Alterations in Anandamide Synthesis and Degradation during Osteoarthritis Progression in an Animal Model

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
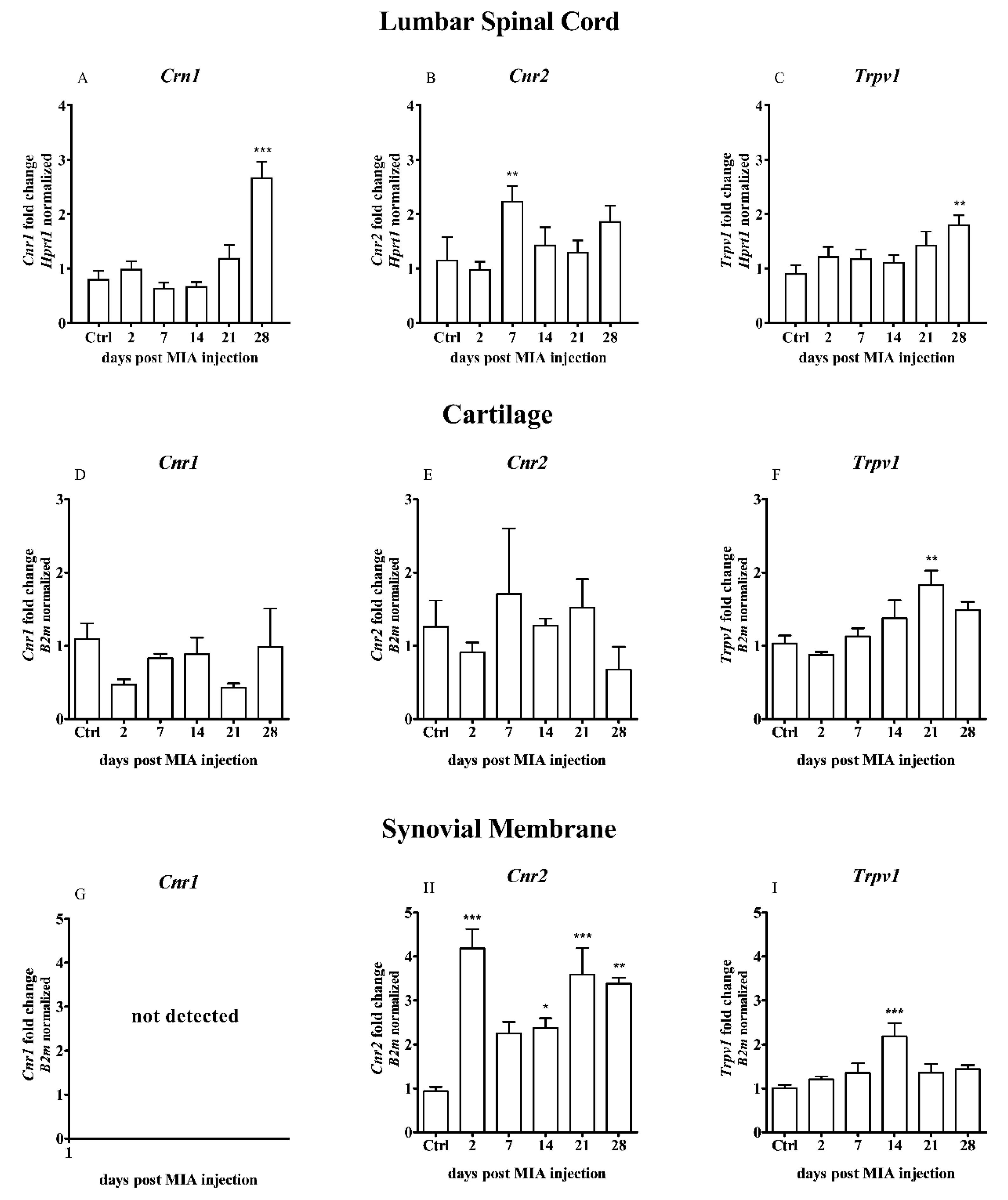
2.1. Changes in the Cnr1, Cnr2 and Trpv1 Gene Expression in the Dorsal Lumbar Spinal Cord and Joint Tissue of Osteoarthritic Rats
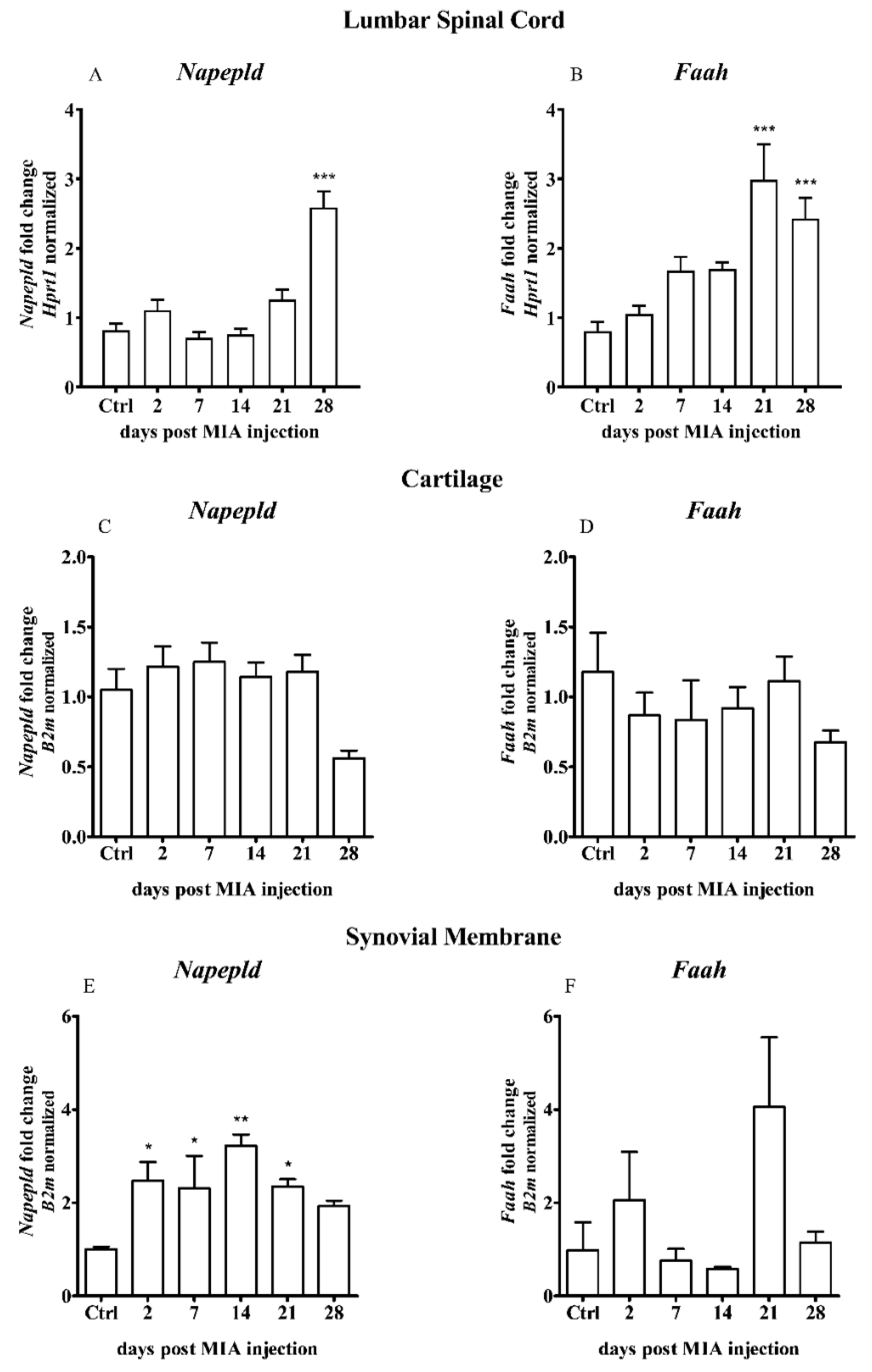
2.2. Expression of the Main Enzymes of AEA Synthesis and Degradation in the Dorsal Lumbar Spinal Cord and Joint Tissue of MIA-Treated Rats
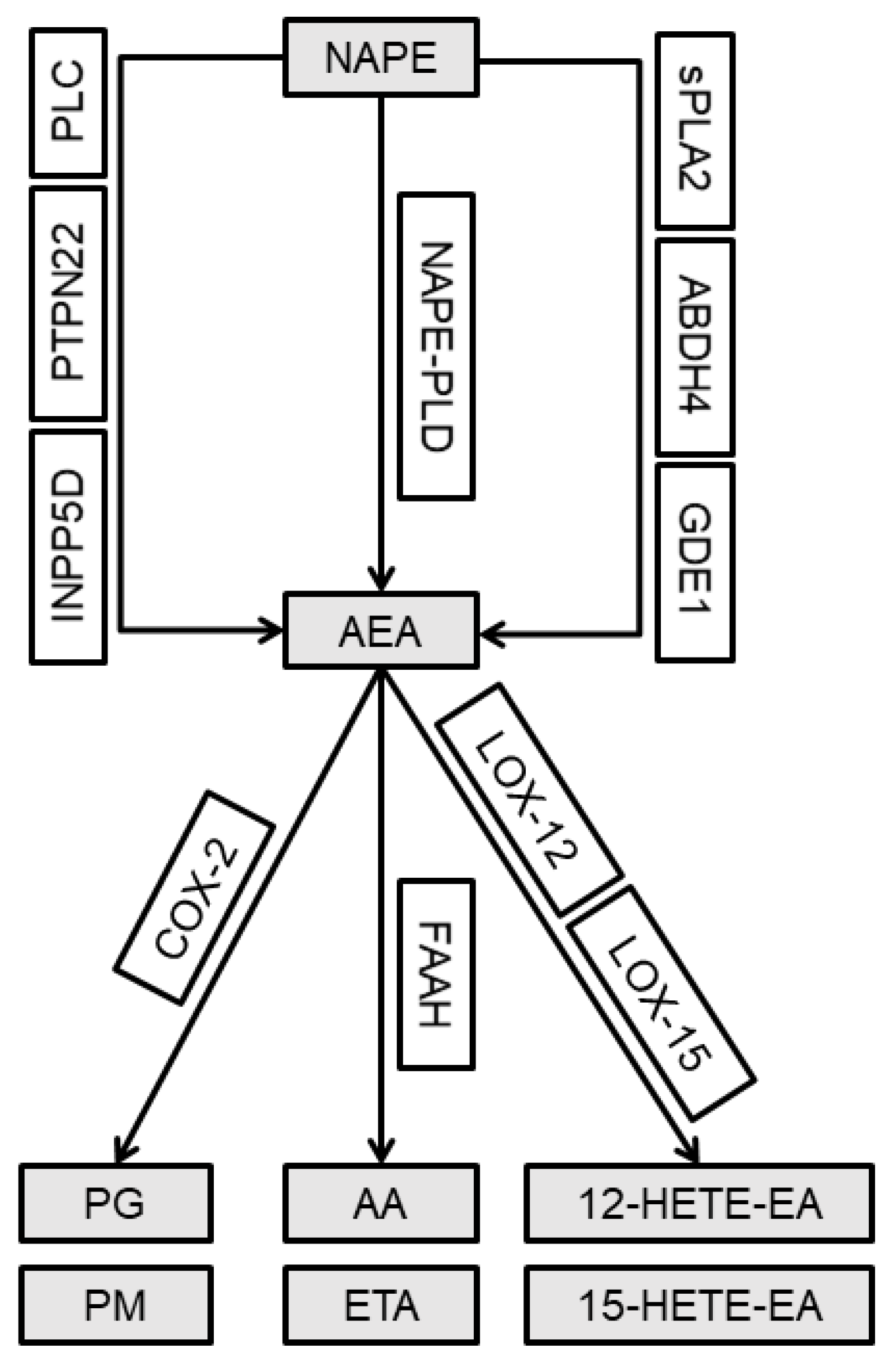
2.3. Alterations in the Gene Expression of the Alternative AEA Synthesis and Degradation Pathways in the Lumbar Spinal Cord and Joint Tissue of Rats after MIA Injection
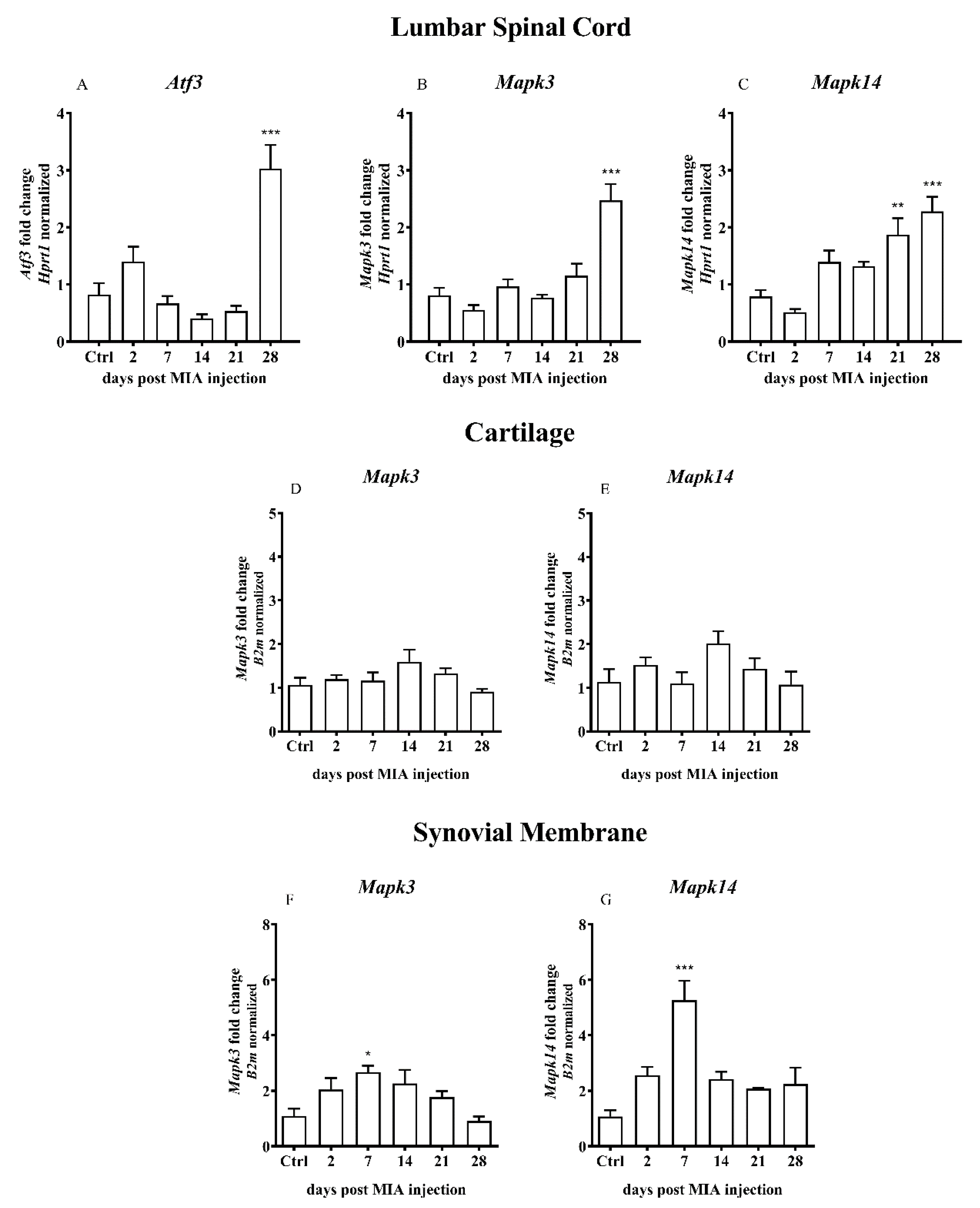
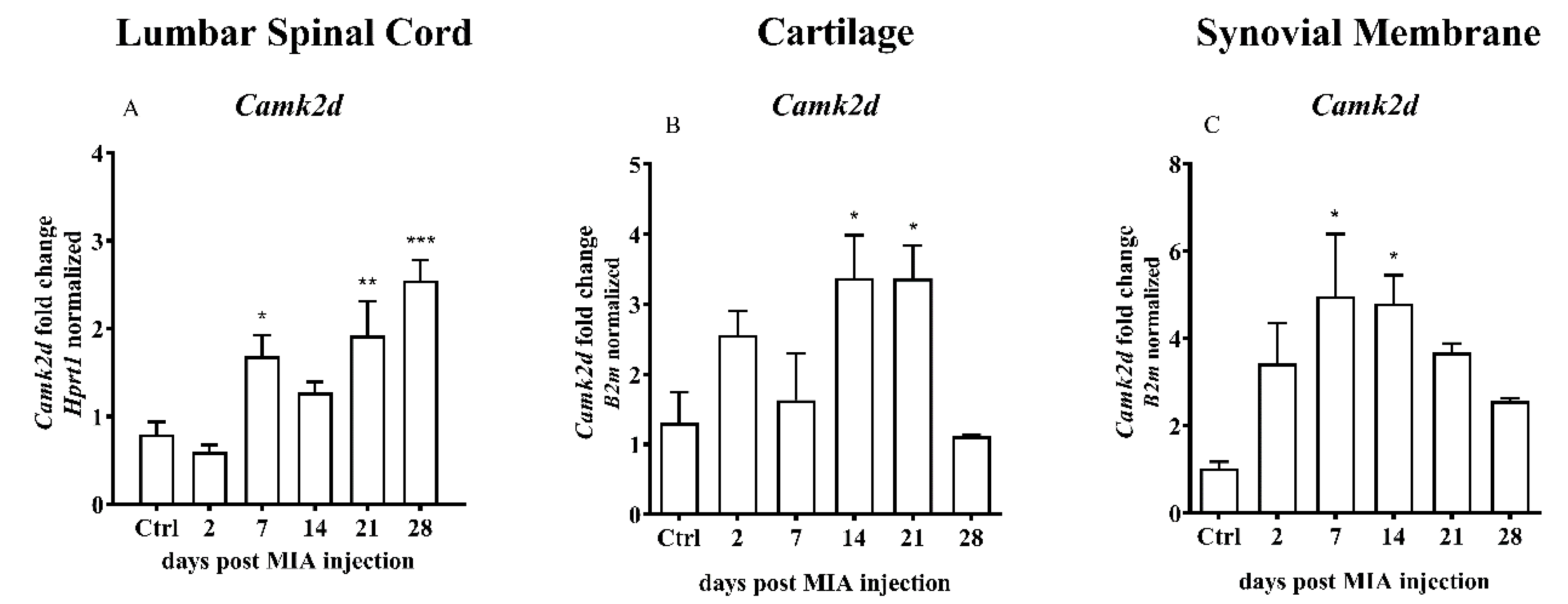
2.4. Overexpression of Calcium/Calmodulin-dependent Protein Kinase II Delta (CaMKII) as a Result of MIA-Induced OA-like Changes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of Osteoarthritis
4.3. RNA Extraction, cDNA Synthesis and Quantitative Real-Time Polymerase Chain Reaction
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 12-HETE-EA | 12-Hydroxyeicosatetraenoyl-ethanolamide |
| 15-HETE-EA | 15-Hydroxyeicosatetraenoyl-ethanolamide |
| AA | Arachidonic acid |
| ABHD4 | α/β Hydrolase domain-containing protein 4 |
| AEA | Anandamide |
| ALOX-12 | Arachidonate 12-lipoxygenase |
| ALOX-15 | Arachidonate 15-lipoxygenase |
| CaMK2 | Ca2+/Calmodulin-dependent protein kinase |
| CB1 | Cannabinoid receptor type 1 |
| CB2 | Cannabinoid receptor type 2 |
| COX-2 | Cyclooxygenase 2 |
| ECS | Endocannabinoid system |
| ETA | Ethanolamine |
| FAAH | Fatty acid amide hydrolase |
| INPP5D | Phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1 |
| MAPK3 | Mitogen activated protein kinase 3 (ERK1) |
| MAPK14 | Mitogen-activated protein kinase (p38) |
| NAPE-PLD | N-Arachidonoyl phosphatidylethanolamine phospholipase D |
| OA | Osteoarthritis |
| PG | Prostaglandins |
| PLC | Phospholipase C |
| PM | Prostamides |
| PRKCA (PKA) | Protein kinase A |
| PRKCG (PKC) | Protein kinase C (isoform gamma) |
| sPLA2 | Secreted phospholipase A2 |
| PTPN22 | Protein tyrosine phosphatase nonreceptor type 22 |
| TRPV1 | Transient receptor potential vanilloid 1 |
References
- Dieppe, P.; Lohmander, L.S.; Lohmander, L.S. Pathogenesis and management of pain in osteoarthritis. Lancet 2005, 365, 965–973. [Google Scholar] [CrossRef]
- Berenbaum, F. Osteoarthritis year 2010 in review: Pharmacological therapies. Osteoarthr. Cart. 2011, 19, 361–365. [Google Scholar] [CrossRef]
- Bjordal, J.M.; Klovning, A.; Ljunggren, A.E.; Slørdal, L. Short-term efficacy of pharmacotherapeutic interventions in osteoarthritic knee pain: A meta-analysis of randomised placebo-controlled trials. Eur. J. Pain 2007, 11, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. FAAH and MAGL inhibitors: Therapeutic opportunities from regulating endocannabinoid levels. Curr. Opin. Investig. Drugs 2010, 11, 51–62. [Google Scholar] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Palazzo, E.; De Novellis, V.; Bisogno, T.; Rossi, F.; Maione, S.; Di Marzo, V. Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology 2007, 52, 415–422. [Google Scholar] [CrossRef]
- Starowicz, K.; Makuch, W.; Korostynski, M.; Malek, N.; Slezak, M.; Zychowska, M.; Petrosino, S.; De Petrocellis, L.; Cristino, L.; Przewlocka, B.; et al. Full Inhibition of Spinal FAAH Leads to TRPV1-Mediated Analgesic Effects in Neuropathic Rats and Possible Lipoxygenase-Mediated Remodeling of Anandamide Metabolism. PLoS ONE 2013, 8, e60040. [Google Scholar] [CrossRef]
- Valastro, C.; Campanile, D.; Marinaro, M.; Franchini, D.; Piscitelli, F.; Verde, R.; Di Marzo, V.; Di Bello, A. Characterization of endocannabinoids and related acylethanolamides in the synovial fluid of dogs with osteoarthritis: A pilot study. BMC Vet. Res. 2017, 13, 309. [Google Scholar] [CrossRef]
- Richardson, D.; Pearson, R.G.; Kurian, N.; Latif, M.L.; Garle, M.J.; A Barrett, D.; Kendall, D.A.; Scammell, B.E.; Reeve, A.J.; Chapman, V. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R43. [Google Scholar] [CrossRef]
- Muccioli, G.G. Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov. Today 2010, 15, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.-Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [PubMed]
- Liu, J.; Wang, L.; Harvey-White, J.; Huang, B.X.; Kim, H.-Y.; Luquet, S.; Palmiter, R.D.; Krystal, G.; Rai, R.; Mahadevan, A.; et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 2008, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-X.; Tsuboi, K.; Okamoto, Y.; Tonai, T.; Murakami, M.; Kudo, I.; Ueda, N. Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochem. J. 2004, 380, 749–756. [Google Scholar] [CrossRef]
- Maccarrone, M.; Attinà, M.; Bari, M.; Cartoni, A.; Ledent, C.; Finazzi-Agrò, A. Anandamide degradation and N-acylethanolamines level in wild-type and CB1 cannabinoid receptor knockout mice of different ages. J. Neurochem. 2001, 78, 339–348. [Google Scholar] [PubMed]
- Smart, D.; Gunthorpe, M.J.; Jerman, J.C.; Nasir, S.; Gray, J.; Muir, A.I.; Chambers, J.K.; Randall, A.D.; Davis, J.B. The Endogenous Lipid Anandamide is a Full Agonist at the Human Vanilloid Receptor (HVR1). Br. J. Pharmacol. 2000, 129, 227–230. [Google Scholar]
- Mezey, E.; Tóth, Z.E.; Cortright, D.N.; Arzubi, M.K.; Krause, J.E.; Elde, R.; Guo, A.; Blumberg, P.M.; Szallasi, A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc. Natl. Acad. Sci. USA 2000, 97, 3655–3660. [Google Scholar]
- Kelly, S.; Chapman, R.J.; Woodhams, S.G.; Sagar, D.R.; Turner, J.; Burston, J.J.; Bullock, C.; Paton, K.; Huang, J.; Wong, A.; et al. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Ann. Rheum. Dis. 2013, 74, 252–259. [Google Scholar] [CrossRef]
- Braucke, A.F.G.V.; Frederiksen, N.L.; Berg, L.C.; Aarsvold, S.; Müller, F.C.; Boesen, M.; Lindegaard, C. Identification and Quantification of Transient Receptor Potential Vanilloid 1 (TRPV1) in Equine Articular Tissue. Animals 2020, 10, 506. [Google Scholar] [CrossRef]
- Hdud, I.M.; El-Shafei, A.A.; Loughna, P.T.; Barrett-Jolley, R.; Mobasheri, A. Expression of Transient Receptor Potential Vanilloid (TRPV) Channels in Different Passages of Articular Chondrocytes. Int. J. Mol. Sci. 2012, 13, 4433–4445. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; De Wilde, G.; A Doherty, S.; Lories, R.; Vaughn, F.L.; Laslett, L.L.; A Maciewicz, R.; Soni, A.; Hart, D.J.; Zhang, W.; et al. The Ile585Val TRPV1 variant is involved in risk of painful knee osteoarthritis. Ann. Rheum. Dis. 2011, 70, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.R.; Beyer, C.E.; Stahl, S.M. TRPV1 Antagonists and Chronic Pain: Beyond Thermal Perception. Pharmaceuticals 2012, 5, 114–132. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Oh, U. Activation and Activators of TRPV1 and Their Pharmaceutical Implication. Curr. Pharm. Des. 2005, 11, 2687–2698. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Shin, J.S.; Lee, S.Y.; Hwang, S.W.; Koo, J.; Cho, H.; Oh, U. Phosphorylation of Vanilloid Receptor 1 by Ca 2+/Calmodulin-Dependent Kinase II Regulates Its Vanilloid Binding. J. Biol. Chem. 2004, 279, 7048–7054. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Mrugala, M.; Makuch, W.; Kolosowska, N.; Przewlocka, B.; Binkowski, M.; Czaja, M.; Morera, E.; Di Marzo, V.; Starowicz, K. A multi-target approach for pain treatment. Pain 2015, 156, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-H.; Lin, J.-P.; Huang, L.-E.; Yang, Y.; Chen, C.-Q.; Li, N.-N.; Su, M.-Y.; Zhao, X.; Zhu, S.-M.; Yao, Y.-X. Silencing of spinal Trpv1 attenuates neuropathic pain in rats by inhibiting CAMKII expression and ERK2 phosphorylation. Sci. Rep. 2019, 9, 2769. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Samad, T.A.; Jin, S.-X.; Schmoll, R.; Woolf, C.J. p38 MAPK Activation by NGF in Primary Sensory Neurons after Inflammation Increases TRPV1 Levels and Maintains Heat Hyperalgesia. Neuron 2002, 36, 57–68. [Google Scholar] [CrossRef]
- Palazzo, E.; Luongo, L.; De Novellis, V.; Rossi, F.; Marabese, I.; Maione, S. Transient receptor potential vanilloid type 1 and pain development. Curr. Opin. Pharm. 2012, 12, 9–17. [Google Scholar] [CrossRef]
- M’Dahoma, S.; Bourgoin, S.; Kayser, V.; Barthélémy, S.; Chevarin, C.; Chali, F.; Orsal, D.; Hamon, M. Spinal Cord Transection-Induced Allodynia in Rats—Behavioral, Physiopathological and Pharmacological Characterization. PLoS ONE 2014, 9, e102027. [Google Scholar]
- Ashwell, M.; Freire, M.; O’Nan, A.; Benito, J.; Hash, J.; McCulloch, R.; Lascelles, B.D.X.; Bdx, B.L. Characterization of gene expression in naturally occurring feline degenerative joint disease-associated pain. Vet. J. 2019, 243, 42–47. [Google Scholar] [CrossRef]
- Yasui, M.; Menjyo, Y.; Tokizane, K.; Shiozawa, A.; Tsuda, M.; Inoue, K.; Ekiyama, H. Hyperactivation of proprioceptors induces microglia-mediated long-lasting pain in a rat model of chronic fatigue syndrome. J. Neuroinflammation 2019, 16, 67. [Google Scholar] [CrossRef]
- Jin, S.-X.; Zhuang, Z.-Y.; Woolf, C.J.; Ji, R.-R. p38 Mitogen-Activated Protein Kinase Is Activated after a Spinal Nerve Ligation in Spinal Cord Microglia and Dorsal Root Ganglion Neurons and Contributes to the Generation of Neuropathic Pain. J. Neurosci. 2003, 23, 4017–4022. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.-Y.; Xu, H.; Clapham, D.E.; Ji, R.-R. Phosphatidylinositol 3-Kinase Activates ERK in Primary Sensory Neurons and Mediates Inflammatory Heat Hyperalgesia through TRPV1 Sensitization. J. Neurosci. 2004, 24, 8300–8309. [Google Scholar] [CrossRef]
- Haudenschild, D.; Carlson, A.K.; Zignego, D.L.; Yik, J.; Hilmer, J.K.; June, R.K. Inhibition of early response genes prevents changes in global joint metabolomic profiles in mouse post-traumatic osteoarthritis. Osteoarthr. Cartil. 2019, 27, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Stensson, N.; Ghafouri, N.; Ernberg, M.; Mannerkorpi, K.; Kosek, E.; Gerdle, B.; Ghafouri, B. The Relationship of Endocannabinoidome Lipid Mediators with Pain and Psychological Stress in Women With Fibromyalgia: A Case-Control Study. J. Pain 2018, 19, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Azim, S.; Nicholson, J.; Rebecchi, M.J.; Galbavy, W.; Feng, T.; Reinsel, R.; Volkow, N.D.; Benveniste, H.; Kaczocha, M. Endocannabinoids and acute pain after total knee arthroplasty. Pain 2015, 156, 341–347. [Google Scholar] [CrossRef]
- Bishay, P.; Häussler, A.; Lim, H.-Y.; Oertel, B.; Galve-Roperh, I.; Ferreirós, N.; Tegeder, I. Anandamide deficiency and heightened neuropathic pain in aged mice. Neuropharmacology 2013, 71, 204–215. [Google Scholar] [CrossRef]
- Sagar, D.R.; Staniaszek, L.E.; Okine, B.N.; Woodhams, S.; Norris, L.M.; Pearson, R.G.; Garle, M.J.; Alexander, S.P.H.; Bennett, A.J.; Barrett, D.A.; et al. Tonic modulation of spinal hyperexcitability by the endocannabinoid receptor system in a rat model of osteoarthritis pain. Arthritis Rheum. 2010, 62, 3666–3676. [Google Scholar] [CrossRef]
- Schuelert, N.; McDougall, J.J. Cannabinoid-mediated antinociception is enhanced in rat osteoarthritic knees. Arthritis Rheum. 2007, 58, 145–153. [Google Scholar] [CrossRef]
- Schuelert, N.; Johnson, M.P.; Oskins, J.L.; Jassal, K.; Chambers, M.G.; McDougall, J.J. Local application of the endocannabinoid hydrolysis inhibitor URB597 reduces nociception in spontaneous and chemically induced models of osteoarthritis. Pain 2011, 152, 975–981. [Google Scholar] [CrossRef]
- Mlost, J.; Kostrzewa, M.; Malek, N.; Starowicz, K. Molecular Understanding of the Activation of CB1 and Blockade of TRPV1 Receptors: Implications for Novel Treatment Strategies in Osteoarthritis. Int. J. Mol. Sci. 2018, 19, 342. [Google Scholar] [CrossRef] [PubMed]
- Burston, J.J.; Mapp, P.I.; Sarmad, S.; Barrett, D.A.; Niphakis, M.J.; Cravatt, B.F.; A Walsh, D.; Chapman, V. Robust anti-nociceptive effects of monoacylglycerol lipase inhibition in a model of osteoarthritis pain. Br. J. Pharmacol. 2016, 173, 3134–3144. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Kucharczyk, M.; Starowicz, K. Alterations in the Anandamide Metabolism in the Development of Neuropathic Pain. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Mathiessen, A.; Conaghan, P. Synovitis in osteoarthritis: Current understanding with therapeutic implications. Arthritis Res. 2017, 19, 18. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997, 74, 129–180. [Google Scholar] [CrossRef]
- Galindo, T.; Reyna, J.; Weyer, A. Evidence for Transient Receptor Potential (TRP) Channel Contribution to Arthritis Pain and Pathogenesis. Pharm. 2018, 11, 105. [Google Scholar] [CrossRef]
- Malek, N.; Starowicz, K. Dual-Acting Compounds Targeting Endocannabinoid and Endovanilloid Systems—A Novel Treatment Option for Chronic Pain Management. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef]
- Turcotte, C.; Chouinard, F.; Lefebvre, J.S.; Flamand, N. Regulation of inflammation by cannabinoids, the endocannabinoids 2-arachidonoyl-glycerol and arachidonoyl-ethanolamide, and their metabolites. J. Leukoc. Biol. 2015, 97, 1049–1070. [Google Scholar] [CrossRef]
- Hammell, D.; Zhang, L.; Ma, F.; Abshire, S.; McIlwrath, S.; Stinchcomb, A.; Westlund, K. Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur. J. Pain 2015, 20, 936–948. [Google Scholar] [CrossRef]
- Philpott, H.T.; OʼBrien, M.; McDougall, J.J. Attenuation of early phase inflammation by cannabidiol prevents pain and nerve damage in rat osteoarthritis. Pain 2017, 158, 2442–2451. [Google Scholar] [CrossRef]
- Lowin, T.; Tingting, R.; Zurmahr, J.; Classen, T.; Schneider, M.; Pongratz, G. Cannabidiol (CBD): A killer for inflammatory rheumatoid arthritis synovial fibroblasts. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Fechtner, S.; Singh, A.K.; Srivastava, I.; Szlenk, C.T.; Muench, T.R.; Natesan, S.; Ahmed, S. Cannabinoid Receptor 2 Agonist JWH-015 Inhibits Interleukin-1β-Induced Inflammation in Rheumatoid Arthritis Synovial Fibroblasts and in Adjuvant Induced Arthritis Rat via Glucocorticoid Receptor. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Zhu, M.; Yu, B.; Bai, J.; Wang, X.; Guo, X.; Liu, Y.; Lin, J.; Hu, S.; Zhang, W.; Tao, Y.; et al. Cannabinoid Receptor 2 Agonist Prevents Local and Systemic Inflammatory Bone Destruction in Rheumatoid Arthritis. J. Bone Miner. Res. 2019, 34, 739–751. [Google Scholar] [CrossRef]
- Bai, J.; Ge, G.; Wang, Y.; Zhang, W.; Wang, Q.; Wang, W.; Guo, X.; Yu, B.; Xu, Y.; Yang, H.; et al. A selective CB2 agonist protects against the inflammatory response and joint destruction in collagen-induced arthritis mice. Biomed. Pharmacother. 2019, 116, 109025. [Google Scholar] [CrossRef] [PubMed]
- Pajak, A.; Kostrzewa, M.; Malek, N.; Korostynski, M.; Starowicz, K. Expression of matrix metalloproteinases and components of the endocannabinoid system in the knee joint are associated with biphasic pain progression in a rat model of osteoarthritis. J. Pain Res. 2017, 10, 1973–1989. [Google Scholar] [CrossRef] [PubMed][Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryk, M.; Chwastek, J.; Kostrzewa, M.; Mlost, J.; Pędracka, A.; Starowicz, K. Alterations in Anandamide Synthesis and Degradation during Osteoarthritis Progression in an Animal Model. Int. J. Mol. Sci. 2020, 21, 7381. https://doi.org/10.3390/ijms21197381
Bryk M, Chwastek J, Kostrzewa M, Mlost J, Pędracka A, Starowicz K. Alterations in Anandamide Synthesis and Degradation during Osteoarthritis Progression in an Animal Model. International Journal of Molecular Sciences. 2020; 21(19):7381. https://doi.org/10.3390/ijms21197381
Chicago/Turabian StyleBryk, Marta, Jakub Chwastek, Magdalena Kostrzewa, Jakub Mlost, Aleksandra Pędracka, and Katarzyna Starowicz. 2020. "Alterations in Anandamide Synthesis and Degradation during Osteoarthritis Progression in an Animal Model" International Journal of Molecular Sciences 21, no. 19: 7381. https://doi.org/10.3390/ijms21197381
APA StyleBryk, M., Chwastek, J., Kostrzewa, M., Mlost, J., Pędracka, A., & Starowicz, K. (2020). Alterations in Anandamide Synthesis and Degradation during Osteoarthritis Progression in an Animal Model. International Journal of Molecular Sciences, 21(19), 7381. https://doi.org/10.3390/ijms21197381