Endothelial-Tumor Cell Interaction in Brain and CNS Malignancies


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Brain and CNS Malignancies
1.1. Gliomas
1.2. Medulloblastomas
1.3. Neuroblastomas
2. Mechanisms of Formation of New Vessels that Promote Further Tumorigenesis in Brain and CNS Tumors
2.1. Vasculogenesis and Recruitment of EC Progenitors from the Bone Marrow
2.2. Sprouting Angiogenesis
2.3. Intussusceptive Angiogenesis
2.4. Vasculogenic Mimicry and Transdifferentiation of Cancer Cells
2.5. Vascular Co-Option
2.6. Role of EMT and EndoMT in Brain Tumors
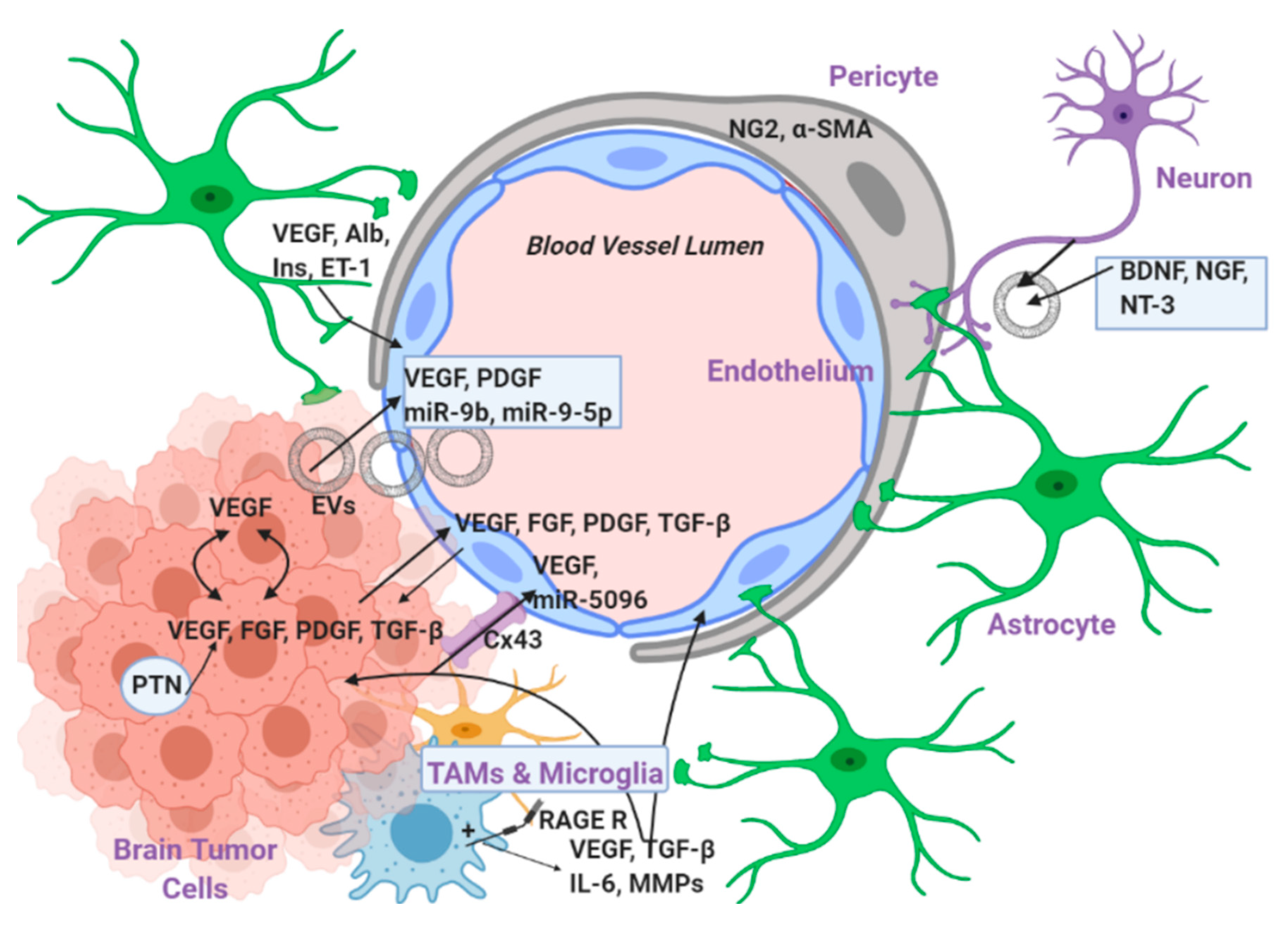
3. Levels of EC–Tumor Cell Interaction
3.1. Interaction with Secreted Factors
3.1.1. VEGF
3.1.2. FGF2
3.1.3. TGF-β
3.1.4. PDFG-B
3.1.5. Pleiotrophin
3.1.6. EVs and miRs
3.2. Direct Interaction via Gap Junctions
3.3. Indirect Interaction via Intermediate Cells
3.3.1. Pericytes
3.3.2. Astrocytes
3.3.3. Resident Microglial Cells and Tumor Associated Macrophages (TAMs)
3.3.4. Neurons
4. Role of Hypoxia, Reactive Oxygen Species, Nitric Oxide, and the Systems that Produce Them
- Hypoxia is an essential environmental cue for the maintenance of the glioblastoma cell population [146].
- By lowering the amount of available oxygen, hypoxia impedes the formation of ROS from the mitochondria (discussed below) which have a well-known cytotoxic role. The formation of ROS is a hallmark cytotoxic mechanism in radiation therapy whose efficacy is thus significantly reduced in the presence of hypoxia [148].
- Hypoxia promotes genomic instability by attenuating the expression of DNA repair enzymes. Therefore, more mutations can be acquired in the DNA which eventually favor the tumor growth even more [149].
- Hypoxia promotes a switch of the cancer cell metabolism to aerobic glycolysis by amplifying glucose uptake and promoting conversion to lactate rather than acetyl-CoA. This aerobic glycolysis creates an intracellular environment where the TCA cycle and oxidative phosphorylation are attenuated and therefore there is an accumulation of glycolytic intermediates which can be then used for anabolic reactions and biosynthesis of nucleotides, phospholipids, and amino-acids which are all absolutely necessary for tumor cell growth and proliferation [150].
- Last but not least, hypoxia supports and favors many of the different mechanisms for new blood vessel formation discussed above such as sprouting angiogenesis, recruitment of EC progenitor cells from the bone marrow and vasculogenic mimicry. In particular, hypoxia stabilizes the HIF-1 family of transcription factors which promote the expression of genes such as VEGF, SDF-1, and angiopoietin from the glioblastoma stem cells [58,151,152].
5. Conventional and New Interventions that Target the Endothelium in Brain and CNS Tumors
6. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| (Eph)A2 | Ephrin A2 |
| ALK1 | Anaplastic Lymphoma Kinase 1 |
| Ang-2 | Angiopoietin-2 |
| Ang-II | Angiotensin II |
| ASIs | Angiotensin System Inhibitors |
| BBB | Blood Brain Barrier |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central Nervous System |
| COX-2 | Cycloxygenase 2 |
| CSC | Glioblastoma Stem Cells |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DLL4 | Delta Like Canonical Notch Ligand 4 |
| EC | Endothelial Cell |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial to Mesenchymal Transition |
| EndoMT | Endothelial to Mesenchymal Transition |
| eNOS | Endothelial NO synthase |
| EPCs | Endothelial Progenitor Cells |
| ERKs | Extracellular Regulated Kinases |
| ET-1 | Endothelin 1 |
| EVs | Extracellular Vesicles |
| FDA | Food and Drug Administration |
| FGF | Fibroblast Growth Factor |
| IL-8 | Interleukin 8 |
| iNOS | Inducible NO synthase |
| MB | Medulloblastoma |
| MDGI | Mammary Derived Growth Inhibitor |
| MMPs | Matrix Metalloproteinases |
| mTOR | Mammalian Target Of Rapamycin |
| MVs | Microvesicles |
| NCC | Neural Crest Cells |
| NG2 | Neuron-Glia Antigen 2 |
| NGF | Nerve Growth Factor |
| nNOS | Neuronal NO synthase |
| NOX | NADPH oxidase |
| NRF2 | Nuclear factor erythroid 2 (NFE2)-related factor 2 |
| NT3 | Neurotrophin 3 |
| NO | Nitric Oxide |
| OS | Overall Survival |
| PDGF | Platelet Derived Growth Factor |
| PECAM-1 | Platelet Endothelial Cell Adhesion Molecule |
| PFS | Progression Free Survival |
| PI3K | Phosphatidyl-inositol 3′ kinase |
| PTEN | Phosphatase and TENsin homolog |
| PTN | Pleiotrophin |
| PTPRz | Protein Tyrosine Phosphatase Receptor Type z |
| RAGE | Receptor for Advanced Glycation End products |
| ROS | Reactive Oxygen Species |
| SDF-1 | Stromal Derived Factor 1 |
| TAMs | Tumor Associated Macrophages |
| TC | Tumor Cell |
| TGF-β | Transforming Growth Factor beta |
| TMZ | Temozolomide |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | VEGF Receptor |
| VM | Vasculogenic Mimicry |
| WHO | World Health Organization |
| XOR | Xanthine Oxidoreductase |
| α-SMA | α Smooth Muscle Actin |
References
- Rouse, C.; Gittleman, H.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Years of potential life lost for brain and CNS tumors relative to other cancers in adults in the United States, 2010. Neuro. Oncol. 2016, 18, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas—implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro. Oncol. 2012, 14, v1-49. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Wang, J.; Garancher, A.; Ramaswamy, V.; Wechsler-Reya, R.J. Medulloblastoma: From Molecular Subgroups to Molecular Targeted Therapies. Annu. Rev. Neurosci. 2018, 41, 207–232. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers 2019, 5, 11. [Google Scholar] [CrossRef]
- Fan, X.; Eberhart, C.G. Medulloblastoma stem cells. J. Clin. Oncol. 2008, 26, 2821–2827. [Google Scholar] [CrossRef] [Green Version]
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2011, 58, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- De Preter, K.; Vandesompele, J.; Heimann, P.; Yigit, N.; Beckman, S.; Schramm, A.; Eggert, A.; Stallings, R.L.; Benoit, Y.; Renard, M.; et al. Human fetal neuroblast and neuroblastoma transcriptome analysis confirms neuroblast origin and highlights neuroblastoma candidate genes. Genome Biol. 2006, 7, R84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Leon, S.P.; Folkerth, R.D.; Black, P.M. Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer 1996, 77, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Jakoniuk, I.; Anderson, S.M.; Li, B.; Pickel, J.; McKay, R.; Nadal-Ginard, B.; Bodine, D.M.; et al. Bone marrow cells regenerate infarcted myocardium. Nature 2001, 410, 701–705. [Google Scholar] [CrossRef]
- Risau, W.; Sariola, H.; Zerwes, H.G.; Sasse, J.; Ekblom, P.; Kemler, R.; Doetschman, T. Vasculogenesis and angiogenesis in embryonic-stem-cell-derived embryoid bodies. Development 1988, 102, 471–478. [Google Scholar] [PubMed]
- Moschetta, M.; Mishima, Y.; Sahin, I.; Manier, S.; Glavey, S.; Vacca, A.; Roccaro, A.M.; Ghobrial, I.M. Role of endothelial progenitor cells in cancer progression. Biochim. Biophys. Acta 2014, 1846, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Yu, C.F.; Hong, J.H.; Tsai, C.S.; Chiang, C.S. Radiation therapy-induced tumor invasiveness is associated with SDF-1-regulated macrophage mobilization and vasculogenesis. PLoS ONE 2013, 8, e69182. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.G.; Cohen, K.S.; Kozin, S.V.; Perentes, J.Y.; Fukumura, D.; Scadden, D.T.; Jain, R.K. Evidence for incorporation of bone marrow-derived endothelial cells into perfused blood vessels in tumors. Blood 2006, 107, 2774–2776. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.P.; Nagpal, S.; Iv, M.; Soltys, S.G.; Bertrand, S.; Pelpola, J.S.; Ball, R.; Yang, J.; Sundaram, V.; Lavezo, J.; et al. Macrophage Exclusion after Radiation Therapy (MERT): A First in Human Phase I/II Trial using a CXCR4 Inhibitor in Glioblastoma. Clin. Cancer Res. 2019, 25, 6948–6957. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Rossler, J.; Geoerger, B.; Laplanche, A.; Hartmann, O.; Vassal, G.; Farace, F. High levels of circulating VEGFR2+ Bone marrow-derived progenitor cells correlate with metastatic disease in patients with pediatric solid malignancies. Clin. Cancer Res. 2009, 15, 4561–4571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [Green Version]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis revisited from a metabolic perspective: Role and therapeutic implications of endothelial cell metabolism. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Kerbel, R.S. Tumor angiogenesis. N Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Crivellato, E. “Sprouting angiogenesis”, a reappraisal. Dev. Biol. 2012, 372, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Hellstrom, M.; Phng, L.K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Scholz, A.; Dumont, D.J. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012, 124, 763–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broholm, H.; Laursen, H. Vascular endothelial growth factor (VEGF) receptor neuropilin-1’s distribution in astrocytic tumors. APMIS 2004, 112, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Du, X.; Wu, H.; Liu, H.; Xie, T.; Tong, H.; Chen, X.; Guo, Y.; Zhang, W. Aberrant glioblastoma neovascularization patterns and their correlation with DCE-MRI-derived parameters following temozolomide and bevacizumab treatment. Sci. Rep. 2017, 7, 13894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamiphak, S.; Seidel, S.; Essmann, C.L.; Wilkinson, G.A.; Pitulescu, M.E.; Acker, T.; Acker-Palmer, A. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 2010, 465, 487–491. [Google Scholar] [CrossRef]
- Thurston, G.; Noguera-Troise, I.; Yancopoulos, G.D. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat. Rev. Cancer 2007, 7, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Makanya, A.N.; Hlushchuk, R.; Djonov, V.G. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis 2009, 12, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Mentzer, S.J.; Konerding, M.A. Intussusceptive angiogenesis: Expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar] [PubMed]
- Wilting, J.; Birkenhager, R.; Eichmann, A.; Kurz, H.; Martiny-Baron, G.; Marme, D.; McCarthy, J.E.; Christ, B.; Weich, H.A. VEGF121 induces proliferation of vascular endothelial cells and expression of flk-1 without affecting lymphatic vessels of chorioallantoic membrane. Dev. Biol. 1996, 176, 76–85. [Google Scholar] [CrossRef]
- Crivellato, E.; Nico, B.; Vacca, A.; Djonov, V.; Presta, M.; Ribatti, D. Recombinant human erythropoietin induces intussusceptive microvascular growth in vivo. Leukemia 2004, 18, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimova, I.; Hlushchuk, R.; Makanya, A.; Styp-Rekowska, B.; Ceausu, A.; Flueckiger, S.; Lang, S.; Semela, D.; Le Noble, F.; Chatterjee, S.; et al. Inhibition of Notch signaling induces extensive intussusceptive neo-angiogenesis by recruitment of mononuclear cells. Angiogenesis 2013, 16, 921–937. [Google Scholar] [CrossRef] [Green Version]
- Dill, M.T.; Rothweiler, S.; Djonov, V.; Hlushchuk, R.; Tornillo, L.; Terracciano, L.; Meili-Butz, S.; Radtke, F.; Heim, M.H.; Semela, D. Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology 2012, 142, 967–977.e2. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Crivellato, E.; Guidolin, D.; Annese, T.; Longo, V.; Finato, N.; Vacca, A.; Ribatti, D. Intussusceptive microvascular growth in human glioma. Clin. Exp. Med. 2010, 10, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Griffith, B.; Alotaibi, F.; Zagzag, D.; Fine, H.; Golfinos, J.; Schultz, L. Glioma Angiogenesis and Perfusion Imaging: Understanding the Relationship between Tumor Blood Volume and Leakiness with Increasing Glioma Grade. Am. J. Neuroradiol. 2015, 36, 2030–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Cortes, M.; Delgado-Bellido, D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 2019, 9, 803. [Google Scholar] [CrossRef] [Green Version]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. APMIS 2004, 112, 508–525. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Gupta, N.; Walcott, B.P.; Snuderl, M.; Kesler, C.T.; Kirkpatrick, N.D.; Heishi, T.; Huang, Y.; Martin, J.D.; Ager, E.; et al. Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J. Natl. Cancer Inst. 2013, 105, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Parodi, F.; Corrias, M.V.; Cinti, R.; Gambini, C.; Pistoia, V. Tumor origin of endothelial cells in human neuroblastoma. J. Clin. Oncol. 2007, 25, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Angara, K.; Rashid, M.H.; Shankar, A.; Ara, R.; Iskander, A.; Borin, T.F.; Jain, M.; Achyut, B.R.; Arbab, A.S. Vascular mimicry in glioblastoma following anti-angiogenic and anti-20-HETE therapies. Histol. Histopathol. 2017, 32, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Yu, L.; Ling, G.Q.; Xiao, S.; Sun, X.L.; Song, Z.H.; Liu, Y.J.; Jiang, X.D.; Cai, Y.Q.; Ke, Y.Q. Vasculogenic mimicry and its clinical significance in medulloblastoma. Cancer Biol. Ther. 2012, 13, 341–348. [Google Scholar] [CrossRef]
- Rong, X.; Huang, B.; Qiu, S.; Li, X.; He, L.; Peng, Y. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget 2016, 7, 83976–83986. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.G.; Xue, X.Y.; Wang, L.; Zhang, X.; Yan, M.; Tu, Y.Y.; Lin, W.; Jiang, X.F.; Ren, H.G.; Zhang, W.; et al. CDH5 is specifically activated in glioblastoma stemlike cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro. Oncol. 2013, 15, 865–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Liu, X.; Zhao, P.; Zhao, H.; Gao, W.; Wang, L. Celastrol Suppresses Glioma Vasculogenic Mimicry Formation and Angiogenesis by Blocking the PI3K/Akt/mTOR Signaling Pathway. Front. Pharm. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Hurlin, P.J. Control of vertebrate development by MYC. Cold Spring Harb. Perspect. Med. 2013, 3, a014332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.S.; Song, Y.K.; Durinck, S.; Chen, Q.R.; Cheuk, A.T.; Tsang, P.; Zhang, Q.; Thiele, C.J.; Slack, A.; Shohet, J.; et al. The MYCN oncogene is a direct target of miR-34a. Oncogene 2008, 27, 5204–5213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Son, M.H.; Cho, H.W.; Ma, Y.E.; Yoo, K.H.; Sung, K.W.; Koo, H.H. Clinical significance of MYCN amplification in patients with high-risk neuroblastoma. Pediatr Blood Cancer 2018, 65, e27257. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Shang, B.; Zhang, G.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumor cell-mediated neovascularization and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim. Biophys. Acta 2013, 1836, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dong, J.; Huang, Q.; Lou, M.; Wang, A.; Lan, Q. Endothelial cell transdifferentiation of human glioma stem progenitor cells in vitro. Brain Res. Bull. 2010, 82, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.S.; Serur, A.; Huang, J.; Manley, C.A.; McCrudden, K.W.; Frischer, J.S.; Soffer, S.Z.; Ring, L.; New, T.; Zabski, S.; et al. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc. Natl. Acad. Sci. USA 2002, 99, 11399–11404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Rankin, S.L.; Larson, J.D.; Zhu, X.; Chow, L.M.; Qu, C.; Zhang, J.; Ellison, D.W.; Baker, S.J. PTEN Signaling in the Postnatal Perivascular Progenitor Niche Drives Medulloblastoma Formation. Cancer Res. 2017, 77, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budde, M.D.; Gold, E.; Jordan, E.K.; Frank, J.A. Differential microstructure and physiology of brain and bone metastases in a rat breast cancer model by diffusion and dynamic contrast enhanced MRI. Clin. Exp. Metastasis 2012, 29, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE 2009, 4, e5857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.; Sontheimer, H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014, 592, 5109–5127. [Google Scholar] [CrossRef]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Le Joncour, V.; Filppu, P.; Hyvonen, M.; Holopainen, M.; Turunen, S.P.; Sihto, H.; Burghardt, I.; Joensuu, H.; Tynninen, O.; Jaaskelainen, J.; et al. Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, O.R.; McKinney, A.; Engler, J.R.; Koshkakaryan, G.; Gong, H.; Robinson, A.E.; Ewald, A.J.; Huillard, E.; David James, C.; Molinaro, A.M.; et al. GBM heterogeneity as a function of variable epidermal growth factor receptor variant III activity. Oncotarget 2016, 7, 79101–79116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Warren, R.Z.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Mechanisms of TGFbeta-Induced Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnan, J.; Isakson, P.; Joel, M.; Cilio, C.; Langmoen, I.A.; Vik-Mo, E.O.; Badn, W. Recruited brain tumor-derived mesenchymal stem cells contribute to brain tumor progression. Stem Cells 2014, 32, 1110–1123. [Google Scholar] [CrossRef]
- Angara, K.; Borin, T.F.; Arbab, A.S. Vascular Mimicry: A Novel Neovascularization Mechanism Driving Anti-Angiogenic Therapy (AAT) Resistance in Glioblastoma. Transl. Oncol. 2017, 10, 650–660. [Google Scholar] [CrossRef]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; Wang, N.; Zhang, T.C. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life 2012, 64, 717–723. [Google Scholar] [CrossRef]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-beta-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, A.R.; Nam, J.K.; Kim, J.M.; Kim, J.Y.; Seo, H.R.; Lee, H.J.; Cho, J.; Lee, Y.J. Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6(+) cancer cell and macrophage polarization. Nat. Commun. 2018, 9, 5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, I.F.; Heese, O.; Black, P.M. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J. NeuroOncol. 2000, 50, 121–137. [Google Scholar] [CrossRef] [PubMed]
- de Vries, C.; Escobedo, J.A.; Ueno, H.; Houck, K.; Ferrara, N.; Williams, L.T. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255, 989–991. [Google Scholar] [CrossRef]
- Terman, B.I.; Dougher-Vermazen, M.; Carrion, M.E.; Dimitrov, D.; Armellino, D.C.; Gospodarowicz, D.; Bohlen, P. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem. Biophys. Res. Commun. 1992, 187, 1579–1586. [Google Scholar] [CrossRef]
- Tate, M.C.; Aghi, M.K. Biology of angiogenesis and invasion in glioma. Neurotherapeutics 2009, 6, 447–457. [Google Scholar] [CrossRef]
- Noell, S.; Ritz, R.; Wolburg-Buchholz, K.; Wolburg, H.; Fallier-Becker, P. An allograft glioma model reveals the dependence of aquaporin-4 expression on the brain microenvironment. PLoS ONE 2012, 7, e36555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auguste, P.; Gursel, D.B.; Lemiere, S.; Reimers, D.; Cuevas, P.; Carceller, F.; Di Santo, J.P.; Bikfalvi, A. Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res. 2001, 61, 1717–1726. [Google Scholar] [PubMed]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Moscatelli, D.; Presta, M.; Rifkin, D.B. Purification of a factor from human placenta that stimulates capillary endothelial cell protease production, DNA synthesis, and migration. Proc. Natl. Acad. Sci. USA 1986, 83, 2091–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignatti, P.; Mazzieri, R.; Rifkin, D.B. Expression of the urokinase receptor in vascular endothelial cells is stimulated by basic fibroblast growth factor. J. Cell Biol. 1991, 113, 1193–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, D.T.; Stoddard, B.L.; Harakas, N.K.; Feder, J. Human fibroblast-derived growth factor is a mitogen and chemoattractant for endothelial cells. Biochem. Biophys. Res. Commun. 1987, 144, 705–712. [Google Scholar] [CrossRef]
- Montesano, R.; Vassalli, J.D.; Baird, A.; Guillemin, R.; Orci, L. Basic fibroblast growth factor induces angiogenesis in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 7297–7301. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. The transforming growth factor-beta family. Annu. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Constam, D.B.; Philipp, J.; Malipiero, U.V.; ten Dijke, P.; Schachner, M.; Fontana, A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 1992, 148, 1404–1410. [Google Scholar]
- Kjellman, C.; Olofsson, S.P.; Hansson, O.; Von Schantz, T.; Lindvall, M.; Nilsson, I.; Salford, L.G.; Sjogren, H.O.; Widegren, B. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int. J. Cancer 2000, 89, 251–258. [Google Scholar] [CrossRef]
- Dieterich, L.C.; Mellberg, S.; Langenkamp, E.; Zhang, L.; Zieba, A.; Salomaki, H.; Teichert, M.; Huang, H.; Edqvist, P.H.; Kraus, T.; et al. Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFbeta2 in vascular abnormalization. J. Pathol. 2012, 228, 378–390. [Google Scholar] [CrossRef]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocana, A.; Penuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, P.; Hu, B.; Gu, W.; Xu, L.; Wang, D.; Huang, H.J.; Cavenee, W.K.; Cheng, S.Y. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am. J. Pathol. 2003, 162, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.H.; Holland, E.C. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett. 2006, 232, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, A. The glioblastoma vasculature as a target for cancer therapy. Biochem. Soc. Trans. 2014, 42, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.; Czupalla, C.J.; Ziegler, N.; Devraj, K.; Zinke, J.; Seidel, S.; Heck, R.; Thom, S.; Macas, J.; Bockamp, E.; et al. Endothelial Wnt/beta-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J. Exp. Med. 2012, 209, 1611–1627. [Google Scholar] [CrossRef]
- Gonzalez-Castillo, C.; Ortuno-Sahagun, D.; Guzman-Brambila, C.; Pallas, M.; Rojas-Mayorquin, A.E. Pleiotrophin as a central nervous system neuromodulator, evidences from the hippocampus. Front. Cell Neurosci. 2014, 8, 443. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Kundu, S.; Feenstra, T.; Li, X.; Jin, C.; Laaniste, L.; El Hassan, T.E.; Ohlin, K.E.; Yu, D.; Olofsson, T.; et al. Pleiotrophin promotes vascular abnormalization in gliomas and correlates with poor survival in patients with astrocytomas. Sci. Signal. 2015, 8, ra125. [Google Scholar] [CrossRef]
- Chiba, R.; Akiya, M.; Hashimura, M.; Oguri, Y.; Inukai, M.; Hara, A.; Saegusa, M. ALK signaling cascade confers multiple advantages to glioblastoma cells through neovascularization and cell proliferation. PLoS ONE 2017, 12, e0183516. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Mondal, A.; Kumari Singh, D.; Panda, S.; Shiras, A. Extracellular Vesicles As Modulators of Tumor Microenvironment and Disease Progression in Glioma. Front. Oncol. 2017, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.D.; Williamson, R.; Almefty, K.K.; Nakaji, P.; Porter, R.; Tse, V.; Kalani, M.Y. The many roles of microRNAs in brain tumor biology. Neurosurg. Focus 2010, 28, E3. [Google Scholar] [CrossRef]
- Visone, R.; Croce, C.M. MiRNAs and cancer. Am. J. Pathol. 2009, 174, 1131–1138. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Lucero, R.; Zappulli, V.; Sammarco, A.; Murillo, O.D.; Cheah, P.S.; Srinivasan, S.; Tai, E.; Ting, D.T.; Wei, Z.; Roth, M.E.; et al. Glioma-Derived miRNA-Containing Extracellular Vesicles Induce Angiogenesis by Reprogramming Brain Endothelial Cells. Cell Rep. 2020, 30, 2065–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayan, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesnil, M.; Aasen, T.; Boucher, J.; Chepied, A.; Cronier, L.; Defamie, N.; Kameritsch, P.; Laird, D.W.; Lampe, P.D.; Lathia, J.D.; et al. An update on minding the gap in cancer. Biochim. Biophys. Acta Biomembr. 2018, 1860, 237–243. [Google Scholar] [CrossRef]
- Lin, J.H.; Takano, T.; Cotrina, M.L.; Arcuino, G.; Kang, J.; Liu, S.; Gao, Q.; Jiang, L.; Li, F.; Lichtenberg-Frate, H.; et al. Connexin 43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J. Neurosci. 2002, 22, 4302–4311. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; DeMattia, J.A.; Song, H.; Couldwell, W.T. Communication between malignant glioma cells and vascular endothelial cells through gap junctions. J. Neurosurg. 2003, 98, 846–853. [Google Scholar] [CrossRef]
- Thuringer, D.; Boucher, J.; Jego, G.; Pernet, N.; Cronier, L.; Hammann, A.; Solary, E.; Garrido, C. Transfer of functional microRNAs between glioblastoma and microvascular endothelial cells through gap junctions. Oncotarget 2016, 7, 73925–73934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artesi, M.; Kroonen, J.; Bredel, M.; Nguyen-Khac, M.; Deprez, M.; Schoysman, L.; Poulet, C.; Chakravarti, A.; Kim, H.; Scholtens, D.; et al. Connexin 30 expression inhibits growth of human malignant gliomas but protects them against radiation therapy. Neuro. Oncol. 2015, 17, 392–406. [Google Scholar] [CrossRef]
- Arun, S.; Ravisankar, S.; Vanisree, A.J. Implication of connexin30 on the stemness of glioma: Connexin30 reverses the malignant phenotype of glioma by modulating IGF-1R, CD133 and cMyc. J. Neurooncol. 2017, 135, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, Y.; Cui, B.; Liu, Z.; Shen, H. Novel insights into astrocyte-mediated signaling of proliferation, invasion and tumor immune microenvironment in glioblastoma. Biomed. Pharm. 2020, 126, 110086. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Reynolds, B.A.; Vescovi, A.; Steindler, D.A.; Deleyrolle, L.P. The origins of glioma: E Pluribus Unum? Glia 2011, 59, 1135–1147. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [Green Version]
- Barlow, K.D.; Sanders, A.M.; Soker, S.; Ergun, S.; Metheny-Barlow, L.J. Pericytes on the tumor vasculature: Jekyll or hyde? Cancer Microenviron. 2013, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Meng, M.B.; Zaorsky, N.G.; Deng, L.; Wang, H.H.; Chao, J.; Zhao, L.J.; Yuan, Z.Y.; Ping, W. Pericytes: A double-edged sword in cancer therapy. Future Oncol. 2015, 11, 169–179. [Google Scholar] [CrossRef]
- Goldstein, G.W. Endothelial cell-astrocyte interactions. A cellular model of the blood-brain barrier. Ann. NY Acad Sci. 1988, 529, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Ostrow, L.W.; Suchyna, T.M.; Sachs, F. Stretch induced endothelin-1 secretion by adult rat astrocytes involves calcium influx via stretch-activated ion channels (SACs). Biochem. Biophys. Res. Commun. 2011, 410, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, K.; Jones, J.; Lwin, Z.; Coward, J.I. Interleukin-6: An angiogenic target in solid tumours. Crit Rev. Oncol. Hematol. 2014, 89, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, L.; Zhang, I.Y.; Liang, J.; Wang, H.; Ouyang, M.; Wu, S.; da Fonseca, A.C.C.; Weng, L.; Yamamoto, Y.; et al. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res. 2014, 74, 7285–7297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R. Do Neural Cells Communicate with Endothelial Cells via Secretory Exosomes and Microvesicles? Cardiovasc Psychiatry Neurol. 2009, 2009, 383086. [Google Scholar] [CrossRef] [Green Version]
- Barami, K. Relationship of neural stem cells with their vascular niche: Implications in the malignant progression of gliomas. J. Clin. Neurosci. 2008, 15, 1193–1197. [Google Scholar] [CrossRef]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro. Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef]
- Beppu, T.; Kamada, K.; Yoshida, Y.; Arai, H.; Ogasawara, K.; Ogawa, A. Change of oxygen pressure in glioblastoma tissue under various conditions. J. Neurooncol. 2002, 58, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Shaffrey, M.E.; Gupta, B.; Larner, J.; Rich, J.N.; Park, D.M. Improving the radiosensitivity of radioresistant and hypoxic glioblastoma. Future Oncol. 2010, 6, 1591–1601. [Google Scholar] [CrossRef]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.P.; Tournaire, R.; Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell Physiol. 2008, 217, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species--the good, the bad and the ugly. Acta Physiol. (Oxf) 2015, 214, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reduction of nitrite to nitric oxide: Insights regarding where, when and how. Nitric Oxide 2013, 34, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, M.; Caffo, M.; Minutoli, L.; Marini, H.; Abbritti, R.V.; Squadrito, F.; Trichilo, V.; Valenti, A.; Barresi, V.; Altavilla, D.; et al. ROS and Brain Gliomas: An Overview of Potential and Innovative Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Han, N.; Yin, T.; Huang, L.; Liu, S.; Liu, D.; Xie, C.; Zhang, M. Lentivirus-mediated Nox4 shRNA invasion and angiogenesis and enhances radiosensitivity in human glioblastoma. Oxid Med. Cell Longev. 2014, 2014, 581732. [Google Scholar] [CrossRef] [Green Version]
- García-Gómez, P.; Dadras, M.; Bellomo, C.; Mezheyeuski, A.; Tzavlaki, K.; Moren, A.; Caja, L. NOX4 regulates TGFβ-induced proliferation and self-renewal in glioblastoma stem cells. bioRxiv 2019. [Google Scholar] [CrossRef]
- Salazar-Ramiro, A.; Ramirez-Ortega, D.; Perez de la Cruz, V.; Hernandez-Pedro, N.Y.; Gonzalez-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of Redox Status in Development of Glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.C.; Park, S.H.; Paek, S.H.; Park, S.Y.; Kwak, M.K.; Choi, H.G.; Yong, C.S.; Yoo, B.K.; Kim, J.A. Chloroquine-induced nitric oxide increase and cell death is dependent on cellular GSH depletion in A172 human glioblastoma cells. Toxicol. Lett. 2008, 178, 52–60. [Google Scholar] [CrossRef]
- Jakubowicz-Gil, J.; Langner, E.; Badziul, D.; Wertel, I.; Rzeski, W. Apoptosis induction in human glioblastoma multiforme T98G cells upon temozolomide and quercetin treatment. Tumour Biol. 2013, 34, 2367–2378. [Google Scholar] [CrossRef] [Green Version]
- Peleli, M.; Zollbrecht, C.; Montenegro, M.F.; Hezel, M.; Zhong, J.; Persson, E.G.; Holmdahl, R.; Weitzberg, E.; Lundberg, J.O.; Carlstrom, M. Enhanced XOR activity in eNOS-deficient mice: Effects on the nitrate-nitrite-NO pathway and ROS homeostasis. Free Radic. Biol. Med. 2016, 99, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Kokoglu, E.; Belce, A.; Ozyurt, E.; Tepeler, Z. Xanthine oxidase levels in human brain tumors. Cancer Lett. 1990, 50, 179–181. [Google Scholar] [CrossRef]
- Kou, B.; Ni, J.; Vatish, M.; Singer, D.R. Xanthine oxidase interaction with vascular endothelial growth factor in human endothelial cell angiogenesis. Microcirculation 2008, 15, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Tietzel, I.; Quick, Q.A. 1’-Acetoxychavicol acetate promotes caspase 3-activated glioblastoma cell death by overcoming enhanced cytokine expression. Oncol. Lett. 2013, 5, 1968–1972. [Google Scholar] [CrossRef] [PubMed]
- Rabender, C.S.; Alam, A.; Sundaresan, G.; Cardnell, R.J.; Yakovlev, V.A.; Mukhopadhyay, N.D.; Graves, P.; Zweit, J.; Mikkelsen, R.B. The Role of Nitric Oxide Synthase Uncoupling in Tumor Progression. Mol. Cancer Res. 2015, 13, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Inducible nitric oxide synthase: What difference does it make? J. Clin. Invest. 1997, 100, 2417–2423. [Google Scholar] [CrossRef]
- Kleinert, H.; Schwarz, P.M.; Forstermann, U. Regulation of the expression of inducible nitric oxide synthase. Biol. Chem. 2003, 384, 1343–1364. [Google Scholar] [CrossRef]
- Broholm, H.; Rubin, I.; Kruse, A.; Braendstrup, O.; Schmidt, K.; Skriver, E.B.; Lauritzen, M. Nitric oxide synthase expression and enzymatic activity in human brain tumors. Clin. Neuropathol. 2003, 22, 273–281. [Google Scholar]
- Erdamar, S.; Bagci, P.; Oz, B.; Dirican, A. Correlation of endothelial nitric oxide synthase and vascular endothelial growth factor expression with malignancy in patients with astrocytic tumors. J. Buon 2006, 11, 213–216. [Google Scholar]
- Iwata, S.; Nakagawa, K.; Harada, H.; Oka, Y.; Kumon, Y.; Sakaki, S. Endothelial nitric oxide synthase expression in tumor vasculature is correlated with malignancy in human supratentorial astrocytic tumors. Neurosurgery 1999, 45, 24–28. [Google Scholar] [CrossRef]
- Ludwig, H.C.; Feiz-Erfan, I.; Bockermann, V.; Behnke-Mursch, J.; Schallock, K.; Markakis, E. Expression of nitric oxide synthase isozymes (NOS I-III) by immunohistochemistry and DNA in situ hybridization. Correlation with macrophage presence, vascular endothelial growth factor (VEGF) and oedema volumetric data in 220 glioblastomas. Anticancer Res. 2000, 20, 299–304. [Google Scholar] [PubMed]
- Bulnes, S.; Argandona, E.G.; Bengoetxea, H.; Leis, O.; Ortuzar, N.; Lafuente, J.V. The role of eNOS in vascular permeability in ENU-induced gliomas. Acta Neurochir. Suppl. 2010, 106, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuraca, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.; Westphal, M.; Quinones-Hinojosa, A. Complications of glioma surgery. Handb Clin. Neurol. 2016, 134, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, N.M.; Lanzino, G. Cerebral vasospasm following tumor resection. J. Neurointerv. Surg. 2013, 5, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Tanaka, S.; Oishi, M.; Miyashita, K.; Misaki, K.; Mohri, M.; Hayashi, Y.; Uchiyama, N.; Watanabe, T.; Hayashi, Y. Cerebral Infarction Related to Carmustine Wafers in Glioblastoma: A Case Report. NMC Case Rep. J. 2015, 2, 36–39. [Google Scholar] [CrossRef]
- Khan, M.Q.; Cirjan, C.; Quadri, N.; Alexopoulos, G.; Coppens, J. Symptomatic cerebral vasospasm in the setting of carmustine wafer placement for glioblastoma: A case presentation and review of literature. Surg. Neurol. Int. 2020, 11, 168. [Google Scholar] [CrossRef]
- Barani, I.J.; Larson, D.A. Radiation therapy of glioblastoma. Cancer Treat. Res. 2015, 163, 49–73. [Google Scholar] [CrossRef]
- Gupta, K.; Burns, T.C. Radiation-Induced Alterations in the Recurrent Glioblastoma Microenvironment: Therapeutic Implications. Front. Oncol. 2018, 8, 503. [Google Scholar] [CrossRef] [Green Version]
- Niyazi, M.; Harter, P.N.; Hattingen, E.; Rottler, M.; von Baumgarten, L.; Proescholdt, M.; Belka, C.; Lauber, K.; Mittelbronn, M. Bevacizumab and radiotherapy for the treatment of glioblastoma: Brothers in arms or unholy alliance? Oncotarget 2016, 7, 2313–2328. [Google Scholar] [CrossRef]
- Lund, E.L.; Hog, A.; Olsen, M.W.; Hansen, L.T.; Engelholm, S.A.; Kristjansen, P.E. Differential regulation of VEGF, HIF1alpha and angiopoietin-1, -2 and -4 by hypoxia and ionizing radiation in human glioblastoma. Int. J. Cancer 2004, 108, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Lumniczky, K.; Szatmari, T.; Safrany, G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vulpen, M.; Kal, H.B.; Taphoorn, M.J.; El-Sharouni, S.Y. Changes in blood-brain barrier permeability induced by radiotherapy: Implications for timing of chemotherapy? (Review). Oncol. Rep. 2002, 9, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanichamy, K.; Chakravarti, A. Combining drugs and radiotherapy: From the bench to the bedside. Curr. Opin. Neurol. 2009, 22, 625–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.F.; de Tribolet, N.; Regli, L.; Wick, W.; Kouwenhoven, M.C.; et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Baisiwala, S.; Auffinger, B.; Caragher, S.P.; Shireman, J.M.; Ahsan, R.; Lee, G.; Hasan, T.; Park, C.; Saathoff, M.R.; Christensen, A.C.; et al. Chemotherapeutic Stress Induces Transdifferentiation of Glioblastoma Cells to Endothelial Cells and Promotes Vascular Mimicry. Stem Cells Int. 2019, 2019, 6107456. [Google Scholar] [CrossRef] [PubMed]
- Virrey, J.J.; Golden, E.B.; Sivakumar, W.; Wang, W.; Pen, L.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C. Glioma-associated endothelial cells are chemoresistant to temozolomide. J. Neurooncol. 2009, 95, 13–22. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Reardon, D.A.; de Groot, J.F.; Wick, W.; Weller, M. Antiangiogenic therapy for glioblastoma: Current status and future prospects. Clin. Cancer Res. 2014, 20, 5612–5619. [Google Scholar] [CrossRef] [Green Version]
- Diaz, R.J.; Ali, S.; Qadir, M.G.; De La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The role of bevacizumab in the treatment of glioblastoma. J. Neurooncol. 2017, 133, 455–467. [Google Scholar] [CrossRef]
- Guo, J.; Shinriki, S.; Su, Y.; Nakamura, T.; Hayashi, M.; Tsuda, Y.; Murakami, Y.; Tasaki, M.; Hide, T.; Takezaki, T.; et al. Hypoxia suppresses cylindromatosis (CYLD) expression to promote inflammation in glioblastoma: Possible link to acquired resistance to anti-VEGF therapy. Oncotarget 2014, 5, 6353–6364. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.G.; Wang, C.; Liu, D.Y.; Zhang, X.; Wang, L.; Yan, M.; Zhang, W.; Zhu, J.; Li, Z.C.; Mi, C.; et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget 2016, 7, 47808–47820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirches, E.; Warich-Kirches, M. 2-methoxyestradiol as a potential cytostatic drug in gliomas? Anticancer Agents Med. Chem. 2009, 9, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Evans, J.W.; Bhupathi, D.; Banica, M.; Lan, L.; Lorente, G.; Duan, J.X.; Cai, X.; Mowday, A.M.; Guise, C.P.; et al. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol. Cancer Ther. 2012, 11, 740–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.J.; Ryu, J.H.; Cho, Y.S.; Chun, Y.S.; Huang, L.E.; Kim, M.S.; Park, J.W. Amphotericin B blunts erythropoietin response to hypoxia by reinforcing FIH-mediated repression of HIF-1. Blood 2006, 107, 916–923. [Google Scholar] [CrossRef]
- Lombardi, G.; Pambuku, A.; Bellu, L.; Farina, M.; Della Puppa, A.; Denaro, L.; Zagonel, V. Effectiveness of antiangiogenic drugs in glioblastoma patients: A systematic review and meta-analysis of randomized clinical trials. Crit. Rev. Oncol. Hematol. 2017, 111, 94–102. [Google Scholar] [CrossRef]
- Gerstner, E.R.; Duda, D.G.; di Tomaso, E.; Ryg, P.A.; Loeffler, J.S.; Sorensen, A.G.; Ivy, P.; Jain, R.K.; Batchelor, T.T. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat. Rev. Clin. Oncol. 2009, 6, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.C.; Roth, I.M.; Wickremesekera, A.C.; Davis, P.F.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells 2019, 8, 1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, V.A.; Chan, J.; Datta, M.; Yee, J.L.; Jain, R.K. Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J. Neurooncol. 2017, 134, 325–330. [Google Scholar] [CrossRef]
- Hu, B.; Guo, P.; Fang, Q.; Tao, H.Q.; Wang, D.; Nagane, M.; Huang, H.J.; Gunji, Y.; Nishikawa, R.; Alitalo, K.; et al. Angiopoietin-2 induces human glioma invasion through the activation of matrix metalloprotease-2. Proc. Natl. Acad. Sci. USA 2003, 100, 8904–8909. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peleli, M.; Moustakas, A.; Papapetropoulos, A. Endothelial-Tumor Cell Interaction in Brain and CNS Malignancies. Int. J. Mol. Sci. 2020, 21, 7371. https://doi.org/10.3390/ijms21197371
Peleli M, Moustakas A, Papapetropoulos A. Endothelial-Tumor Cell Interaction in Brain and CNS Malignancies. International Journal of Molecular Sciences. 2020; 21(19):7371. https://doi.org/10.3390/ijms21197371
Chicago/Turabian StylePeleli, Maria, Aristidis Moustakas, and Andreas Papapetropoulos. 2020. "Endothelial-Tumor Cell Interaction in Brain and CNS Malignancies" International Journal of Molecular Sciences 21, no. 19: 7371. https://doi.org/10.3390/ijms21197371