Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease

Abstract
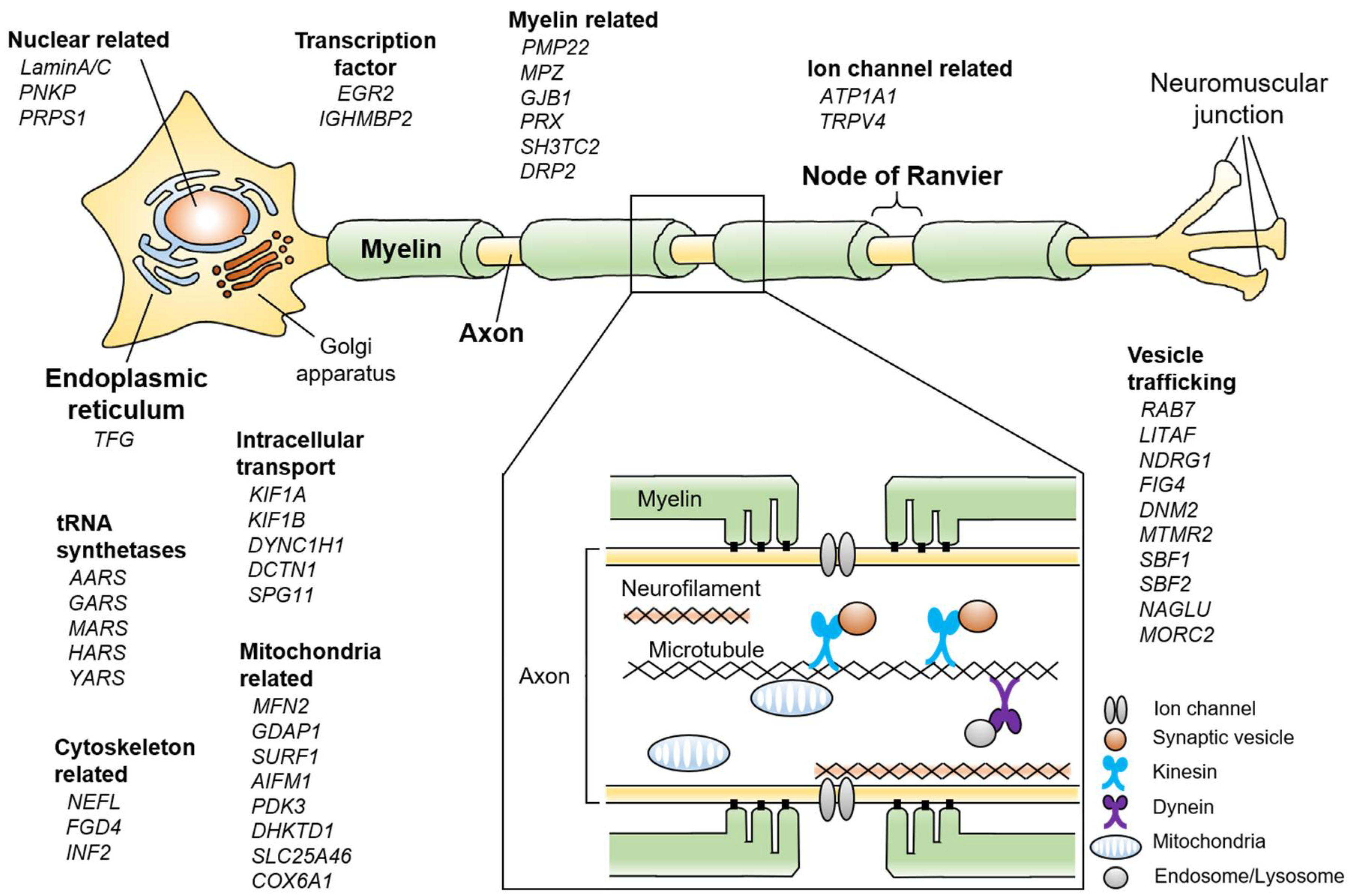
:1. Introduction
1.1. Clinical Features of CMT
1.2. Classification of CMT
1.3. Various CMT Models
2. Drosophila CMT Models for Investigating Aberrant Mitochondrial Dynamics
2.1. MFN2
2.2. GDAP1
2.3. SLC25A46
3. Drosophila CMT Models for Investigating Membrane Trafficking Defects
3.1. FIG4
3.2. RAB7
4. Drosophila CMT Models for Investigating Mutant Aminoacyl-tRNA Synthetases
5. Drosophila CMT Models for Investigating Impaired Axonal Transport
6. Drosophila CMT Model for Investigating Mutant Sorbitol Dehydrogenase
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CMT | Charcot-Marie-Tooth disease |
| HMSN | Hereditary motor and sensory neuropathy |
| ALS | Amyotrophic lateral sclerosis |
| NMJ AZ PSDs | Neuromuscular junction Active zone Postsynaptic densities |
References
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Saveri, P.; Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017, 30, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Barreto, L.C.; Oliveira, F.S.; Nunes, P.S.; de Franca Costa, I.M.; Garcez, C.A.; Goes, G.M.; Neves, E.L.; de Souza Siqueira Quintans, J.; de Souza Araujo, A.A. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology 2016, 46, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Lefter, S.; Hardiman, O.; Ryan, A.M. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology 2017, 88, 304–313. [Google Scholar] [CrossRef]
- Lousa, M.; Vazquez-Huarte-Mendicoa, C.; Gutierrez, A.J.; Saavedra, P.; Navarro, B.; Tugores, A. Genetic epidemiology, demographic, and clinical characteristics of Charcot-Marie-tooth disease in the island of Gran Canaria (Spain). J. Peripher. Nerv. Syst. 2019, 24, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Choi, Y.C.; Oh, J.W.; Yi, S.W. Prevalence, Mortality, and Cause of Death in Charcot-Marie-Tooth Disease in Korea: A Nationwide, Population-Based Study. Neuroepidemiology 2020, 54, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, M.; Mathis, S.; Richard, L.; Magdelaine, C.; Corcia, P.; Nouioua, S.; Tazir, M.; Magy, L.; Vallat, J.M. Nerve Biopsy Is Still Useful in Some Inherited Neuropathies. J. Neuropathol. Exp. Neurol. 2018, 77, 88–99. [Google Scholar] [CrossRef]
- Pareyson, D.; Marchesi, C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009, 8, 654–667. [Google Scholar] [CrossRef]
- Lee, M.; Park, C.H.; Chung, H.K.; Kim, H.J.; Choi, Y.; Yoo, J.H.; Yoon, Y.C.; Hong, Y.B.; Chung, K.W.; Choi, B.O.; et al. Cerebral white matter abnormalities in patients with charcot-marie-tooth disease. Ann. Neurol. 2017, 81, 147–151. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Lyu, H.; Jin, S.Q.; Zuo, Y.H.; Liu, J.; Wang, Z.X.; Zhang, W.; Yuan, Y. Clinical and Genetic Features of Chinese X-linked Charcot-Marie-Tooth Type 1 Disease. Chin. Med. J. (Engl) 2017, 130, 1049–1054. [Google Scholar] [CrossRef]
- Hu, G.; Zhang, L.; Zhang, M.; Yang, C.; Nie, X.; Xiang, F.; Chen, L.; Dong, Z.; Yu, S. Novel gap junction protein beta-1 gene mutation associated with a stroke-like syndrome and central nervous system involvement in patients with X-linked Charcot-Marie-Tooth Type 1: A case report and literature review. Clin. Neurol. Neurosurg. 2019, 180, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Saifi, G.M.; Szigeti, K.; Snipes, G.J.; Garcia, C.A.; Lupski, J.R. Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. J. Investig. Med. 2003, 51, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Yamamoto, M.; Yoshihara, T.; Koike, H.; Nakagawa, M.; Yoshikawa, H.; Ohnishi, A.; Hayasaka, K.; Onodera, O.; Baba, M.; et al. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): A clinicopathological study of 205 Japanese patients. Brain 2003, 126 Pt 1, 134–151. [Google Scholar] [CrossRef] [Green Version]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Carr, A.S.; Devine, H.; Chandrashekar, H.; Pelayo-Negro, A.L.; Pareyson, D.; Shy, M.E.; Scherer, S.S.; Reilly, M.M. Peripheral neuropathy in complex inherited diseases: An approach to diagnosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 846–863. [Google Scholar] [CrossRef]
- Murphy, S.M.; Laura, M.; Fawcett, K.; Pandraud, A.; Liu, Y.T.; Davidson, G.L.; Rossor, A.M.; Polke, J.M.; Castleman, V.; Manji, H.; et al. Charcot-Marie-Tooth disease: Frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry 2012, 83, 706–710. [Google Scholar] [CrossRef]
- Saporta, A.S.; Sottile, S.L.; Miller, L.J.; Feely, S.M.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef]
- Sivera, R.; Sevilla, T.; Vilchez, J.J.; Martinez-Rubio, D.; Chumillas, M.J.; Vazquez, J.F.; Muelas, N.; Bataller, L.; Millan, J.M.; Palau, F.; et al. Charcot-Marie-Tooth disease: Genetic and clinical spectrum in a Spanish clinical series. Neurology 2013, 81, 1617–1625. [Google Scholar] [CrossRef] [Green Version]
- Manganelli, F.; Tozza, S.; Pisciotta, C.; Bellone, E.; Iodice, R.; Nolano, M.; Geroldi, A.; Capponi, S.; Mandich, P.; Santoro, L. Charcot-Marie-Tooth disease: Frequency of genetic subtypes in a Southern Italy population. J. Peripher. Nerv. Syst. 2014, 19, 292–298. [Google Scholar] [CrossRef]
- Gess, B.; Schirmacher, A.; Boentert, M.; Young, P. Charcot-Marie-Tooth disease: Frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul. Disord. 2013, 23, 647–651. [Google Scholar] [CrossRef]
- Schroder, J.M. Neuropathology of Charcot-Marie-Tooth and related disorders. Neuromol. Med. 2006, 8, 23–42. [Google Scholar] [CrossRef]
- Patel, P.I.; Roa, B.B.; Welcher, A.A.; Schoener-Scott, R.; Trask, B.J.; Pentao, L.; Snipes, G.J.; Garcia, C.A.; Francke, U.; Shooter, E.M.; et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992, 1, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Baets, J.; Deconinck, T.; De Vriendt, E.; Zimon, M.; Yperzeele, L.; Van Hoorenbeeck, K.; Peeters, K.; Spiegel, R.; Parman, Y.; Ceulemans, B.; et al. Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain 2011, 134 Pt 9, 2664–2676. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V.; Said, G. Dejerine-Sottas disease and hereditary demyelinating polyneuropathy of infancy. Muscle Nerve 2002, 26, 608–621. [Google Scholar] [CrossRef]
- Tazir, M.; Bellatache, M.; Nouioua, S.; Vallat, J.M. Autosomal recessive Charcot-Marie-Tooth disease: From genes to phenotypes. J. Peripher. Nerv. Syst. 2013, 18, 113–129. [Google Scholar] [CrossRef]
- Nicot, A.S.; Laporte, J. Endosomal phosphoinositides and human diseases. Traffic 2008, 9, 1240–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccari, I.; Carbone, A.; Previtali, S.C.; Mironova, Y.A.; Alberizzi, V.; Noseda, R.; Rivellini, C.; Bianchi, F.; Del Carro, U.; D’Antonio, M.; et al. Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum. Mol. Genet. 2015, 24, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; McCollum, M.; Ravi, V.; Arpag, S.; Moiseev, D.; Castoro, R.; Mobley, B.; Burnette, B.; Siskind, C.; Day, J.; et al. Myelin abnormality in Charcot-Marie-Tooth type 4J recapitulates features of acquired demyelination. Ann. Neurol. 2018, 83, 756–770. [Google Scholar] [CrossRef]
- Shy, M.E.; Siskind, C.; Swan, E.R.; Krajewski, K.M.; Doherty, T.; Fuerst, D.R.; Ainsworth, P.J.; Lewis, R.A.; Scherer, S.S.; Hahn, A.F. CMT1X phenotypes represent loss of GJB1 gene function. Neurology 2007, 68, 849–855. [Google Scholar] [CrossRef]
- Ionasescu, V.V.; Trofatter, J.; Haines, J.L.; Summers, A.M.; Ionasescu, R.; Searby, C. X-linked recessive Charcot-Marie-Tooth neuropathy: Clinical and genetic study. Muscle Nerve 1992, 15, 368–373. [Google Scholar] [CrossRef]
- Kanhangad, M.; Cornett, K.; Brewer, M.H.; Nicholson, G.A.; Ryan, M.M.; Smith, R.L.; Subramanian, G.M.; Young, H.K.; Zuchner, S.; Kennerson, M.L.; et al. Unique clinical and neurophysiologic profile of a cohort of children with CMTX3. Neurology 2018, 90, e1706–e1710. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hong, S.H.; Ki, C.S.; Kim, B.J.; Shim, J.S.; Cho, S.H.; Park, J.H.; Kim, J.W. A novel locus for X-linked recessive CMT with deafness and optic neuropathy maps to Xq21.32-q24. Neurology 2005, 64, 1964–1967. [Google Scholar] [CrossRef] [PubMed]
- Kennerson, M.L.; Yiu, E.M.; Chuang, D.T.; Kidambi, A.; Tso, S.C.; Ly, C.; Chaudhry, R.; Drew, A.P.; Rance, G.; Delatycki, M.B.; et al. A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. Hum. Mol. Genet. 2013, 22, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Vondracek, P.; Seeman, P.; Hermanova, M.; Fajkusova, L. X-linked Charcot-Marie-Tooth disease: Phenotypic expression of a novel mutation Ile127Ser in the GJB1 (connexin 32) gene. Muscle Nerve 2005, 31, 252–255. [Google Scholar] [CrossRef]
- Bergoffen, J.; Scherer, S.S.; Wang, S.; Scott, M.O.; Bone, L.J.; Paul, D.L.; Chen, K.; Lensch, M.W.; Chance, P.F.; Fischbeck, K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 1993, 262, 2039–2042. [Google Scholar] [CrossRef]
- Paulson, H.L.; Garbern, J.Y.; Hoban, T.F.; Krajewski, K.M.; Lewis, R.A.; Fischbeck, K.H.; Grossman, R.I.; Lenkinski, R.; Kamholz, J.A.; Shy, M.E. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann. Neurol. 2002, 52, 429–434. [Google Scholar] [CrossRef]
- Hayasaka, K.; Himoro, M.; Sato, W.; Takada, G.; Uyemura, K.; Shimizu, N.; Bird, T.D.; Conneally, P.M.; Chance, P.F. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat. Genet. 1993, 5, 31–34. [Google Scholar] [CrossRef]
- Martini, R.; Zielasek, J.; Toyka, K.V.; Giese, K.P.; Schachner, M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat. Genet. 1995, 11, 281–286. [Google Scholar] [CrossRef]
- Huxley, C.; Passage, E.; Manson, A.; Putzu, G.; Figarella-Branger, D.; Pellissier, J.F.; Fontes, M. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum. Mol. Genet. 1996, 5, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Sereda, M.; Griffiths, I.; Puhlhofer, A.; Stewart, H.; Rossner, M.J.; Zimmerman, F.; Magyar, J.P.; Schneider, A.; Hund, E.; Meinck, H.M.; et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron 1996, 16, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Magyar, J.P.; Martini, R.; Ruelicke, T.; Aguzzi, A.; Adlkofer, K.; Dembic, Z.; Zielasek, J.; Toyka, K.V.; Suter, U. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J. Neurosci. 1996, 16, 5351–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzini, P.; Neuberg, D.H.; Schachner, M.; Nelles, E.; Willecke, K.; Zielasek, J.; Toyka, K.V.; Suter, U.; Martini, R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J. Neurosci. 1997, 17, 4545–4551. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, C.S.; Sherman, D.L.; Fleetwood-Walker, S.M.; Cottrell, D.F.; Tait, S.; Garry, E.M.; Wallace, V.C.; Ure, J.; Griffiths, I.R.; Smith, A.; et al. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 2000, 26, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.M.; Perea, J.; McGuigan, A.; King, R.H.; Muddle, J.R.; Gabreels-Festen, A.A.; Thomas, P.K.; Huxley, C. Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J. Anat. 2002, 200, 377–390. [Google Scholar] [CrossRef]
- Grandis, M.; Leandri, M.; Vigo, T.; Cilli, M.; Sereda, M.W.; Gherardi, G.; Benedetti, L.; Mancardi, G.; Abbruzzese, M.; Nave, K.A.; et al. Early abnormalities in sciatic nerve function and structure in a rat model of Charcot-Marie-Tooth type 1A disease. Exp. Neurol. 2004, 190, 213–223. [Google Scholar] [CrossRef]
- Dequen, F.; Filali, M.; Lariviere, R.C.; Perrot, R.; Hisanaga, S.; Julien, J.P. Reversal of neuropathy phenotypes in conditional mouse model of Charcot-Marie-Tooth disease type 2E. Hum. Mol. Genet. 2010, 19, 2616–2629. [Google Scholar] [CrossRef] [Green Version]
- Fledrich, R.; Schlotter-Weigel, B.; Schnizer, T.J.; Wichert, S.P.; Stassart, R.M.; Meyer zu Horste, G.; Klink, A.; Weiss, B.G.; Haag, U.; Walter, M.C.; et al. A rat model of Charcot-Marie-Tooth disease 1A recapitulates disease variability and supplies biomarkers of axonal loss in patients. Brain 2012, 135 Pt 1, 72–87. [Google Scholar] [CrossRef]
- d’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef]
- Chapman, A.L.; Bennett, E.J.; Ramesh, T.M.; De Vos, K.J.; Grierson, A.J. Axonal Transport Defects in a Mitofusin 2 Loss of Function Model of Charcot-Marie-Tooth Disease in Zebrafish. PLoS ONE 2013, 8, e67276. [Google Scholar] [CrossRef] [Green Version]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Storkebaum, E.; Leitao-Goncalves, R.; Godenschwege, T.; Nangle, L.; Mejia, M.; Bosmans, I.; Ooms, T.; Jacobs, A.; Van Dijck, P.; Yang, X.L.; et al. Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proc. Nat. Acad. Sci. USA 2009, 106, 11782–11787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschenbacher, W.H.; Song, M.; Chen, Y.; Bhandari, P.; Zhao, P.; Jowdy, C.C.; Engelhard, J.T.; Dorn, G.W., 2nd. Two rare human mitofusin 2 mutations alter mitochondrial dynamics and induce retinal and cardiac pathology in Drosophila. PLoS ONE 2012, 7, e44296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.S.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, R.; Imamura, K.; Morii, F.; Egawa, N.; Tsukita, K.; Enami, T.; Shibukawa, R.; Mizuno, T.; Nakagawa, M.; Inoue, H. Modeling Drug-Induced Neuropathy Using Human iPSCs for Predictive Toxicology. Clin. Pharmacol. Ther. 2017, 101, 754–762. [Google Scholar] [CrossRef]
- Kitani-Morii, F.; Imamura, K.; Kondo, T.; Ohara, R.; Enami, T.; Shibukawa, R.; Yamamoto, T.; Sekiguchi, K.; Toguchida, J.; Mizuno, T.; et al. Analysis of neural crest cells from Charcot-Marie-Tooth disease patients demonstrates disease-relevant molecular signature. Neuroreport 2017, 28, 814–821. [Google Scholar] [CrossRef]
- Juneja, M.; Burns, J.; Saporta, M.A.; Timmerman, V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 2019, 90, 58–67. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Kaminska, J.; Kabzinska, D.; Binieda, K.; Kochanski, A. A Yeast-Based Model for Hereditary Motor and Sensory Neuropathies: A Simple System for Complex, Heterogeneous Diseases. Int. J. Mol. Sci. 2020, 21, 4277. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Grunwald, D.J.; Eisen, J.S. Headwaters of the zebrafish—Emergence of a new model vertebrate. Nat. Rev. Genet. 2002, 3, 717–724. [Google Scholar] [CrossRef]
- Venken, K.J.; Bellen, H.J. Emerging technologies for gene manipulation in Drosophila melanogaster. Nat. Rev. Genet. 2005, 6, 167–178. [Google Scholar] [CrossRef]
- Nichols, C.D.; Becnel, J.; Pandey, U.B. Methods to assay Drosophila behavior. J. Vis. Exp 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bourg, E.; Lints, F.A. Hypergravity and aging in Drosophila melanogaster. 4. Climbing activity. Gerontology 1992, 38, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Min, V.A.; Condron, B.G. An assay of behavioral plasticity in Drosophila larvae. J. Neurosci. Methods 2005, 145, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef]
- Misko, A.L.; Sasaki, Y.; Tuck, E.; Milbrandt, J.; Baloh, R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012, 32, 4145–4155. [Google Scholar] [CrossRef] [Green Version]
- Debattisti, V.; Pendin, D.; Ziviani, E.; Daga, A.; Scorrano, L. Reduction of endoplasmic reticulum stress attenuates the defects caused by Drosophila mitofusin depletion. J. Cell Biol. 2014, 204, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, T.; Pendin, D.; Montagna, A.; Bova, S.; Ghelli, A.M.; Daga, A. Manipulation of Mitochondria Dynamics Reveals Separate Roles for Form and Function in Mitochondria Distribution. Cell Rep. 2018, 23, 1742–1753. [Google Scholar] [CrossRef] [Green Version]
- El Fissi, N.; Rojo, M.; Aouane, A.; Karatas, E.; Poliacikova, G.; David, C.; Royet, J.; Rival, T. Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT2A neuropathy. EMBO Rep. 2018, 19, e45241. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Celardo, I.; Costa, A.C.; Lehmann, S.; Loh, S.H.Y.; Martins, L.M. Enhancing folic acid metabolism suppresses defects associated with loss of Drosophila mitofusin. Cell Death Dis. 2019, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Bouhouche, A.; Birouk, N.; Azzedine, H.; Benomar, A.; Durosier, G.; Ente, D.; Muriel, M.P.; Ruberg, M.; Slassi, I.; Yahyaoui, M.; et al. Autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2): Phenotype-genotype correlations in 13 Moroccan families. Brain 2007, 130 Pt 4, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Zimon, M.; Baets, J.; Fabrizi, G.M.; Jaakkola, E.; Kabzinska, D.; Pilch, J.; Schindler, A.B.; Cornblath, D.R.; Fischbeck, K.H.; Auer-Grumbach, M.; et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 2011, 77, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Del Amo, V.; Seco-Cervera, M.; Garcia-Gimenez, J.L.; Whitworth, A.J.; Pallardo, F.V.; Galindo, M.I. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: Implications for the Charcot-Marie-Tooth neuropathy. Hum. Mol. Genet. 2015, 24, 21–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Del Amo, V.; Palomino-Schatzlein, M.; Seco-Cervera, M.; Garcia-Gimenez, J.L.; Pallardo, F.V.; Pineda-Lucena, A.; Galindo, M.I. A Drosophila model of GDAP1 function reveals the involvement of insulin signalling in the mitochondria-dependent neuromuscular degeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 801–809. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Y.; Hufnagel, R.B.; Hu, Y.C.; Zhao, C.; Queme, L.F.; Khuchua, Z.; Driver, A.M.; Dong, F.; Lu, Q.R.; et al. Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice. Hum. Mol. Genet. 2017, 26, 3776–3791. [Google Scholar] [CrossRef]
- Suda, K.; Ueoka, I.; Azuma, Y.; Muraoka, Y.; Yoshida, H.; Yamaguchi, M. Novel Drosophila model for mitochondrial diseases by targeting of a solute carrier protein SLC25A46. Brain Res. 2018, 1689, 30–44. [Google Scholar] [CrossRef]
- Ali, M.S.; Suda, K.; Kowada, R.; Ueoka, I.; Yoshida, H.; Yamaguchi, M. Neuron-specific knockdown of solute carrier protein SLC25A46a induces locomotive defects, an abnormal neuron terminal morphology, learning disability, and shortened lifespan. IBRO Rep. 2020, 8, 65–75. [Google Scholar] [CrossRef]
- Suda, K.; Muraoka, Y.; Ortega-Yanez, A.; Yoshida, H.; Kizu, F.; Hochin, T.; Kimura, H.; Yamaguchi, M. Reduction of Rpd3 suppresses defects in locomotive ability and neuronal morphology induced by the knockdown of Drosophila SLC25A46 via an epigenetic pathway. Exp. Cell Res. 2019, 385, 111673. [Google Scholar] [CrossRef]
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.E.; Li, J.; Zhang, X.; et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, A.J.; Zhang, Y.; Weisman, L.S. Phosphatidylinositol 3,5-bisphosphate: Low abundance, high significance. Bioessays 2014, 36, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyotani, A.; Azuma, Y.; Yamamoto, I.; Yoshida, H.; Mizuta, I.; Mizuno, T.; Nakagawa, M.; Tokuda, T.; Yamaguchi, M. Knockdown of the Drosophila FIG4 induces deficient locomotive behavior, shortening of motor neuron, axonal targeting aberration, reduction of life span and defects in eye development. Exp. Neurol. 2016, 277, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, R.; Cunningham, K.M.; Zhang, K.; Lloyd, T.E. FIG4 regulates lysosome membrane homeostasis independent of phosphatase function. Hum. Mol. Genet. 2016, 25, 681–692. [Google Scholar] [CrossRef]
- Muraoka, Y.; Nakamura, A.; Tanaka, R.; Suda, K.; Azuma, Y.; Kushimura, Y.; Lo Piccolo, L.; Yoshida, H.; Mizuta, I.; Tokuda, T.; et al. Genetic screening of the genes interacting with Drosophila FIG4 identified a novel link between CMT-causing gene and long noncoding RNAs. Exp. Neurol. 2018, 310, 1–13. [Google Scholar] [CrossRef]
- Shimada, S.; Muraoka, Y.; Ibaraki, K.; Takano-Shimizu-Kouno, T.; Yoshida, H.; Yamaguchi, M. Identification of CR43467 encoding a long non-coding RNA as a novel genetic interactant with dFIG4, a CMT-causing gene. Exp. Cell Res. 2020, 386, 111711. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; De Jonghe, P.; Wagner, K.; Verhoeven, K.; Hartung, H.P.; Timmerman, V. Phenotype-genotype correlations in a CMT2B family with refined 3q13-q22 locus. Neurology 2000, 55, 1552–1557. [Google Scholar] [CrossRef]
- Langemeyer, L.; Frohlich, F.; Ungermann, C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef]
- Janssens, K.; Goethals, S.; Atkinson, D.; Ermanoska, B.; Fransen, E.; Jordanova, A.; Auer-Grumbach, M.; Asselbergh, B.; Timmerman, V. Human Rab7 mutation mimics features of Charcot-Marie-Tooth neuropathy type 2B in Drosophila. Neurobiol. Dis. 2014, 65, 211–219. [Google Scholar] [CrossRef]
- Cioni, J.M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.Q.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003, 72, 1293–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordanova, A.; Irobi, J.; Thomas, F.P.; Van Dijck, P.; Meerschaert, K.; Dewil, M.; Dierick, I.; Jacobs, A.; De Vriendt, E.; Guergueltcheva, V.; et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006, 38, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latour, P.; Thauvin-Robinet, C.; Baudelet-Mery, C.; Soichot, P.; Cusin, V.; Faivre, L.; Locatelli, M.C.; Mayencon, M.; Sarcey, A.; Broussolle, E.; et al. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2010, 86, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Vester, A.; Velez-Ruiz, G.; McLaughlin, H.M.; Program, N.C.S.; Lupski, J.R.; Talbot, K.; Vance, J.M.; Zuchner, S.; Roda, R.H.; Fischbeck, K.H.; et al. A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum. Mutat. 2013, 34, 191–199. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, H.M.; Sakaguchi, R.; Liu, C.; Igarashi, T.; Pehlivan, D.; Chu, K.; Iyer, R.; Cruz, P.; Cherukuri, P.F.; Hansen, N.F.; et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet. 2010, 87, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, M.; McLaughlin, H.; Houlden, H.; Guo, M.; Yo-Tsen, L.; Hadjivassilious, M.; Speziani, F.; Yang, X.L.; Antonellis, A.; Reilly, M.M.; et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1247–1249. [Google Scholar] [CrossRef] [Green Version]
- Ermanoska, B.; Motley, W.W.; Leitao-Goncalves, R.; Asselbergh, B.; Lee, L.H.; De Rijk, P.; Sleegers, K.; Ooms, T.; Godenschwege, T.A.; Timmerman, V.; et al. CMT-associated mutations in glycyl- and tyrosyl-tRNA synthetases exhibit similar pattern of toxicity and share common genetic modifiers in Drosophila. Neurobiol. Dis. 2014, 68, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Grice, S.J.; Liu, J.L.; Webber, C. Synergistic interactions between Drosophila orthologues of genes spanned by de novo human CNVs support multiple-hit models of autism. PLoS Genet. 2015, 11, e1004998. [Google Scholar] [CrossRef]
- Niehues, S.; Bussmann, J.; Steffes, G.; Erdmann, I.; Kohrer, C.; Sun, L.; Wagner, M.; Schafer, K.; Wang, G.; Koerdt, S.N.; et al. Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat. Commun. 2015, 6, 7520. [Google Scholar] [CrossRef] [Green Version]
- Bussmann, J.; Storkebaum, E. Molecular pathogenesis of peripheral neuropathies: Insights from Drosophila models. Curr. Opin. Genet. Dev. 2017, 44, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, S.; Wei, N.; Erfurth, M.L.; Yusein-Myashkova, S.; Ermanoska, B.; Mateiu, L.; Asselbergh, B.; Blocquel, D.; Kakad, P.; Penserga, T.; et al. Transcriptional dysregulation by a nucleus-localized aminoacyl-tRNA synthetase associated with Charcot-Marie-Tooth neuropathy. Nat. Commun. 2019, 10, 5045. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y. Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Exp. Cell Res. 2015, 334, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Takita, J.; Tanaka, Y.; Setou, M.; Nakagawa, T.; Takeda, S.; Yang, H.W.; Terada, S.; Nakata, T.; Takei, Y.; et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 2001, 105, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Riviere, J.B.; Ramalingam, S.; Lavastre, V.; Shekarabi, M.; Holbert, S.; Lafontaine, J.; Srour, M.; Merner, N.; Rochefort, D.; Hince, P.; et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet. 2011, 89, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Kern, J.V.; Zhang, Y.V.; Kramer, S.; Brenman, J.E.; Rasse, T.M. The kinesin-3, unc-104 regulates dendrite morphogenesis and synaptic development in Drosophila. Genetics 2013, 195, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.V.; Hannan, S.B.; Stapper, Z.A.; Kern, J.V.; Jahn, T.R.; Rasse, T.M. The Drosophila KIF1A Homolog unc-104 Is Important for Site-Specific Synapse Maturation. Front. Cell Neurosci. 2016, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Baumann, S.; Komissarov, A.; Gili, M.; Ruprecht, V.; Wieser, S.; Maurer, S.P. A reconstituted mammalian APC-kinesin complex selectively transports defined packages of axonal mRNAs. Sci. Adv. 2020, 6, eaaz1588. [Google Scholar] [CrossRef] [Green Version]
- El-Kabbani, O.; Darmanin, C.; Chung, R.P. Sorbitol dehydrogenase: Structure, function and ligand design. Curr. Med. Chem. 2004, 11, 465–476. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Serrano, A.M.; Duarte, J.M.N. Brain Metabolism Alterations in Type 2 Diabetes: What Did We Learn From Diet-Induced Diabetes Models? Front. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, A.; Zhu, Y.; Rebelo, A.P.; Negri, S.; Courel, S.; Abreu, L.; Bacon, C.J.; Bai, Y.; Bis-Brewer, D.M.; Bugiardini, E.; et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020, 52, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Luque, T.; Hjelmqvist, L.; Marfany, G.; Danielsson, O.; El-Ahmad, M.; Persson, B.; Jornvall, H.; Gonzalez-Duarte, R. Sorbitol dehydrogenase of Drosophila. Gene, protein, and expression data show a two-gene system. J. Biol. Chem. 1998, 273, 34293–34301. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| CMT Subtype | Genomic Locus | Gene Symbol | Biological Functions | Drosophila Homolog | Phenotype MIM |
|---|---|---|---|---|---|
| CMT1 (Demyelinating, autosomal dominant) | |||||
| CMT1A | 17p12 | PMP22 | Myelin protein | - | 118220 |
| CMT1B | 1q23 | MPZ | Myelin protein | - | 118200 |
| CMT1C | 16p13.3 | LITAF | Regulation of endosomal trafficking | CG13510, CG13559, CG32280 | 601098 |
| CMT1D | 10q21 | EGR2 | Transcription factor | sr | 607678 |
| CMT1E | 17p12 | PMP22 | Myelin protein | - | 118300 |
| CMT1F | 8p21 | NEFL | Neurofilament protein | - | 607684 |
| CMT1G | 8q21 | PMP2 | Myelin protein | - | 618279 |
| HNPP | 17p12 | PMP22 | Myelin protein | - | 162500 |
| CMT2 (Axonal, autosomal dominant) | |||||
| CMT2A1 | 1p36 | KIF1B | Intracellular transport | unc-104 | 118210 |
| CMT2A2 | 1p36 | MFN2 | Mitochondrial dynamics | Marf | 609260 |
| CMT2B | 3q21 | RAB7 | Regulation of vesicular transport | Rab7 | 600882 |
| CMT2C | 12q24 | TRPV4 | Regulation of calcium ion influx | nan, iav | 606071 |
| CMT2D | 7p14 | GARS | Protein translation | gars, GlyRS | 601472 |
| CMT2E | 8p21 | NEFL | Neurofilament protein | - | 607684 |
| CMT2F | 7q11 | HSPB1 | Microtubule regulator and chaperon activity | heat shock protein family B member | 606595 |
| CMT2I/J | 1q22 | MPZ | Myelin protein | - | 607736 |
| CMT2K | 8q21 | GDAP1 | Mitochondrial dynamics | dGdap1 | 607831 |
| CMT2L | 12q24 | HSPB8 | Microtubule regulator and chaperon activity | - | 608673 |
| CMT2M | 19q13 | DNM2 | Endocytosis and regulation of cell motility | shi | 606482 |
| CMT2N | 16q22 | AARS | Protein translation | - | 613287 |
| CMT2O | 14q32 | DYNC1H1 | Intracellular transport | dynein heavy chain 64C | 614228 |
| CMT2P | 9q33 | LRSAM1 | E3 ubiquitin ligase | - | 614436 |
| CMT2Q | 10p14 | DHKTD1 | Mitochondrial biogenesis | CG1544 | 615025 |
| CMT2U | 12q13 | MARS | Protein translation | mars, MetRS | 616280 |
| CMT2V | 17q21 | NAGLU | Lysosomal enzyme | CG13397 | 616491 |
| CMT2W | 5q31 | HARS | Protein translation | hars, HisRS | 616625 |
| CMT2Y | 9q13 | VCP | Regulation of autophagy | TER94 | 616687 |
| CMT2Z | 22q12 | MORC2 | Fatty acid metabolism | - | 616688 |
| CMT2DD | 1p13 | ATP1A1 | Ion channel at Ranvier nodes | - | 618036 |
| HMSN-P | 3q12 | TFG | ER vesicle trafficking | - | 604484 |
| CMT2 (Axonal, autosomal recessive) | |||||
| AR-CMT2A | 1q22 | LaminA/C | Nuclear membrane protein | Lam | 605588 |
| AR-CMT2B | 19q13 | PNKP | Regulation of phosphorylation of nucleic acids | CG9601 | 605589 |
| AR-CMT2F | 7q11 | HSPB1 | Microtubule regulator and chaperon activity | heat shock protein family B member | 606595 |
| AR-CMT2K | 8q21 | GDAP1 | Mitochondrial dynamics | dGdap1 | 607831 |
| AR-CMT2P | 9q33 | LRSAM1 | E3 ubiquitin ligase | - | 614436 |
| AR-CMT2R | 4q31 | TRIM2 | E3 ubiquitin ligase | - | 615490 |
| AR-CMT2S | 11q13 | IGHMBP2 | Transcription factor | CG30094 | 616155 |
| AR-CMT2T | 3q25 | MME | Neutral endopeptidase | Nep1, Nep2 | 617017 |
| AR-CMT2X | 15q21 | SPG11 | Membrane associated | CG13531 | 616668 |
| AR-CMT2A2B | 1p36 | MFN2 | Mitochondrial dynamics | Marf | 617087 |
| HMSN6B | 5q22 | SLC25A46 | Mitochondrial dynamics | Slc25A46b | 616505 |
| SCAN3 | 1p32.3 | COA7 | Mitochondrial biogenesis | Coa7 | 618387 |
| HSMN IIC | 2q37 | KIF1A | Intracellular transport | unc-104 | 614213 |
| CMT4 (Demyelinating, Autosomal recessive) | |||||
| CMT4A | 8q13-q21.1 | GDAP1 | Mitochondrial dynamics | dGdap1 | 214400 |
| CMT4B1 | 11q22 | MTMR2 | Regulation of phosphorylation of Phosphatidylinositol | mtm | 601382 |
| CMT4B2 | 11p15 | SBF2 | Signaling pathway | Sbf | 604563 |
| CMT4B3 | 22q13 | SBF1 | Signaling pathway | Sbf | 615284 |
| CMT4C | 5q23-q33 | SH3TC2 | Myelin maturation | - | 601596 |
| CMT4D | 8q24 | NDRG1 | Vesicle transport | MESK2 | 601455 |
| CMT4E | 10q21-q22 | EGR2 | Transcription factor | - | 605253 |
| CMT4F | 19q13 | PRX | Myelin maturation | - | 614895 |
| CMT4G | 10q22 | HK1 | Glucose metabolism | Hex-A | 605285 |
| CMT4H | 12p11.2 | FGD4 | Regulation of actin fibers | - | 609311 |
| CMT4J | 6p21 | FIG4 | Endo-lysosomal trafficking | dFig4 | 611228 |
| CMT4K | 9q34 | SURF1 | Mitochondrial biogenesis | Surf1 | 616684 |
| X-linked CMT | |||||
| Dominant | |||||
| CMTX1 | Xq13 | GJB1 | Gap junction formation | - | 302800 |
| CMTX3 | Xq27 | - | - | - | 302802 |
| Semi-dominant | |||||
| CMTX6 | Xp22 | PDK3 | Mitochondrial biogenesis | Pdk | 300905 |
| Recessive | |||||
| CMTX2 | Xp22.2 | - | - | - | 302801 |
| CMTX4 | Xq26 | AIFM1 | Mitochondrial biogenesis | AIF | 310490 |
| CMTX5 | Xq22 | PRPS1 | Nucleotide biosynthesis | Prps | 311070 |
| CMT (Intermediate NCV, autosomal dominant) | |||||
| CMT-DIA | 10q24 | - | - | - | 606483 |
| CMT-IB | 19p13 | DNM2 | Regulation of cellular proliferation | - | 606482 |
| CMT-DIC | 1p35 | YARS | Protein translation | yars, TyrRS | 608323 |
| CMT-DID | 1q22 | MPZ | Myelin protein | - | 607791 |
| CMT-DIE | 14q32 | INF2 | Regulation of actin fibers | form3 | 614455 |
| CMT-DIF | 3q26 | GNB4 | Signaling pathway | Gβ13F | 615185 |
| CMT-DIG | 8p21 | NEFL | Neurofilament protein | - | 617882 |
| CMT (Intermediate NCV, autosomal recessive) | |||||
| CMT RIA | 8q21.1 | GDAP1 | Mitochondrial dynamics | dGdap1 | 608340 |
| CMT RIB | 16q23 | KARS | Protein translation | kars, LysRS | 613641 |
| CMT RIC | 1p36 | PLEKHG5 | Signaling pathway | CG42674 | 615376 |
| CMT RID | 12q24 | COX6A1 | Mitochondrial biogenesis | levy, COX6AL, CG14077 | 616039 |
| CMTXI | Xq22 | DRP2 | Myelin maturation | - | 300052 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitani-Morii, F.; Noto, Y.-i. Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease. Int. J. Mol. Sci. 2020, 21, 7419. https://doi.org/10.3390/ijms21197419
Kitani-Morii F, Noto Y-i. Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease. International Journal of Molecular Sciences. 2020; 21(19):7419. https://doi.org/10.3390/ijms21197419
Chicago/Turabian StyleKitani-Morii, Fukiko, and Yu-ichi Noto. 2020. "Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease" International Journal of Molecular Sciences 21, no. 19: 7419. https://doi.org/10.3390/ijms21197419
APA StyleKitani-Morii, F., & Noto, Y.-i. (2020). Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease. International Journal of Molecular Sciences, 21(19), 7419. https://doi.org/10.3390/ijms21197419