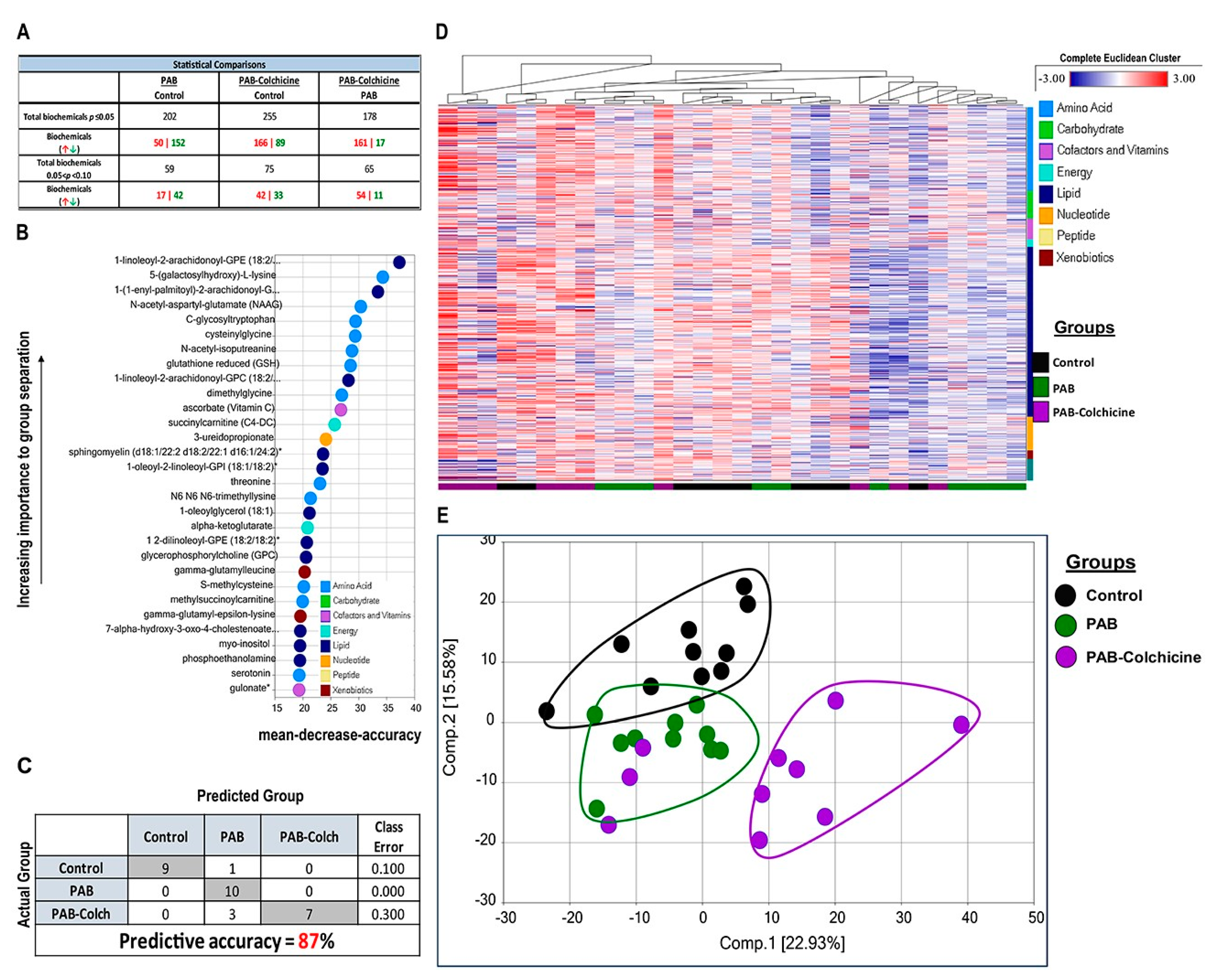
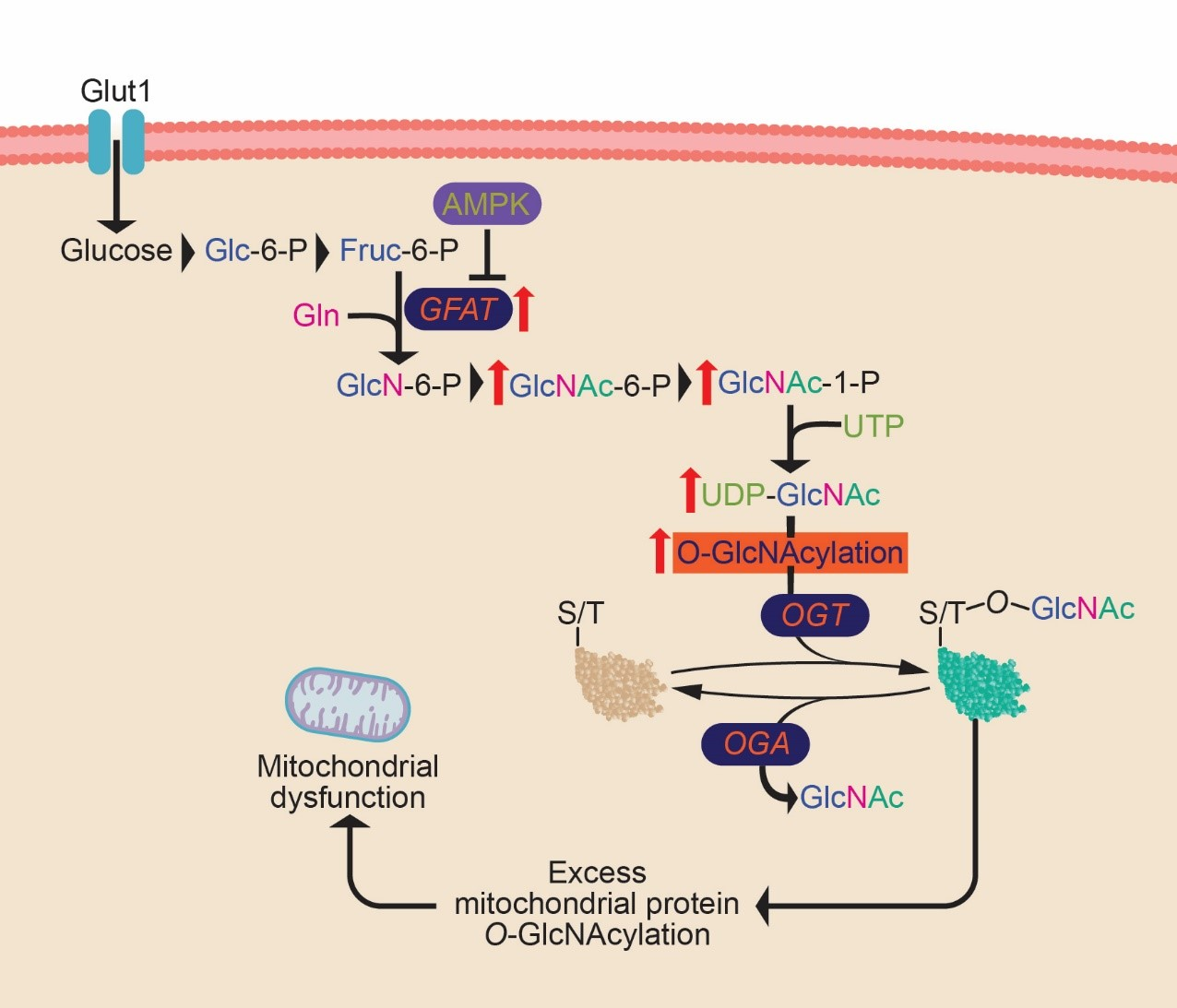
2. Results
3. Discussion
4. Materials and Methods
4.1. Rat Models of RV Pressure Overload
4.2. Colchicine Treatment
4.3. Western Blot Analysis
4.4. Antibodies
4.5. Global Metabolomics
4.6. Rodent Echocardiography
4.7. O-GlcNAcylated Protein Immunoprecipitation (IP)
4.8. Quantitative Mass Spectrometry
4.9. Cellular Fractionation
4.10. Cell Culture
4.11. Mitochondrial Respiration Measurements
4.12. PAH Patient Cohort
4.13. RV Contractility Analysis
4.14. Quantitative Immunofluorescence of Human RV Samples
4.15. Relationship Between HBP Intermediates, O-GlcNAcylation, and RV Function
4.16. Statistical Analysis
5. Limitation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thenappan, T.; Shah, S.J.; Rich, S.; Tian, L.; Archer, S.L.; Gomberg-Maitland, M. Survival in pulmonary arterial hypertension: A reappraisal of the NIH risk stratification equation. Eur. Respir. J. 2010, 35, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benza, R.L.; Miller, D.P.; Gomberg-Maitland, M.; Frantz, R.P.; Foreman, A.J.; Coffey, C.S.; Frost, A.; Barst, R.J.; Badesch, D.B.; Elliott, C.G.; et al. Predicting survival in pulmonary arterial hypertension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010, 122, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Sitbon, O.; Yaïci, A.; Montani, D.; O’Callaghan, D.S.; Jaïs, X.; Parent, F.; Savale, L.; Natali, D.; Günther, S.; et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur. Respir. J. 2010, 36, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Fang, Y.H.; Ryan, J.J.; Piao, L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm. Circ. 2013, 3, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung ¹⁸F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Wang, L.; Xiong, C.M.; He, J.G.; Zhang, Y.; Gu, Q.; Zhao, Z.H.; Ni, X.H.; Fang, W.; Liu, Z.H. The ratio of (18)F-FDG activity uptake between the right and left ventricle in patients with pulmonary hypertension correlates with the right ventricular function. Clin. Nucl. Med. 2014, 39, 426–430. [Google Scholar] [CrossRef]
- Fang, W.; Zhao, L.; Xiong, C.M.; Ni, X.H.; He, Z.X.; He, J.G.; Wilkins, M.R. Comparison of 18F-FDG uptake by right ventricular myocardium in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with congenital heart disease. Pulm. Circ. 2012, 2, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Ohira, H.; deKemp, R.; Pena, E.; Davies, R.A.; Stewart, D.J.; Chandy, G.; Contreras-Dominguez, V.; Dennie, C.; Mc Ardle, B.; Mc Klein, R.; et al. Shifts in myocardial fatty acid and glucose metabolism in pulmonary arterial hypertension: A potential mechanism for a maladaptive right ventricular response. Eur. Heart J. Cardiovasc Imaging 2016, 17, 1424–1431. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Facundo, H.T.; Zafir, A.; Jones, S.P. O-GlcNAc signaling in the cardiovascular system. Circ. Res. 2010, 107, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Gélinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, S.; Oshiro, N.; Miyamoto, T.; Yoshino, K.; Okamoto, S.; Ono, T.; Kikkawa, U.; Yonezawa, K. AMP-activated protein kinase phosphorylates glutamine: Fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells 2009, 14, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.S.; Ma, J.; Hart, G.W. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 6050–6055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducheix, S.; Magré, J.; Cariou, B.; Prieur, X. Chronic O-GlcNAcylation and Diabetic Cardiomyopathy: The Bitterness of Glucose. Front. Endocrinol. (Lausanne) 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Banerjee, P.; Whelan, S.A.; Liu, T.; Wei, A.C.; Ramirez-Correa, G.; McComb, M.E.; Costello, C.E.; O’Rourke, B.; Murphy, A.; et al. Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. J. Proteome Res. 2016, 15, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Suarez, J.; Fricovsky, E.; Wang, H.; Scott, B.T.; Trauger, S.A.; Han, W.; Oyeleye, M.O.; Dillmann, W.H. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J. Biol. Chem. 2009, 284, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.P.; McGreal, S.R.; Graw, S.; Tessman, R.; Koppel, S.J.; Dhakal, P.; Zhang, Z.; Machacek, M.; Zachara, N.E.; Koestler, D.C.; et al. Sustained. J. Biol. Chem. 2017, 292, 14940–14962. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Fang, Y.H.; Parikh, K.; Ryan, J.J.; Toth, P.T.; Archer, S.L. Cardiac glutaminolysis: A maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J. Mol. Med. (Berl.) 2013, 91, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [Green Version]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef]
- Potus, F.; Hindmarch, C.C.T.; Dunham-Snary, K.J.; Stafford, J.; Archer, S.L. Transcriptomic Signature of Right Ventricular Failure in Experimental Pulmonary Arterial Hypertension: Deep Sequencing Demonstrates Mitochondrial, Fibrotic, Inflammatory and Angiogenic Abnormalities. Int. J. Mol. Sci. 2018, 19, 2730. [Google Scholar] [CrossRef] [Green Version]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.I.; Gomez-Arroyo, J.; Dumur, C.I.; Kraskauskas, D.; Natarajan, R.; Bogaard, H.J.; Fawcett, P.; Voelkel, N.F. Chronic carvedilol treatment partially reverses the right ventricular failure transcriptional profile in experimental pulmonary hypertension. Physiol. Genom. 2013, 45, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, E.D.; Hauton, D.; Boyle, J.; Egginton, S.; Steele, D.S.; White, E. Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System. Int. J. Mol. Sci. 2019, 20, 1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Liu, T.; Wei, A.C.; Banerjee, P.; O’Rourke, B.; Hart, G.W. O-GlcNAcomic Profiling Identifies Widespread O-Linked β-N-Acetylglucosamine Modification (O-GlcNAcylation) in Oxidative Phosphorylation System Regulating Cardiac Mitochondrial Function. J. Biol. Chem. 2015, 290, 29141–29153. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.J.; Archer, S.L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Bullen, J.W.; Balsbaugh, J.L.; Chanda, D.; Shabanowitz, J.; Hunt, D.F.; Neumann, D.; Hart, G.W. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 10592–10606. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, P.; Clark, P.M.; Mason, D.E.; Peters, E.C.; Hsieh-Wilson, L.C.; Baltimore, D. Activation of the transcriptional function of the NF-κB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 2013, 6, ra75. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.W.; Tian, L.; Heresi, G.A.; Farver, C.F.; Asosingh, K.; Comhair, S.A.; Aulak, K.S.; Dweik, R.A. O-linked β-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 2015, 131, 1260–1268. [Google Scholar] [CrossRef] [Green Version]
- Benson, L.; Brittain, E.L.; Pugh, M.E.; Austin, E.D.; Fox, K.; Wheeler, L.; Robbins, I.M.; Hemnes, A.R. Impact of diabetes on survival and right ventricular compensation in pulmonary arterial hypertension. Pulm. Circ. 2014, 4, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Roifman, I.; Ghugre, N.; Zia, M.I.; Farkouh, M.E.; Zavodni, A.; Wright, G.A.; Connelly, K.A. Diabetes is an independent predictor of right ventricular dysfunction post ST-elevation myocardial infarction. Cardiovasc. Diabetol. 2016, 15, 34. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.W.; Tian, L.; Wu, D.; Thenappan, T.; Metzger, J.M.; Archer, S.L. Colchicine Depolymerizes Microtubules, Increases Junctophilin-2, and Improves Right Ventricular Function in Experimental Pulmonary Arterial Hypertension. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tochinai, R.; Suzuki, K.; Nagata, Y.; Ando, M.; Hata, C.; Komatsu, K.; Suzuki, T.; Uchida, K.; Kado, S.; Kaneko, K.; et al. Cardiotoxic changes of colchicine intoxication in rats: Electrocardiographic, histopathological and blood chemical analysis. J. Toxicol. Pathol. 2014, 27, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Caporizzo, M.A.; Bedi, K.; Vite, A.; Bogush, A.I.; Robison, P.; Heffler, J.G.; Salomon, A.K.; Kelly, N.A.; Babu, A.; et al. Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat. Med. 2018, 24, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Caporizzo, M.A.; Chen, C.Y.; Salomon, A.K.; Margulies, K.B.; Prosser, B.L. Microtubules Provide a Viscoelastic Resistance to Myocyte Motion. Biophys. J. 2018, 115, 1796–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robison, P.; Caporizzo, M.A.; Ahmadzadeh, H.; Bogush, A.I.; Chen, C.Y.; Margulies, K.B.; Shenoy, V.B.; Prosser, B.L. Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 2016, 352, aaf0659. [Google Scholar] [CrossRef] [Green Version]
- Lahm, T.; Douglas, I.S.; Archer, S.L.; Bogaard, H.J.; Chesler, N.C.; Haddad, F.; Hemnes, A.R.; Kawut, S.M.; Kline, J.A.; Kolb, T.M.; et al. Assessment of Right Ventricular Function in the Research Setting: Knowledge Gaps and Pathways Forward. An Official American Thoracic Society Research Statement. Am. J. Respir. Crit. Care Med. 2018, 198, e15–e43. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.W.; Thenappan, T.; Weir, E.K.; Kalra, R.; Pritzker, M.; Archer, S.L. Repurposing Medications for Treatment of Pulmonary Arterial Hypertension: What’s Old Is New Again. J. Am. Heart Assoc. 2019, 8, e011343. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.W.; Asp, M.L.; Zhang, H.; Wang, W.; Metzger, J.M. Microtubule-Mediated Misregulation of Junctophilin-2 Underlies T-Tubule Disruptions and Calcium Mishandling in mdx Mice. JACC Basic Transl. Sci. 2016, 1, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.C. Molecular therapeutic target for type-2 diabetes. J. Proteome Res. 2004, 3, 1284–1288. [Google Scholar] [CrossRef]
- Urboniene, D.; Haber, I.; Fang, Y.H.; Thenappan, T.; Archer, S.L. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L401–L412. [Google Scholar] [CrossRef] [Green Version]
- Thu, Y.M.; Van Riper, S.K.; Higgins, L.; Zhang, T.; Becker, J.R.; Markowski, T.W.; Nguyen, H.D.; Griffin, T.J.; Bielinsky, A.K. Slx5/Slx8 Promotes Replication Stress Tolerance by Facilitating Mitotic Progression. Cell Rep. 2016, 15, 1254–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihama, Y.; Rappsilber, J.; Mann, M. Modular stop and go extraction tips with stacked disks for parallel and multidimensional Peptide fractionation in proteomics. J. Proteome Res. 2006, 5, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.W.; Weir, E.K.; Archer, S.L.; Markowitz, J.; Rose, L.; Pritzker, M.; Madlon-Kay, R.; Thenappan, T. Pulmonary pulse wave transit time is associated with right ventricular-pulmonary artery coupling in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Hsu, S.; Houston, B.A.; Tampakakis, E.; Bacher, A.C.; Rhodes, P.S.; Mathai, S.C.; Damico, R.L.; Kolb, T.M.; Hummers, L.K.; Shah, A.A.; et al. Right Ventricular Functional Reserve in Pulmonary Arterial Hypertension. Circulation 2016, 133, 2413–2422. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.W.; Rose, L.; Archer, S.L.; Pritzker, M.; Weir, E.K.; Olson, M.D.; Thenappan, T. Clinical Determinants and Prognostic Implications of Right Ventricular Dysfunction in Pulmonary Hypertension Caused by Chronic Lung Disease. J. Am. Heart Assoc. 2019, 8, e011464. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.; Kokkonen-Simon, K.M.; Kirk, J.A.; Kolb, T.M.; Damico, R.L.; Mathai, S.C.; Mukherjee, M.; Shah, A.A.; Wigley, F.M.; Margulies, K.B.; et al. Right Ventricular Myofilament Functional Differences in Humans with Systemic Sclerosis-Associated Versus Idiopathic Pulmonary Arterial Hypertension. Circulation 2018, 137, 2360–2370. [Google Scholar] [CrossRef]
- Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 2019, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Manzari, B.; Kudlow, J.E.; Fardin, P.; Merello, E.; Ottaviano, C.; Puppo, M.; Eva, A.; Varesio, L. Induction of macrophage glutamine: Fructose-6-phosphate amidotransferase expression by hypoxia and by picolinic acid. Int. J. Immunopathol. Pharmacol. 2007, 20, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and new. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Protein | Control Levels (n = 3) | MCT Levels (n = 3) | MCT-Colch Levels (n = 4) | Previously Identified as O-GlcNAcylated | Citation |
|---|---|---|---|---|---|---|
| HADHA | Trifunctional enzyme subunit alpha | 1.0 ± 0.1 | 2.0 ± 1.2 | 1.2 ± 0.1 | Yes | [14,24] |
| SLC25A3 | Phosphate carrier protein | 1.0 ± 0.1 | 1.6 ± 0.3 | 1.4 ± 0.2 | Yes | [14,24] |
| CHCHD3 | MICOS complex subunit | 1.0 ± 0.2 | 1.6 ± 0.2 | 1.3 ± 0.2 | Yes | [14,24] |
| ATP5F1A | ATP synthase subunit alpha | 1.0 ± 0.1 | 1.3 ± 0.2 | 1.2 ± 0.2 | Yes | [14,24] |
| IMMT | MICOS complex subunit MIC60 | 1.0 ± 0.1 | 1.4 ± 0.2 | 1.1 ± 0.1 | Yes | [14,24] |
| HADHB | Trifunctional enzyme subunit beta | 1.0 ± 0.1 | 1.9 ± 1.1 | 1.1 ± 0.1 | Yes | [14,24] |
| NDUFS1 | NADH-ubiquinone oxidoreductase 75 kDa subunit | 1.0 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.2 | Yes | [14,24] |
| UQCRC2 | Cytochrome b-c1 complex subunit 2 | 1.0 ± 0.2 | 1.5 ± 0.5 | 1.5 ± 0.3 | Yes | [14,24] |
| MRPL34 | Mitochondrial ribosomal protein L34 | 1.0 ± 0.3 | 3.2 ± 0.5 | 2.5 ± 0.4 | No | NA |
| SUCLG2 | Succinate-CoA ligase subunit beta | 1.0 ± 0.2 | 1.5 ± 0.5 | 1.5 ± 0.4 | No | NA |
| Characteristics | Total Cohort (n = 45) | Nondiabetic (n = 31) | Diabetic (n = 14) | p-Value |
|---|---|---|---|---|
| Female, n (%) | 33 (73) | 23 (75) | 10 (71) | 1.0 |
| Age | 53.7 ± 16.0 | 50.0 ± 15.5 | 61.9 ± 14.3 | 0.019 |
| BMI, kg/m2 | 31.6 ± 8.1 | 30.3 ± 8.0 | 34.7 ± 7.9 | 0.089 |
| WHO Group 1 Etiology n (%) | ||||
| Associated | 23 (51) | 15 (48) | 8 (57) | 0.75 |
| Idiopathic | 17 (38) | 13 (42) | 4 (29) | 0.51 |
| Drug/toxin | 4 (9) | 3 (10) | 1 (7) | 1.0 |
| Heritable | 1 (2) | 0 (0) | 1 (7) | 0.31 |
| Medications, n (%) | ||||
| Oxygen | 12 (27) | 9 (29) | 3 (21) | 0.73 |
| Diuretics | 30 (67) | 19 (61) | 11 (79) | 0.32 |
| Digoxin | 3 (7) | 1 (3) | 2 (14) | 0.22 |
| Warfarin | 5 (11) | 3 (10) | 2 (14) | 0.64 |
| Calcium channel blockers | 4 (9) | 2 (6) | 2 (14) | 0.58 |
| Phosphodiesterase-5-inhibitors | 17 (38) | 12 (39) | 5 (36) | 1.0 |
| Endothelin receptor antagonists | 8 (18) | 7 (23) | 1 (7) | 0.40 |
| Prostacyclins | 7 (16) | 6 (19) | 1 (7) | 0.41 |
| Six minute walk test | ||||
| Distance, meters | 333 ± 157 | 368 ± 146 | 200 ± 133 | 0.03 |
| Rest oxygen saturation, % | 96 ± 2 | 96 ± 2 | 96 ± 2 | 0.53 |
| Peak exercise oxygen saturation, % | 91 ± 4 | 90 ± 5 | 91 ± 4 | 0.70 |
| Borg dyspnea score | 4 ± 1 | 4 ± 2 | 5 ± 1 | 0.33 |
| Hemodynamics | ||||
| Heart rate, beats/min | 76 ± 15 | 77 ± 17 | 74 ± 9 | 0.47 |
| Mean right atrial, mm Hg | 9 ± 5 | 8 ± 6 | 10 ± 4 | 0.37 |
| Mean PAP, mm Hg | 48 ± 14 | 48 ± 16 | 47 ± 12 | 0.80 |
| PCWP, mm Hg | 10 ± 3 | 10 ± 3 | 10 ± 4 | 0.60 |
| Cardiac output, L/min | 5.1 ± 1.9 | 5.1 ± 2.0 | 5.3 ± 2.0 | 0.72 |
| Cardiac Index, L/min/m2 | 2.6 ± 1.0 | 2.6 ± 1.0 | 2.6 ± 2.0 | 0.99 |
| PVR, WU | 8.9 ± 5.5 | 9.2 ± 5.5 | 8.6 ± 5.8 | 0.75 |
| PAC, mL/mm Hg | 1.8 ± 1.1 | 1.8 ± 1.2 | 1.9 ± 1.1 | 0.74 |
| Measures of RV Function | ||||
| NT pro-BNP, pg/mL | 1529 ± 2454 | 1125 ± 1279 | 2737 ± 3969 | 0.16 |
| TAPSE, cm | 1.9 ± 0.5 | 1.9 ± 0.4 | 1.9 ± 0.6 | 0.96 |
| RVFAC, % | 33 ± 9 | 33 ± 10 | 31 ± 7 | 0.49 |
| S’, cm/s | 10.6 ± 2.3 | 10.7 ± 2.4 | 9.9 ± 1.8 | 0.44 |
| RVEDP, mm Hg | 11 ± 6 | 10 ± 6 | 12 ± 5 | 0.58 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prisco, S.Z.; Rose, L.; Potus, F.; Tian, L.; Wu, D.; Hartweck, L.; Al-Qazazi, R.; Neuber-Hess, M.; Eklund, M.; Hsu, S.; et al. Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 7278. https://doi.org/10.3390/ijms21197278
Prisco SZ, Rose L, Potus F, Tian L, Wu D, Hartweck L, Al-Qazazi R, Neuber-Hess M, Eklund M, Hsu S, et al. Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2020; 21(19):7278. https://doi.org/10.3390/ijms21197278
Chicago/Turabian StylePrisco, Sasha Z., Lauren Rose, Francois Potus, Lian Tian, Danchen Wu, Lynn Hartweck, Ruaa Al-Qazazi, Monica Neuber-Hess, Megan Eklund, Steven Hsu, and et al. 2020. "Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 21, no. 19: 7278. https://doi.org/10.3390/ijms21197278
APA StylePrisco, S. Z., Rose, L., Potus, F., Tian, L., Wu, D., Hartweck, L., Al-Qazazi, R., Neuber-Hess, M., Eklund, M., Hsu, S., Thenappan, T., Archer, S. L., & Prins, K. W. (2020). Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 21(19), 7278. https://doi.org/10.3390/ijms21197278