Gene Editing by Extracellular Vesicles

 ,
, 

Abstract
:1. Introduction
2. Extracellular Vesicles (EV) as Drug Delivery Vehicles
2.1. General Characteristics
2.2. Extracellular Vesicles (EV) as Drug Delivery Vehicles
2.3. Safety of EVs
2.4. Biodistribution of EVs
3. Engineering the Surface of EVs for Improved and Targeted Delivery
3.1. Genetic Engineering
3.2. Chemical Methods
3.2.1. Click-Chemistry
3.2.2. Painting EVs with Targeting Peptides
3.2.3. Displaying Targeting Ligands by Co-Incubation with Liposomes or Synthetic Peptides
4. Packaging CRISPR/Cas Protein and RNA Components into EVs
4.1. Cas Protein Packaging
4.1.1. WW-Ndfip1 Interaction and Post-Translational Modifications
4.1.2. Arrestin-Domain Containing Protein 1 (ARRDC1)-Mediated EVs
4.1.3. Nanoblades
4.1.4. VEsiCas
4.1.5. Gesicle System
4.2. sgRNA Loading into EVs
4.2.1. Endogenous Packaging of RNAs for CRISPR/Cas Applications
The Use of EV-Associated Motifs
Overexpression Strategy
Light-Inducible MS2-MCP Interaction of sgRNAs with EVs-Constitutive Proteins
Synthetic Constructs for Loading sgRNAs into EVs
EXOtic RNA-Packaging Device
4.2.2. Exogenous Loading Approaches
Hydrophobically Modified RNAs
Fusion with RNA-Loaded Liposomes
Physical Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbrevations
| CRISPR/Cas | Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein |
| EVs | Extracellular vesicles |
| sgRNA | Single guide RNA |
| dCas9 | Dead or deactivated Cas9 |
| CRISPRa | CRISPR-activation |
| CRISPRi | CRISPR-interference |
| RNPs | Ribonucleoprotein complexes |
| MSCs | Mesenchymal stem/stromal cells |
| MHC | Major histocompatibility complex |
| iPSCs | induced pluripotent stem cells |
| ESCs | embryonic stem cells |
| RVG | rabies viral glycoprotein |
| MSP | muscle-specific peptide |
| POI | proteins of interest |
| GPI | glycosylphosphatidylinositol |
| VSV | vesicular stomatitis virus |
| GEDEX | Genome editing with designed extracellular vesicles |
| ARRDC1 | Arrestin domain containing protein 1 |
| ARMMs | ARRDC1- mediated microvesicles |
| MLV | murine leukemia virus |
| LID | light-induced dimerization |
| CID | chemically-induced dimerization |
| MCP | MS2 bacteriophage coat protein |
| HH | self-cleaving ribozymes hammerhead |
| HDV | hepatitis D virus ribozyme |
| EXOtic | exosomal transfer into cells |
References
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [Green Version]
- Savić, N.; Schwank, G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl. Res. 2016, 168, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; He, Z.; Wei, X.; Gao, G.-P.; Wei, Y. Challenges in CRISPR/CAS9 Delivery: Potential Roles of Nonviral Vectors. Hum. Gene Ther. 2015, 26, 452–462. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-X.; Li, M.; Lee, C.M.; Chakraborty, S.; Kim, H.-W.; Bao, G.; Leong, K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017, 117, 9874–9906. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, S.; Chen, X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 2018, 171, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Alphonse, M.; Liu, Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, 1609. [Google Scholar] [CrossRef] [PubMed]
- Mout, R.; Ray, M.; Lee, Y.-W.; Scaletti, F.; Rotello, V.M. In Vivo Delivery of CRISPR/Cas9 for Therapeutic Gene Editing: Progress and Challenges. Bioconjug. Chem. 2017, 28, 880–884. [Google Scholar] [CrossRef] [Green Version]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [Green Version]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Kim, S.; Koo, T.; Jee, H.-G.; Cho, H.-Y.; Lee, G.; Lim, D.-G.; Shin, H.S.; Kim, J.H. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 2018, 28, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.L.; Amini, L.; Wendering, D.J.; Burkhardt, L.-M.; Akyüz, L.; Reinke, P.; Volk, H.-D.; Schmueck-Henneresse, M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2018, 25, 242–248. [Google Scholar] [CrossRef]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, J.; Zhu, M.; Yang, Y.; Shen, J.; Gentile, E.; Paolino, D.; Fresta, M.; Nie, G.; Chen, C.; Shen, H.; et al. Safety of Nanoparticles in Medicine. Curr. Drug Targets 2015, 16, 1671–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Genet. 2019, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Cradick, T.J.; Bao, G. The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther. 2016, 24, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef] [PubMed]
- Burstein, D.; Harrington, L.B.; Strutt, S.C.; Probst, A.J.; Anantharaman, K.; Thomas, B.C.; Doudna, J.A.; Banfield, J.F. New CRISPR–Cas systems from uncultivated microbes. Nat. Cell Biol. 2016, 542, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brezgin, S.; Kostyusheva, A.; Kostyushev, D.; Chulanov, V. Dead Cas Systems: Types, Principles, and Applications. Int. J. Mol. Sci. 2019, 20, 6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA–guided activation of endogenous human genes. Nat. Chem. Biol. 2013, 10, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nat. Cell Biol. 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nat. Cell Biol. 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nat. Cell Biol. 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-R.; Yang, H.-C.; Kuo, Y.-T.; Liu, C.-J.; Yang, T.-Y.; Sung, K.-C.; Lin, Y.-Y.; Wang, H.-Y.; Wang, C.-C.; Shen, Y.-C.; et al. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef]
- Kostyushev, D.S.; Brezgin, S.; Kostyusheva, A.; Zarifyan, D.; Goptar, I.; Chulanov, V. Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell. Mol. Life Sci. 2019, 76, 1779–1794. [Google Scholar] [CrossRef] [PubMed]
- Price, A.A.; Sampson, T.R.; Ratner, H.K.; Grakoui, A.; Weiss, D.S. Cas9-mediated targeting of viral RNA in eukaryotic cells. Proc. Natl. Acad. Sci. USA 2015, 112, 6164–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, srep02510. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.-K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.-J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci. Rep. 2016, 6, 1–15. [Google Scholar]
- Hu, Z.; Yu, L.; Zhu, D.; Ding, W.; Wang, X.; Zhang, C.; Wang, L.; Jiang, X.; Shen, H.; He, D.; et al. Disruption of HPV16-E7 by CRISPR/Cas System Induces Apoptosis and Growth Inhibition in HPV16 Positive Human Cervical Cancer Cells. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876.e12. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2014, 33, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.K.; Kwon, K.; Ryu, J.S.; Lee, H.N.; Park, C.; Chung, H.J. Nonviral Genome Editing Based on a Polymer-Derivatized CRISPR Nanocomplex for Targeting Bacterial Pathogens and Antibiotic Resistance. Bioconjug. Chem. 2017, 28, 957–967. [Google Scholar] [CrossRef]
- Kooijmans, S.; Fliervoet, L.; Van Der Meel, R.; Fens, M.; Heijnen, H.; Henegouwen, P.V.B.E.; Vader, P.; Schiffelers, R. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Wohlford-Lenane, C.; Kandimalla, S.; Sartre, G.; Meyerholz, D.K.; Théberge, V.; Hallée, S.; Duperré, A.-M.; Guidice, T.E.; Lepetit-Stoffaes, J.-P.; et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019, 10, 4906–4912. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Wang, J.; Hu, Q.; Zhou, X.; Khademhosseini, A.; Gu, Z. CRISPR-Cas12a delivery by DNA-mediated bioresponsive editing for cholesterol regulation. Sci. Adv. 2020, 6, eaba2983. [Google Scholar] [CrossRef]
- Sun, W.; Ji, W.; Hall, J.M.; Hu, Q.; Wang, C.; Beisel, C.L.; Gu, Z. Self-Assembled DNA Nanoclews for the Efficient Delivery of CRISPR-Cas9 for Genome Editing. Angew. Chem. Int. Ed. 2015, 54, 12029–12033. [Google Scholar] [CrossRef]
- Mout, R.; Ray, M.; Tonga, G.Y.; Lee, Y.-W.; Tay, T.; Sasaki, K.; Rotello, V.M. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano 2017, 11, 2452–2458. [Google Scholar] [CrossRef] [Green Version]
- Lostalé-Seijo, I.; Louzao, I.; Juanes, M.; Montenegro, J. Peptide/Cas9 nanostructures for ribonucleoprotein cell membrane transport and gene edition. Chem. Sci. 2017, 8, 7923–7931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhang, L.; Zheng, W.; Cong, L.; Guo, Z.; Xie, Y.; Wang, L.; Tang, R.; Feng, Q.; Hamada, Y.; et al. Thermo-triggered Release of CRISPR-Cas9 System by Lipid-Encapsulated Gold Nanoparticles for Tumor Therapy. Angew. Chem. Int. Ed. 2018, 57, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhou, X.; Cheng, M.; Xing, D. Graphene oxide-mediated Cas9/sgRNA delivery for efficient genome editing. Nanoscale 2018, 10, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2013, 24, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Merienne, N.; Vachey, G.; De Longprez, L.; Meunier, C.; Zimmer, V.; Perriard, G.; Canales, M.; Mathias, A.; Herrgott, L.; Beltraminelli, T.; et al. The Self-Inactivating KamiCas9 System for the Editing of CNS Disease Genes. Cell Rep. 2017, 20, 2980–2991. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Lee, C.M.; Hurley, A.E.; Jarrett, K.E.; De Giorgi, M.; Lu, W.; Balderrama, K.S.; Doerfler, A.M.; Deshmukh, H.; Ray, A.; et al. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2019, 12, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Kelkar, A.; Zhu, Y.; Groth, T.; Stolfa, G.; Stablewski, A.B.; Singhi, N.; Nemeth, M.; Neelamegham, S. Doxycycline-Dependent Self-Inactivation of CRISPR-Cas9 to Temporally Regulate On- and Off-Target Editing. Mol. Ther. 2020, 28, 29–41. [Google Scholar] [CrossRef]
- E Dow, L.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nat. Cell Biol. 2012, 493, 429–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, A.P.; Rousseau, G.M.; Agudelo, D.; Goulet, A.; Amigues, B.; Loehr, J.; Romero, D.A.; Fremaux, C.; Horvath, P.; Doyon, Y.; et al. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat. Commun. 2018, 9, 2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R.; Bowie, A.G. Immune Sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.-W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting Edge: Ku70 Is a Novel Cytosolic DNA Sensor That Induces Type III Rather Than Type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenova, N.; Bosnjak, M.; Markelc, B.; Znidar, K.; Cemazar, M.; Heller, L. Multiple cytosolic DNA sensors bind plasmid DNA after transfection. Nucleic Acids Res. 2019, 47, 10235–10246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Znidar, K.; Bosnjak, M.; Semenova, N.; Pakhomova, O.; Heller, L.C.; Cemazar, M. Tumor cell death after electrotransfer of plasmid DNA is associated with cytosolic DNA sensor upregulation. Oncotarget 2018, 9, 18665–18681. [Google Scholar] [CrossRef] [Green Version]
- Klompe, S.E.; Vo, P.L.H.; Halpin-Healy, T.S.; Sternberg, S.H. Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nat. Cell Biol. 2019, 571, 219–225. [Google Scholar] [CrossRef]
- Strecker, J.; Ladha, A.; Gardner, Z.; Schmid-Burgk, J.L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019, 365, 48–53. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlake, T.; Thess, A.; Thran, M.; Jordan, I. mRNA as novel technology for passive immunotherapy. Cell. Mol. Life Sci. 2018, 76, 301–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luther, D.C.; Lee, Y.; Nagaraj, H.; Scaletti, F.; Rotello, V. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opin. Drug Deliv. 2018, 15, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- DeWitt, M.A.; Corn, J.E.; Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods 2017, 121, 9–15. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lee, B.; Lee, A.Y.-F.; Modzelewski, A.J.; He, L. Highly Efficient Mouse Genome Editing by CRISPR Ribonucleoprotein Electroporation of Zygotes. J. Boil. Chem. 2016, 291, 14457–14467. [Google Scholar] [CrossRef] [Green Version]
- Ferdosi, S.R.; Ewaisha, R.; Moghadam, F.; Krishna, S.; Park, J.G.; Ebrahimkhani, M.R.; Kiani, S.; Anderson, K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019, 10, 1842. [Google Scholar] [CrossRef] [Green Version]
- Wignakumar, T.; Fairchild, P.J. Evasion of Pre-Existing Immunity to Cas9: A Prerequisite for Successful Genome Editing In Vivo? Curr. Transplant. Rep. 2019, 6, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Buzas, E.I.; György, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef]
- Sterzenbach, U.; Putz, U.; Low, L.-H.; Silke, J.; Tan, S.-S.; Howitt, J. Engineered Exosomes as Vehicles for Biologically Active Proteins. Mol. Ther. 2017, 25, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Morishita, M.; Takahashi, Y.; Nishikawa, M.; Takakura, Y. Pharmacokinetics of Exosomes—An Important Factor for Elucidating the Biological Roles of Exosomes and for the Development of Exosome-Based Therapeutics. J. Pharm. Sci. 2017, 106, 2265–2269. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Gratton, S.E.A.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Barua, S.; Mitragotri, S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Van Der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 2015, 277, 277–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, P.A. Hyaluronan Regulation of Endothelial Barrier Function in Cancer. Adv. Cancer Res. 2014, 123, 191–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshad, F.; Wang, L.; Sy, C.; Avraham, S.; Avraham, H.K. Blood-Brain Barrier Integrity and Breast Cancer Metastasis to the Brain. Pathol. Res. Int. 2010, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Wang, Z.; Yue, T.; Su, G.; Teng, C.; Yan, B. Crossing Biological Barriers by Engineered Nanoparticles. Chem. Res. Toxicol. 2020, 33, 1055–1060. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, Z.; Seven, E.S.; Leblanc, R.M. Crossing the blood-brain barrier with nanoparticles. J. Control. Release 2018, 270, 290–303. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Engin, A.B.; Nikitovic, D.; Neagu, M.; Henrich-Noack, P.; Docea, A.O.; Shtilman, M.; Golokhvast, K.; Tsatsakis, A. Mechanistic understanding of nanoparticles’ interactions with extracellular matrix: The cell and immune system. Part. Fibre Toxicol. 2017, 14, 22. [Google Scholar] [CrossRef]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Nakase, I.; Futaki, S. Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep. 2015, 5, 10112. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.K.; Pan, H.; Schlesinger, P.H.; Wickline, S.A. A role for peptides in overcoming endosomal entrapment in siRNA delivery — A focus on melittin. Biotechnol. Adv. 2015, 33, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervia, L.D.; Chang, C.-C.; Wang, L.; Yuan, F. Distinct effects of endosomal escape and inhibition of endosomal trafficking on gene delivery via electrotransfection. PLoS ONE 2017, 12, e0171699. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Zhou, W.; Prabha, S.; Sahoo, S.K.; Labhasetwar, V. Rapid endo-lysosomal escape of poly(DL-lactide- co glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002, 16, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Verdera, H.C.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.-L.; Zhou, Y.-Y.; Liang, G.-F.; Wang, Y.-Y.; Hu, F.-H.; Xiao, Z.-D. Exosome Uptake through Clathrin-mediated Endocytosis and Macropinocytosis and Mediating miR-21 Delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [Green Version]
- Stremersch, S.; Vandenbroucke, R.; Van Wonterghem, E.; Hendrix, A.; De Smedt, S.C.; Raemdonck, K. Comparing exosome-like vesicles with liposomes for the functional cellular delivery of small RNAs. J. Control. Release 2016, 232, 51–61. [Google Scholar] [CrossRef]
- Mack, M.; Kleinschmidt, A.; Brühl, H.; Klier, C.; Nelson, P.J.; Cihak, J.; Plachý, J.; Stangassinger, M.; Erfle, V.; Schlöndorff, D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 2000, 6, 769–775. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Li, T.; Yan, Y.; Wang, B.; Qian, H.; Zhang, X.; Shen, L.; Wang, M.; Zhou, Y.; Zhu, W.; Li, W.; et al. Exosomes Derived from Human Umbilical Cord Mesenchymal Stem Cells Alleviate Liver Fibrosis. Stem Cells Dev. 2012, 22, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.-C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nat. Cell Biol. 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, 99263. [Google Scholar] [CrossRef] [PubMed]
- Bahadar, H.; Maqbool, F.; Niaz, K.; Abdollahi, M. Toxicity of Nanoparticles and an Overview of Current Experimental Models. Iran. Biomed. J. 2015, 20, 1–11. [Google Scholar] [PubMed]
- Nauta, A.J.; Westerhuis, G.; Kruisselbrink, A.B.; Lurvink, E.G.A.; Willemze, R.; Fibbe, W.E. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood 2006, 108, 2114–2120. [Google Scholar] [CrossRef]
- Oliveira, R.L.; Chagastelles, P.C.; Sesterheim, P.; Pranke, P. In Vivo Immunogenic Response to Allogeneic Mesenchymal Stem Cells and the Role of Preactivated Mesenchymal Stem Cells Cotransplanted with Allogeneic Islets. Stem Cells Int. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Eliopoulos, N.; Stagg, J.; Lejeune, L.; Pommey, S.; Galipeau, J. Allogeneic marrow stromal cells are immune rejected by MHC class I– and class II–mismatched recipient mice. Blood 2005, 106, 4057–4065. [Google Scholar] [CrossRef]
- Ge, Q.; Rao, V.P.; Cho, B.K.; Eisen, H.N.; Chen, J. Dependence of lymphopenia-induced T cell proliferation on the abundance of peptide/ MHC epitopes and strength of their interaction with T cell receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 1728–1733. [Google Scholar] [CrossRef] [Green Version]
- Ankrum, J.A.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Elahi, F.M.; Farwell, D.G.; Nolta, J.A.; Anderson, J.D. Preclinical translation of exosomes derived from mesenchymal stem/stromal cells. Stem Cells 2019, 38, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; La Perle, K.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef]
- Bai, L.; Shao, H.; Wang, H.; Zhang, Z.; Su, C.; Dong, L.; Yu, B.; Chen, X.; Li, X.-R.; Zhang, X. Effects of Mesenchymal Stem Cell-Derived Exosomes on Experimental Autoimmune Uveitis. Sci. Rep. 2017, 7, 4323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.D.; Johansson, H.J.; Graham, C.S.; Vesterlund, M.; Pham, M.T.; Bramlett, C.S.; Montgomery, E.N.; Mellema, M.S.; Bardini, R.L.; Contreras, Z.; et al. Comprehensive Proteomic Analysis of Mesenchymal Stem Cell Exosomes Reveals Modulation of Angiogenesis via Nuclear Factor-KappaB Signaling. Stem Cells 2016, 34, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, C.R.; Fellabaum, C.; Markovic, B.S.; Arsenijevic, A.; Volarevic, V. Therapeutic Potential of “Exosomes Derived Multiple Allogeneic Proteins Paracrine Signaling: Exosomes d-MAPPS” is Based on the Effects of Exosomes, Immunosuppressive and Trophic Factors. Serbian J. Exp. Clin. Res. 2019, 20, 189–197. [Google Scholar] [CrossRef] [Green Version]
- He, J.-G.; Xie, Q.-L.; Li, B.-B.; Zhou, L.; Yan, D. Exosomes Derived from IDO1-Overexpressing Rat Bone Marrow Mesenchymal Stem Cells Promote Immunotolerance of Cardiac Allografts. Cell Transplant. 2018, 27, 1657–1683. [Google Scholar] [CrossRef]
- Shigemoto-Kuroda, T.; Oh, J.Y.; Kim, D.-K.; Jeong, H.J.; Park, S.Y.; Lee, H.J.; Park, J.W.; Kim, T.W.; An, S.Y.; Prockop, D.J.; et al. MSC-derived Extracellular Vesicles Attenuate Immune Responses in Two Autoimmune Murine Models: Type 1 Diabetes and Uveoretinitis. Stem Cell Rep. 2017, 8, 1214–1225. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Morelli, A.E. Extracellular vesicle-mediated MHC cross-dressing in immune homeostasis, transplantation, infectious diseases, and cancer. Semin. Immunopathol. 2018, 40, 477–490. [Google Scholar] [CrossRef]
- He, C.; Zheng, S.; Luo, Y.; Wang, B. Exosome Theranostics: Biology and Translational Medicine. Theranostics 2018, 8, 237–255. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef]
- Rahman, M.A.; Barger, J.F.; Lovat, F.; Gao, M.; Otterson, G.A.; Nana-Sinkam, P. Lung cancer exosomes as drivers of epithelial mesenchymal transition. Oncotarget 2016, 7, 54852–54866. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Goulet, C.R.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGFβ Signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anel, A.; Gallego-Lleyda, A.; De Miguel, D.; Naval, J.; Martínez-Lostao, L. Role of Exosomes in the Regulation of T-cell Mediated Immune Responses and in Autoimmune Disease. Cells 2019, 8, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, M.E.; Zaffaroni, N.; Novara, F.; Cometa, A.M.; Avanzini, M.A.; Moretta, A.; Montagna, D.; Maccario, R.; Villa, R.; Daidone, M.G.; et al. Human Bone Marrow–Derived Mesenchymal Stem Cells Do Not Undergo Transformation after Long-termIn vitroCulture and Do Not Exhibit Telomere Maintenance Mechanisms. Cancer Res. 2007, 67, 9142–9149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Z.; Chi, Y.; Zhang, Q.; Xu, F.; Yang, Z.; Meng, L.; Yang, S.; Mao, A.; Zhang, J.; et al. Long-term cultured mesenchymal stem cells frequently develop genomic mutations but do not undergo malignant transformation. Cell Death Dis. 2013, 4, e950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, W.; El-Ansary, M.; Sabry, D.; Mostafa, M.A.; Fayad, T.; Kotb, E.; Temraz, M.; Saad, A.-N.; Essa, W.; Adel, H. Umbilical cord mesenchymal stem cells derived extracellular vesicles can safely ameliorate the progression of chronic kidney diseases. Biomater. Res. 2016, 20, 21. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Huang, L.; Li, Y.; Zhang, X.; Gu, J.; Yan, Y.; Xu, X.; Wang, M.; Qian, H.; Xu, W. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012, 315, 28–37. [Google Scholar] [CrossRef]
- Qi, J.; Zhou, Y.; Jiao, Z.; Wang, X.; Zhao, Y.; Li, Y.; Chen, H.; Yang, L.; Zhu, H.; Li, Y. Exosomes Derived from Human Bone Marrow Mesenchymal Stem Cells Promote Tumor Growth Through Hedgehog Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2242–2254. [Google Scholar] [CrossRef]
- Yang, Y.; Bucan, V.; Baehre, H.; Von Der Ohe, J.; Otte, A.; Hass, R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int. J. Oncol. 2015, 47, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.-T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell–derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles Derived from Human Bone Marrow Mesenchymal Stem Cells Inhibit Tumor Growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef]
- Lee, J.-K.; Park, S.-R.; Jung, B.-K.; Jeon, Y.-K.; Lee, Y.-S.; Kim, M.-K.; Kim, Y.-G.; Jang, J.-Y.; Kim, C.W. Exosomes Derived from Mesenchymal Stem Cells Suppress Angiogenesis by Down-Regulating VEGF Expression in Breast Cancer Cells. PLoS ONE 2013, 8, e84256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.P.; Khan, S.; Gilligan, K.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.H.; Nguyen, T.D.; Nguyen, H.-P.; Nguyen, X.-H.; Do, P.T.X.; Dang, V.D.; Dam, P.T.M.; Bui, H.T.H.; Trinh, M.Q.; Vu, D.M.; et al. Differential Wound Healing Capacity of Mesenchymal Stem Cell-Derived Exosomes Originated From Bone Marrow, Adipose Tissue and Umbilical Cord Under Serum- and Xeno-Free Condition. Front. Mol. Biosci. 2020, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in Exosome Isolation and Analysis in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.D.; Shah, S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 2015, 87, 3–10. [Google Scholar] [CrossRef]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [Green Version]
- Whitford, W.; Guterstam, P. Exosome manufacturing status. Futur. Med. Chem. 2019, 11, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Nishikawa, M.; Shinotsuka, H.; Matsui, Y.; Ohara, S.; Imai, T.; Takakura, Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J. Biotechnol. 2013, 165, 77–84. [Google Scholar] [CrossRef]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A Novel Nanoparticle Drug Delivery System: The Anti-inflammatory Activity of Curcumin Is Enhanced When Encapsulated in Exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.; Vigliotti, C.; Mosca, T.; Cammarota, M.; Capone, D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. Int. J. Mol. Sci. 2017, 18, 1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.-L.; Zhuang, X.; Sriwastva, M.K.; Mu, J.; Teng, Y.; Deng, Z.; Zhang, L.; Sundaram, K.; Kumar, A.; Miller, N.; et al. Blood exosomes regulate the tissue distribution of grapefruit-derived nanovector via CD36 and IGFR1 pathways. Theranostics 2018, 8, 4912–4924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Benito-Martin, A.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Denzer, K.; Van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; De Groot, C.; Geuze, H.J. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.-I.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Otero-Ortega, L.; De Frutos, M.C.G.; Laso-García, F.; Rodríguez-Frutos, B.; Gutiérrez-Fernández, M.; Lopez, J.A.; Vázquez, J.; Díez-Tejedor, E.; Gutiérrez-Fernández, M. Exosomes promote restoration after an experimental animal model of intracerebral hemorrhage. Br. J. Pharmacol. 2017, 38, 767–779. [Google Scholar] [CrossRef]
- Grange, C.; Tapparo, M.; Bruno, S.; Chatterjee, D.; Quesenberry, P.J.; Tetta, C.; Camussi, G. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int. J. Mol. Med. 2014, 33, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Pomatto, M.A.C.; Bussolati, B.; D’Antico, S.; Ghiotto, S.; Tetta, C.; Brizzi, M.F.; Camussi, G. Improved Loading of Plasma-Derived Extracellular Vesicles to Encapsulate Antitumor miRNAs. Mol. Ther. Methods Clin. Dev. 2019, 13, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zeng, Q.; Han, Q.; Xia, W. Effect of pH, temperature and freezing-thawing on quantity changes and cellular uptake of exosomes. Protein Cell 2018, 10, 295–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skliar, M.; Chernyshev, V.S.; Belnap, D.M.; Sergey, G.V.; Al-Hakami, S.M.; Bernard, P.S.; Stijleman, I.J.; Rachamadugu, R. Membrane proteins significantly restrict exosome mobility. Biochem. Biophys. Res. Commun. 2018, 501, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Charoenviriyakul, C.; Takahashi, Y.; Morishita, M.; Nishikawa, M.; Takakura, Y. Role of Extracellular Vesicle Surface Proteins in the Pharmacokinetics of Extracellular Vesicles. Mol. Pharm. 2018, 15, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Lee, J.H.; Kim, S.-Y.; Pack, C.G.; Ha, D.H.; Park, S.R.; Youn, J.; Cho, B.S. Advances in Analysis of Biodistribution of Exosomes by Molecular Imaging. Int. J. Mol. Sci. 2020, 21, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antes, T.J.; Middleton, R.C.; Luther, K.M.; Ijichi, T.; Peck, K.A.; Liu, W.J.; Valle, J.; Echavez, A.K.; Marbán, E. Targeting extracellular vesicles to injured tissue using membrane cloaking and surface display. J. Nanobiotechnol. 2018, 16, 61. [Google Scholar] [CrossRef]
- Shanmuganathan, M.; Vughs, J.; Noseda, M.; Emanueli, C. Exosomes: Basic Biology and Technological Advancements Suggesting Their Potential as Ischemic Heart Disease Therapeutics. Front. Physiol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Sancho-Albero, M.; Navascués, N.; Mendoza, G.; Sebastián, V.; Arruebo, M.; Martín-Duque, P.; Santamaría, J. Exosome origin determines cell targeting and the transfer of therapeutic nanoparticles towards target cells. J. Nanobiotechnol. 2019, 17, 16. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Bai, M.; Wang, J.; Zhu, K.; Liu, R.; Ge, S.; Li, J.; Ning, T.; Deng, T.; et al. Exosomes serve as nanoparticles to suppress tumor growth and angiogenesis in gastric cancer by delivering hepatocyte growth factor siRNA. Cancer Sci. 2018, 109, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles — Endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta (BBA) Rev. Cancer. 2014, 1846, 75–87. [Google Scholar] [CrossRef]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The Majority of MicroRNAs Detectable in Serum and Saliva Is Concentrated in Exosomes. PLoS ONE 2012, 7, e30679. [Google Scholar] [CrossRef] [Green Version]
- Evitt, N.H.; Mascharak, S.; Altman, R.B. Human Germline CRISPR-Cas Modification: Toward a Regulatory Framework. Am. J. Bioeth. 2015, 15, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcayre, A.; Estelles, A.; Sperinde, J.; Roulon, T.; Paz, P.; Aguilar, B.; Villanueva, J.; Khine, S.; Le Pecq, J.-B. Exosome Display technology: Applications to the development of new diagnostics and therapeutics. Blood Cells Mol. Dis. 2005, 35, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Ruiss, R.; Jochum, S.; Mocikat, R.; Hammerschmidt, W.; Zeidler, R. EBV-gp350 Confers B-Cell Tropism to Tailored Exosomes and Is a Neo-Antigen in Normal and Malignant B Cells—A New Option for the Treatment of B-CLL. PLoS ONE 2011, 6, e25294. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nat. Cell Biol. 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Men, Y.; Yelick, J.; Jin, S.; Tian, Y.; Chiang, M.S.R.; Higashimori, H.; Brown, E.; Jarvis, R.; Yang, Y. Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; Yue, S.; Stadel, D.; Zöller, M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef]
- Williams, C.; Royo, F.; Aizpurua-Olaizola, O.; Pazos, R.; Boons, G.-J.; Reichardt, N.; Falcon-Perez, J.M. Glycosylation of extracellular vesicles: Current knowledge, tools and clinical perspectives. J. Extracell. Vesicles 2018, 7, 1442985. [Google Scholar] [CrossRef]
- Christianson, H.C.; Svensson, K.J.; Van Kuppevelt, T.H.; Li, J.-P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Brigstock, D.R. Integrins and heparan sulfate proteoglycans on hepatic stellate cells (HSC) are novel receptors for HSC-derived exosomes. FEBS Lett. 2016, 590, 4263–4274. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yun, N.; Mun, D.; Kang, J.-Y.; Lee, S.-H.; Park, H.; Park, H.; Joung, B. Cardiac-specific delivery by cardiac tissue-targeting peptide-expressing exosomes. Biochem. Biophys. Res. Commun. 2018, 499, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.E.; Leonard, J.N. Stabilization of Exosome-targeting Peptides via Engineered Glycosylation. J. Biol. Chem. 2015, 290, 8166–8172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stickney, Z.; Losacco, J.; McDevitt, S.; Zhang, Z.; Lu, B. Development of exosome surface display technology in living human cells. Biochem. Biophys. Res. Commun. 2016, 472, 53–59. [Google Scholar] [CrossRef]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [Green Version]
- Kooijmans, S.A.A.; Aleza, C.G.; Roffler, S.R.; Van Solinge, W.W.; Vader, P.; Schiffelers, R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles 2016, 5, 31053. [Google Scholar] [CrossRef]
- Cheng, Q.; Shi, X.; Han, M.; Smbatyan, G.; Lenz, H.-J.; Zhang, Y. Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity. J. Am. Chem. Soc. 2018, 140, 16413–16417. [Google Scholar] [CrossRef]
- Qi, H.; Liu, C.; Long, L.; Ren, Y.; Zhang, S.; Chang, X.; Qian, X.; Jia, H.; Zhao, J.; Sun, J.; et al. Blood Exosomes Endowed with Magnetic and Targeting Properties for Cancer Therapy. ACS Nano 2016, 10, 3323–3333. [Google Scholar] [CrossRef]
- Wang, M.; Altinoglu, S.; Takeda, Y.S.; Xu, Q. Integrating Protein Engineering and Bioorthogonal Click Conjugation for Extracellular Vesicle Modulation and Intracellular Delivery. PLoS ONE 2015, 10, e0141860. [Google Scholar] [CrossRef]
- Tian, T.; Zhang, H.-X.; He, C.-P.; Fan, S.; Zhu, Y.-L.; Qi, C.; Huang, N.-P.; Xiao, Z.-D.; Lu, Z.-H.; Tannous, B.A.; et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [Google Scholar] [CrossRef]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface Functionalization of Exosomes Using Click Chemistry. Bioconjug. Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Ran, N.; Dong, X.; Zuo, B.; Yang, R.; Zhou, Q.; Moulton, H.M.; Seow, Y.; Yin, H. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018, 10, eaat0195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakase, I.; Noguchi, K.; Aoki, A.; Takatani-Nakase, T.; Fujii, I.; Futaki, S. Arginine-rich cell-penetrating peptide-modified extracellular vesicles for active macropinocytosis induction and efficient intracellular delivery. Sci. Rep. 2017, 7, 1991. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.-A.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakase, I.; Kobayashi, N.B.; Takatani-Nakase, T.; Yoshida, T. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci. Rep. 2015, 5, srep10300. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Uemoto, S.; Tabata, Y. Augmented liver targeting of exosomes by surface modification with cationized pullulan. Acta Biomater. 2017, 57, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Losacco, J.; Stickney, Z.; Li, L.; Marriott, G.; Lu, B. Pseudotyping exosomes for enhanced protein delivery in mammalian cells. Int. J. Nanomed. 2017, 12, 3153–3170. [Google Scholar] [CrossRef] [Green Version]
- Longatti, A.; Schindler, C.; Collinson, A.; Jenkinson, L.; Matthews, C.; Fitzpatrick, L.; Blundy, M.; Minter, R.; Vaughan, T.; Shaw, M.; et al. High affinity single-chain variable fragments are specific and versatile targeting motifs for extracellular vesicles. Nanoscale 2018, 10, 14230–14244. [Google Scholar] [CrossRef] [Green Version]
- Pi, F.; Binzel, D.W.; Lee, T.J.; Li, Z.; Sun, M.; Rychahou, P.; Li, H.; Haque, F.; Wang, S.; Croce, C.M.; et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2017, 13, 82–89. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Tian, B.; Liu, J.; Yang, L.; Zeng, L.; Chen, T.; Hong, A.; Wang, X. Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer. Theranostics 2017, 7, 1360–1372. [Google Scholar] [CrossRef]
- Pütz, U.; Howitt, J.; Doan, A.; Goh, C.-P.; Low, L.-H.; Silke, J.; Tan, S.-S. The Tumor Suppressor PTEN Is Exported in Exosomes and Has Phosphatase Activity in Recipient Cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Schorey, J. Targeting soluble proteins to exosomes using a ubiquitin tag. Biotechnol. Bioeng. 2015, 113, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Giovannone, A.J.; Reales, E.; Bhattaram, P.; Fraile-Ramos, A.; Weimbs, T. Monoubiquitination of syntaxin 3 leads to retrieval from the basolateral plasma membrane and facilitates cargo recruitment to exosomes. Mol. Biol. Cell 2017, 28, 2843–2853. [Google Scholar] [CrossRef] [PubMed]
- Gauvreau, M.-E.; Côté, M.-H.; Bourgeois-Daigneault, M.-C.; Rivard, L.-D.; Xiu, F.; Brunet, A.; Shaw, A.; Steimle, V.; Ethibodeau, J. Sorting of MHC Class II Molecules into Exosomes through a Ubiquitin-Independent Pathway. Traffic 2009, 10, 1518–1527. [Google Scholar] [CrossRef]
- Moreno-Gonzalo, O.; Fernández-Delgado, I.; Sánchez-Madrid, F. Post-translational add-ons mark the path in exosomal protein sorting. Cell. Mol. Life Sci. 2017, 75, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Wu, N.; Yang, J.-M.; Gould, S.J. Protein Targeting to Exosomes/Microvesicles by Plasma Membrane Anchors. J. Biol. Chem. 2011, 286, 14383–14395. [Google Scholar] [CrossRef] [Green Version]
- Yim, N.; Ryu, S.-W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.-H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein–protein interaction module. Nat. Commun. 2016, 7, 12277. [Google Scholar] [CrossRef]
- Gee, P.; Lung, M.S.Y.; Okuzaki, Y.; Sasakawa, N.; Iguchi, T.; Makita, Y.; Hozumi, H.; Miura, Y.; Yang, L.F.; Iwasaki, M.; et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lainšček, D.; Kadunc, L.; Manček-Keber, M.; Hafner-Bratkovič, I.; Romih, R.; Jerala, R. Delivery of an Artificial Transcription Regulator dCas9-VPR by Extracellular Vesicles for Therapeutic Gene Activation. ACS Synth. Biol. 2018, 7, 2715–2725. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- E Mangeot, P.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massourides, E.; Sohier, T.J.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagna, C.; Petris, G.; Casini, A.; Maule, G.; Franceschini, G.M.; Zanella, I.; Conti, L.; Arnoldi, F.; Burrone, O.R.; Zentilin, L.; et al. VSV-G-Enveloped Vesicles for Traceless Delivery of CRISPR-Cas9. Mol. Ther. Nucleic Acids 2018, 12, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, L.A.; Coke, L.M.; Richie, C.T.; Fortuno, L.V.; Park, A.Y.; Harvey, B.K. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol. Ther. 2019, 27, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crescitelli, R.; Lässer, C.; Szabó, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzás, E.I.; Lötvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2, 2. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 1–22. [Google Scholar] [CrossRef]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.W.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nat. Cell Biol. 2010, 465, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-S.; Maurin, T.; Lai, E.C. Functional parameters of Dicer-independent microRNA biogenesis. RNA 2012, 18, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Reshke, R.; Taylor, J.A.; Savard, A.; Guo, H.; Rhym, L.H.; Kowalski, P.S.; Trung, M.T.; Campbell, C.; Little, W.; Anderson, D.G.; et al. Reduction of the therapeutic dose of silencing RNA by packaging it in extracellular vesicles via a pre-microRNA backbone. Nat. Biomed. Eng. 2020, 4, 52–68. [Google Scholar] [CrossRef]
- Ragusa, M.; Barbagallo, C.; Cirnigliaro, M.; Battaglia, R.; Brex, D.; Caponnetto, A.; Barbagallo, D.; Di Pietro, C.; Purrello, M. Asymmetric RNA Distribution among Cells and Their Secreted Exosomes: Biomedical Meaning and Considerations on Diagnostic Applications. Front. Mol. Biosci. 2017, 4, 66. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, K.; Balter, M. Crystal Structure of an Early Protein-RNA Assembly Complex of the Signal Recognition Particle. Science 2001, 294, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Katsuda, T.; Gailhouste, L.; Kosaka, N.; Ochiya, T. Commitment of Annexin A2 in recruitment of microRNAs into extracellular vesicles. FEBS Lett. 2015, 589, 4071–4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collén, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santangelo, L.; Giurato, G.; Cicchini, C.; Montaldo, C.; Mancone, C.; Tarallo, R.; Battistelli, C.; Alonzi, T.; Weisz, A.; Tripodi, M. The RNA-Binding Protein SYNCRIP Is a Component of the Hepatocyte Exosomal Machinery Controlling MicroRNA Sorting. Cell Rep. 2016, 17, 799–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Huang, H.; Liu, H.; Xi, J.; Ning, J.; Zeng, W.; Shen, C.; Zhang, T.; Yu, G.; Xu, Q.; et al. Friend or Foe? Evidence Indicates Endogenous Exosomes Can Deliver Functional gRNA and Cas9 Protein. Small 2019, 15, e1902686. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.E.; Leonard, J.N. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J. Extracell. Vesicles 2016, 5, 31027. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Cai, B. Blue light-induced apoptosis of A2E-containing RPE: Involvement of caspase-3 and protection by Bcl-2. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1356–1362. [Google Scholar]
- King, A.; Gottlieb, E.; Brooks, D.G.; Murphy, M.P.; Dunaief, J.L. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem. Photobiol. 2004, 79, 470–475. [Google Scholar] [CrossRef]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, V.J.; Catarino, S.; Pinho, J.M.; Zuzarte, M.; Anjo, S.I.; Manadas, B.; Sluijter, J.P.G.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [Green Version]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.-E.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolukbasi, M.F.; Mizrak, A.; Ozdener, G.B.; Madlener, S.; Ströbel, T.; Erkan, E.P.; Fan, J.-B.; Breakefield, X.O.; Saydam, O. miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles. Mol. Ther. Nucleic Acids 2012, 1, e10. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, K.; Halvorsen, M.; Abrahamyan, L.; Chatel-Chaix, L.; Poupon, V.; Gordon, H.; DesGroseillers, L.; Gatignol, A.; Mouland, A.J. Trafficking of HIV-1 RNA is Mediated by Heterogeneous Nuclear Ribonucleoprotein A2 Expression and Impacts on Viral Assembly. Traffic 2006, 7, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Gu, N.; Zhang, X.-E.; Wang, D.-B. Light-Inducible Exosome-Based Vehicle for Endogenous RNA Loading and Delivery to Leukemia Cells. Adv. Funct. Mater. 2019, 29, 1807189. [Google Scholar] [CrossRef]
- Biscans, A.; Haraszti, R.A.; Echeverria, D.; Miller, R.; Didiot, M.-C.; Nikan, M.; Roux, L.; Aronin, N.; Khvorova, A. Hydrophobicity of Lipid-Conjugated siRNAs Predicts Productive Loading to Small Extracellular Vesicles. Mol. Ther. 2018, 26, 1520–1528. [Google Scholar] [CrossRef] [Green Version]
- Didiot, M.-C.; Haraszti, R.; Aronin, N.; Khvorova, A. Loading of Extracellular Vesicles with Hydrophobically Modified siRNAs. Methods Mol. Biol. 2018, 1740, 199–214. [Google Scholar]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. 2018, 5, 1700611. [Google Scholar] [CrossRef]
- Piffoux, M.; Silva, A.K.A.; Wilhelm, C.; Gazeau, F.; Tareste, D. Modification of Extracellular Vesicles by Fusion with Liposomes for the Design of Personalized Biogenic Drug Delivery Systems. ACS Nano 2018, 12, 6830–6842. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lee, H.; Zhu, Z.; Minhas, J.K.; Jin, Y. Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L110–L121. [Google Scholar] [CrossRef]
- Jeyaram, A.; Lamichhane, T.N.; Wang, S.; Zou, L.; Dahal, E.; Kronstadt, S.M.; Levy, D.; Parajuli, B.; Knudsen, D.R.; Chao, W.; et al. Enhanced Loading of Functional miRNA Cargo via pH Gradient Modification of Extracellular Vesicles. Mol. Ther. 2020, 28, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Podolak, I.; Galanty, A.; Sobolewska, D. Saponins as cytotoxic agents: A review. Phytochem. Rev. 2010, 9, 425–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.E.; Taussig, D.; Steinfeld, I.; Phadnis, S.M.; Lunstad, B.D.; Singh, M.; Vuong, X.; Okochi, K.D.; McCaffrey, R.; Olesiak, M.; et al. Improving CRISPR–Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2017, 46, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Taemaitree, L.; Shivalingam, A.; El-Sagheer, A.H.; Brown, T. An artificial triazole backbone linkage provides a split-and-click strategy to bioactive chemically modified CRISPR sgRNA. Nat. Commun. 2019, 10, 1610. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell. Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef]

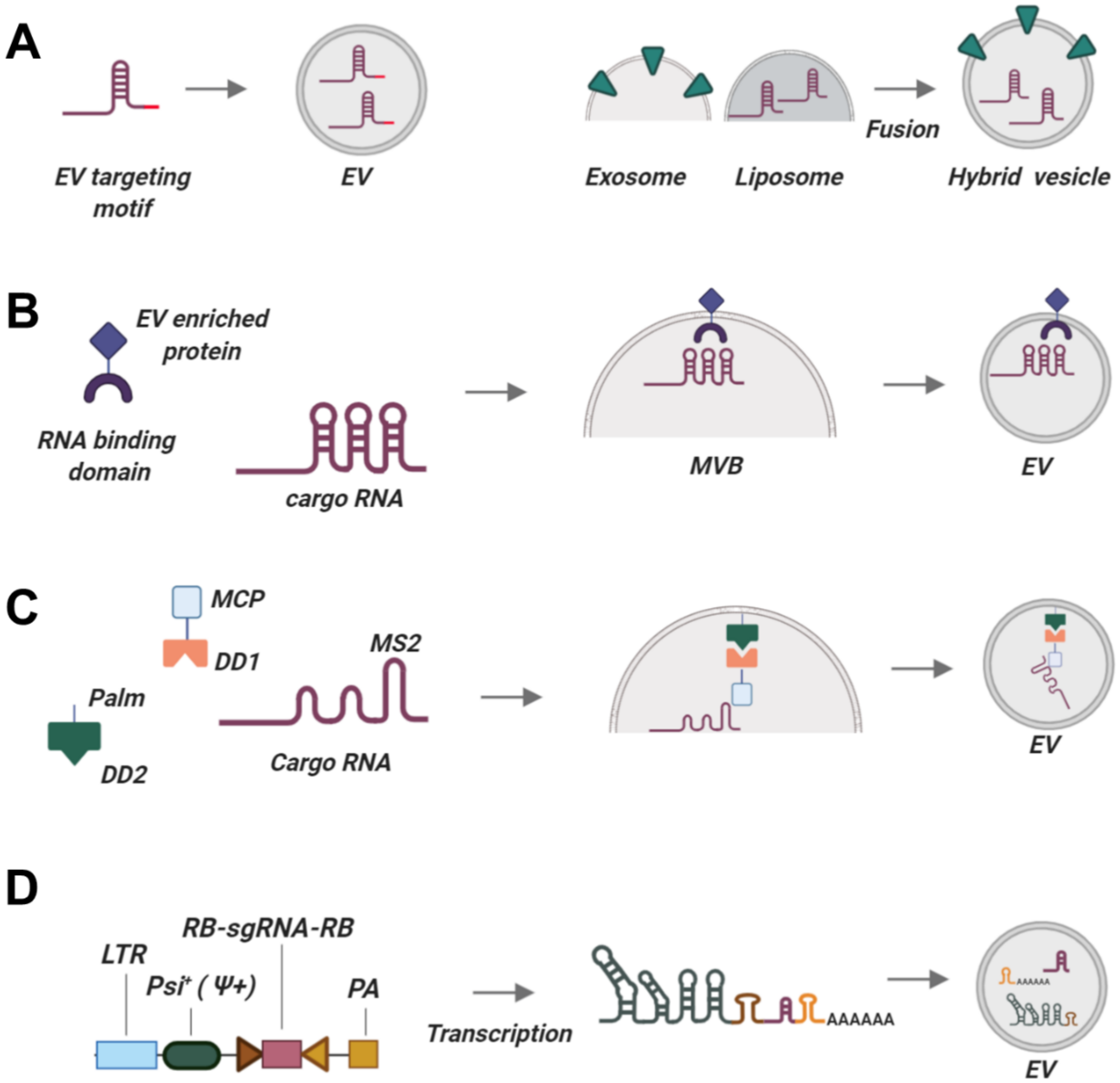

{kind=link}
{kind=link}
| Name | Type of Cas Package | Cas Packaging System | Type of sgRNA Package | sgRNA Packaging System | Vesicle Producing Cell Line | Number of Cas:sgRNA RNP Complexes Per Vesicle | Notes |
|---|---|---|---|---|---|---|---|
| EXPLORs [200] | Optogenetic dimerization system | CRY2 interacting with CIB1 module via blue light illumination and transient docking of CRY2-POI proteins to EVs (schematically depicted in Figure 1C) | NA | NA | HEK293T | Unknown | Induced by blue light illumination |
| NanoMEDIC [201] | Chemical ligand-dependent dimerization | -FRB-SpCas9:FKBP12--GagHIV (schematically depicted in Figure 1C) | Packaging signal | HIV Ψ packaging signal with HH and HDV self-cleaving ribozymes | HEK293T (adherent and in suspension) | 3,5-7,9 | Production in xeno-free conditions is 30% less efficient |
| Tags for post-translational modification [202] | Post-translational modification | -Ubiquitination -Myristoylation -Palmitoylation | NA | NA | Cancer cells | -Not specified -May be cell type-specific | -Release and functionality of POI in target cells is unclear -Efficiency is unclear |
| Genome editing with designed extracellular vesicles (GEDEX) or stochastic packaging [203] | Stochastic | Overexpression | Stochastic | Overexpression | -HEK293 -HepAD38 -HeLa -Huh7 | 10 µg of EVs contain 100 ng of Cas9 protein | Tested in vivo and in vitro |
| WW-Ndfip1 interaction [87] | Ubiquitination of the target protein | Fusion WW domain linked to POI | NA | NA | MEFs | Unknown | -Ndfip1 overexpression is required for packaging -Ndfip1 is toxic to cells |
| Arrestin domain containing protein 1- (ARRDC1) mediated microvesicles (ARMMs) [204] | Fusion of Cas9 with 2–4 ITCH domains | ARRDC1:WW-Cas9 (ITCH WW domains) | Packaging signal | ARRDC1-Tat: TAR-RNA co-transfection | HEK293T | 540 protein molecules | -Delivers cargo to many organs in vivo -Vesicle targeting and tissue specificity need to be tested |
| NanoBlades [205] | GagMLV fusion | SpCas9- GagMLV supplemented by Gag-PolMLV | Unclear, depends on Cas9-Gag interaction | Depends both on interaction with Cas9 and Gag proteins, but is not elucidated | Adherent HEK293T | Unknown | NanoBlades are shed vesicles with unknown characteristics |
| VEsiCas [206] | Stochastic incorporation with VSV-G assistance | VSV-G-assisted accumulation at cell periphery and vesicle packaging | Transcription of sgRNAs in the cytoplasm | T 7 RNA Pol-driven transcription. HDV ribozymes between the sgRNA and T7 terminator generate mature sgRNAs with unmodified 3′-constant regions | Adherent HEK293T | 1.5–2% of the total protein content of VEsiCas | VEsiCas are shed vesicles with unknown composition |
| Gesicle system [207] | Chemical-induced incorporation | -CherryPicker membrane-anchoring DmrA proteins associate with DmrC domain of Cas9-DmrC via A/C heterodimerizer molecule | Stochastic or mediated by interaction with Cas9 | NA | Adherent HEK293FT | <1% of gesicles contain Cas9:sgRNA RNPs | -Very inefficient packaging -sgRNA package not enforced -Cas9 half-life reduced |
| Name | Advantages | Drawbacks | Prospects |
|---|---|---|---|
| EXPLORs [201] | -Highly efficient -Utilize reversible protein-protein interaction modules -Transient protein docking into EVs | -Have not been used for CRISPR/Cas9 -sgRNA delivery is not addressed -Blue light is toxic to the cells | -Can be coupled with other light-induced dimerization (LID) or chemically-induced dimerization (CID) systems for sgRNA packaging -Cycles of blue light may be less toxic to producer cells |
| NanoMEDIC [202] | -Very first demonstration of successful exosome engineering for packaging and delivering CRISPR/Cas9 -Very efficient packaging of both Cas9 and sgRNA-Very efficient in vitro and in vivo genome editing -Proven activity in vivo-Cleared in vivo within 3 days -Scalable system in chemically-defined media with suspension cell culture | -Use HEK293T, a transformed cell line -Transformed cell lines produce exosomes with pro-oncogenic properties -Use rapamycin, an immunosuppressive drug with a number of potential adverse effects, to induce dimerization of domains. Rapamycin may potentially be packaged into EVs or alter exosome composition -Tissue-specific targeting upon systemic delivery has not been investigated -Reliance on HIV-1 Tat/Gag to drive sgRNA expression imposes the risks of toxicity both to producer cell lines and target cells -HIV-1 Tat/Gag may alter exosome composition -Co-produces Cas9 and sgRNA in the same cell | -Can be potentially expanded to clinically relevant EV-producing cell lines -Packaging of rapamycin into EVs and its effects on exosomes still needs to be defined -Any type of CRISPR/Cas system can be packaged |
| Tags for post-translational modification [203] | -Simple and feasible even for large proteins | -Have not been used for CRISPR/Cas9 -Most likely cell type-specific -Efficiency is unclear-Functionality in target cells unclear | -Simple and feasible -Applicability for Cas proteins needs to be defined |
| GEDEX or stochastic packaging [204] | -Very first demonstration of CRISPR/Cas9 RNP stochastic packaging into exosomes -Packaging of both Cas and sgRNAs -Efficient in vitro and in vivo genome editing -Scalable | -Utilize transformed cell lines -Transformed cell lines produce exosomes with pro-oncogenic properties -Tissue-specific targeting upon systemic delivery has not been investigated -Co-produce Cas9 and sgRNA in the same cell | -Very simple (overexpression of CRISPR/Cas components) -Any type of CRISPR/Cas system can be packaged |
| WW-Ndfip1 interaction [87] | -Efficiently delivers Cre-recombinase to target cells -Tested in vivo -Very simple (very short fusion peptides) | -Has not been used for CRISPR/Cas9 -Utilize mouse cells; not studied in human cells -Not studied with CRISPR/Cas packaging -Do not contribute to sgRNA packaging -Overexpressed Ndfip1 is required -Ndfip1 is toxic to producer cells -Ndfip1 interacts with numerous pro-oncogenic and pro-apoptotic factors | -Ndfip1 is toxic to producer cells -Ndfip1-WW interaction needs to be rationally engineered |
| ARMMs [205] | -Simple loading of protein and RNA cargo into vesicles -Efficient packaging of CRISPR/Cas RNPs -Efficient genome editing -Scalable -ARMMs may enter cells by direct fusion -Cargo bypasses endolysosomal pathway | -Use transformed cell lines -Transformed cell lines may produce vesicles with pro-oncogenic properties -Tissue-specific targeting upon systemic delivery has not been investigated -Co-produces Cas9 and sgRNA in the same cell | -Very simple packaging -Effects of ARRDC1 expression on producer cells and vesicle composition need to be addressed -Benefits of ARMMs over exosomes in terms of scalability and production need to be addressed |
| Nanoblades [206] | -Very limited carry-over of cellular proteins or overexpressed RNAs -Can potentially be produced from non-transformed cell lines -Have been combined with BaEV and VSV-G for improved delivery -Tested in vivo -Complex homologous DNA templates to generate knock-ins | -Carry-over of cellular RNAs (including those with pro-oncogenic potential) has not been investigated -Virus-like particles (viral origin) with membrane-associated proteins -Competition between HahMLV and Gag-PolMLV potentially reduces Cas packaging per particle -MLV protease may non-specifically cleave SpCas9 and reduce activity -Cas9 and sgRNA co-produced in the same cell | -Any type of CRISPR/Cas system can be packaged -Demonstrated for SpCas9 and dCas9-VPR |
| VEsiCas [207] | -Efficient Cas9 and sgRNA packaging -Very simple and easy-to-use fusion of Cas9-VSV-G and sgRNA-expressing constructs -Efficient, on-target genome editing -Tested in vivo | -Use HEK293T, a transformed cell line -Generated EVs are not exosomes; their properties and interaction with target cells need to be determined -Tissue-specific targeting upon systemic delivery has not been investigated -Quantity needed and quality of VEsiCas remain to be investigated -Composition of VEsiCas and co-packaging of potentially toxic proteins is not clear -Cas9 and sgRNA co-produced in the same cell | -Can be potentially expanded to clinically relevant EV-producing cell lines -Any type of CRISPR/Cas system can be packaged |
| Gesicles [208] | -Transfer Cas9:sgRNA RNPs -Efficient genome editing in target cells -Simple packaging system | -Use HEK293FT, a transformed cell line -Evidently less effective than NanoMEDIC -Cas9 protein half-life is reduced -<1% of produced gesicles contain RNPs -Carry-over of producer proteins and RNAs is possible -Use potentially toxic A/C heterodimerizer -Cytotoxicity and immunogenicity have not been studied -Not tested in vivo -Cas9 and sgRNA co-produced in the same cell -No tissue-specific targeting reported | -Potentially consist of a vesicle population mixed with cell waste as evidenced by increased gesicle formation following transfection |
| Type of Packaging | Mechanism | Type of RNA | Advantages | Disadvantages | Used Previously for sgRNA Targeting? |
|---|---|---|---|---|---|
| Insertion of exosome-targeting motifs | -miR451 stem loop and its structural mimics [216] | -miRNA -May be suitable for sgRNAs | -Many thousand-fold enrichment in different cell types | -Enrichment is cell type-specific -Inefficient | No |
| -EXOmotifs: GGAG in the 3′-half of RNA [217] -C/UCCU/G anywhere in RNA [217] -CTGCC motif [218] -Depend on hnRNPA2B1 | miRNA | -Exosome-specific motifs | -Never used to load sgRNAs -Requires several motifs -Enrichment in EVs may depend on trans-acting factors, sequence context, secondary and tertiary structures -Efficacy is unclear | No | |
| -Insertion of HIV sequences -A2RE sequences present in Gag and vpr ORFs [219] -Depend on hnRNPA2B1 | -Short RNAs | -Exosome-specific motifs | -Has never used for programmed loading -Efficacy unclear -May be cell-type specific | No | |
| -Secretion motifs: -ACCAGCCU -CAGUGAGC -UAAUCCCA | -RNAs -Non-coding RNAs | -Exosome-specific motifs | -Motifs may not be sufficient for transporting RNA into exosomes -May be cell-type specific -Requires a combination of different motifs | No | |
| -AnxA2-interacting motifs [220] -Putative binding motif is 5′-AA(C/G)(A/U)G | mRNAs | -Exosome-specific motifs | -Requires high-order RNA structures for interaction -May require two AnxA2-binding motifs -Depending on AnxA2 protein, may be cell-type specific | No | |
| GEDEX or stochastic packaging [204] | -Stochastic packaging | sgRNAs | -Efficient -Proven delivery in vitro and in vivo in several disease models -Suitable for mass-scale production | -Packaging is most likely cell type-specific -Produced in transformed cell lines -Safety issues | Yes209,226 |
| Insertion of exosome-targeting motifs [202] | -Ψ+-RGR HH ribozyme-sgRNA-HDV ribozyme-pA -Ψ+ interacts with expressed HIV Gag protein to package HH-sgRNA-HDV into exosomes -HH and HDV self-cleave to release sgRNA -HIV Tat/Tar interaction is required for EV packaging | RNAs | ~4-times more efficient at loading sgRNAs than stochastic loading from U6-sgRNA -Proven as a NanoMEDIC CRISPR-loading platform | -Requires several HIV proteins -HIV Gag and Tat proteins are associated with oxidative stress and may be toxic to cells [Oxidative Stress during HIV Infection: Mechanisms and Consequences] | Yes, as a component of NanoMEDIC208 |
| EXOtic RNA packaging devices [221] | -Archaeal ribosomal protein L7Ae binding to C/Dbox RNA structure -Fused CD63-L7A3 interacts with 3′-UTR C/Dbox-containing RNA -Number of C/Dbox-moieties affects efficacy -Connexin 43 (Cc43) acts as a cytosolic RNA delivery helper | -mRNAs -May be suitable for short RNAs | -Efficient -Can be adapted for sgRNAs packaging -Shown to be functional in human MSCs | -Release of RNA in target cells needs to be clarified | No |
| TAMEL platform [222] | -EV-enriched protein fused with an RNA-binding domain (MS2 protein dimer) -EV-enriched proteins: Lamp2b, CD63, Hspa8 | -mRNA -May be suitable for short RNAs | -Very efficient for RNA loading -Can be adapted for sgRNAs packaging | -Efficiency of RNA release unclear (no mRNA translation seen in target cell) -RNA is not released or is degraded by lysosomes | No |
| LID RNA binding [223] | -Palmytoylation sequence-EGFP-CIBN CYR2-mCherry-MCP BFP-miR-21Sponge-6×MS2-PolyA | -miRNA -May be used for short RNAs | -Very efficient (~14-fold enrichment) -Reversible -Can be adapted for sgRNAs packaging | -Requires blue light illumination (may be toxic to the producer cells) | No |
| Chemical RNA modification [224,225] | -Covalent conjugation of RNAs to hydrophobic moieties: -Docosanoic acid (efficient packaging) -Cholesterol (efficient packaging) -Tocopheryl succinate (vitamin E) (most efficient packaging) -TEG linker for attaching hydrophobic moiety to RNA is most efficient -RNA must be modified to resist phosphatase and nuclease | -Shown for siRNA -Can be used for sgRNAs and other short RNAs | -Thousands of copies packaged per vesicle -Very efficient -Very useful for mass-scale production -No need to be intracellularly produced, can be mixed with EVs | -Substantial portion is attached to the surface of EVs -Unknown which RNAs (surface-bound or luminal) are functionally active -Highest loading EV efficacy shown is 43% -The level of loading may be cell type-specific -Release of RNAs from EV membrane is not clear | No |
| RNA transfer by making hybrid exosomes [193,226,227] | -Incubation of exosomes with RNA-loaded liposomes for 12 h at 37 °C | -DNA -May be useful for RNA loading | -Incubation makes most exosomes form hybrids with liposomes -Very efficient -No need for intracellular RNA production -Very useful for large-scale production | -Long-term incubation at 37 °C may deteriorate RNA -Hybrid liposomes are toxic to cells (modification of liposomes is essential) | -Yes -Tested for CRISPR/Cas9-expressing DNA plasmids |
| Physical methods | -Electroporation of EV-producing cells [98] | Any nucleic acids | -Very efficient and reproducible -Can deliver both Cas and sgRNAs into EVs simultaneously | -Damaging to EVs (large holes in membranes) | -Yes -Not suitable for clinical applications |
| -Sonicating EVs for RNA loading [228] | -siRNA -Suitable for other short RNAs | -No significant aggregation of RNAs or EVs -Very efficient | -Degradation of RNA with prolonged sonication -Damaging to EVs – impaired functional properties -May damage Cas proteins pre-packaged into EVs | No | |
| Heat shocking EVs [229] | -miRNAs -Suitable for other short RNAs | -Efficient RNA loading -Suitable for large-scale production | -RNA deterioration -May result in degradation of pre-packaged Cas proteins -Impairment of EV membranes | No | |
| pH gradient modification of EVs [230] | -miRNAs -Suitable for other short RNAs | -Efficient -Suitable for large-scale production | -Evident protein degradation in EVs (decreased total protein content) | No | |
| Freezing-thawing [193] | -Mostly used for engineering EV surfaces | -Likely results in CRISPR/Cas RNPs packaging | -Freezing-thawing may destroy CRISPR/Cas components -Damaging to EVs | No | |
| Extrusion [231] | -Disrupts EV membranes | -Potentially able to load sgRNAs into EVs -Resulting EVs are not toxic | -Damages EV membranes | No | |
| Incubation with membrane permeabilizers | Saponin [228] | -Damages EV membranes | -Potentially able to load sgRNAs | -Saponin is a cytotoxic agent [232] -Saponin induces hemolysis [232] | No |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Smirnov, V.; Volchkova, E.; Lukashev, A.; Chulanov, V. Gene Editing by Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 7362. https://doi.org/10.3390/ijms21197362
Kostyushev D, Kostyusheva A, Brezgin S, Smirnov V, Volchkova E, Lukashev A, Chulanov V. Gene Editing by Extracellular Vesicles. International Journal of Molecular Sciences. 2020; 21(19):7362. https://doi.org/10.3390/ijms21197362
Chicago/Turabian StyleKostyushev, Dmitry, Anastasiya Kostyusheva, Sergey Brezgin, Valery Smirnov, Elena Volchkova, Alexander Lukashev, and Vladimir Chulanov. 2020. "Gene Editing by Extracellular Vesicles" International Journal of Molecular Sciences 21, no. 19: 7362. https://doi.org/10.3390/ijms21197362
APA StyleKostyushev, D., Kostyusheva, A., Brezgin, S., Smirnov, V., Volchkova, E., Lukashev, A., & Chulanov, V. (2020). Gene Editing by Extracellular Vesicles. International Journal of Molecular Sciences, 21(19), 7362. https://doi.org/10.3390/ijms21197362