Recent Advances in Lipopolysaccharide Recognition Systems



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
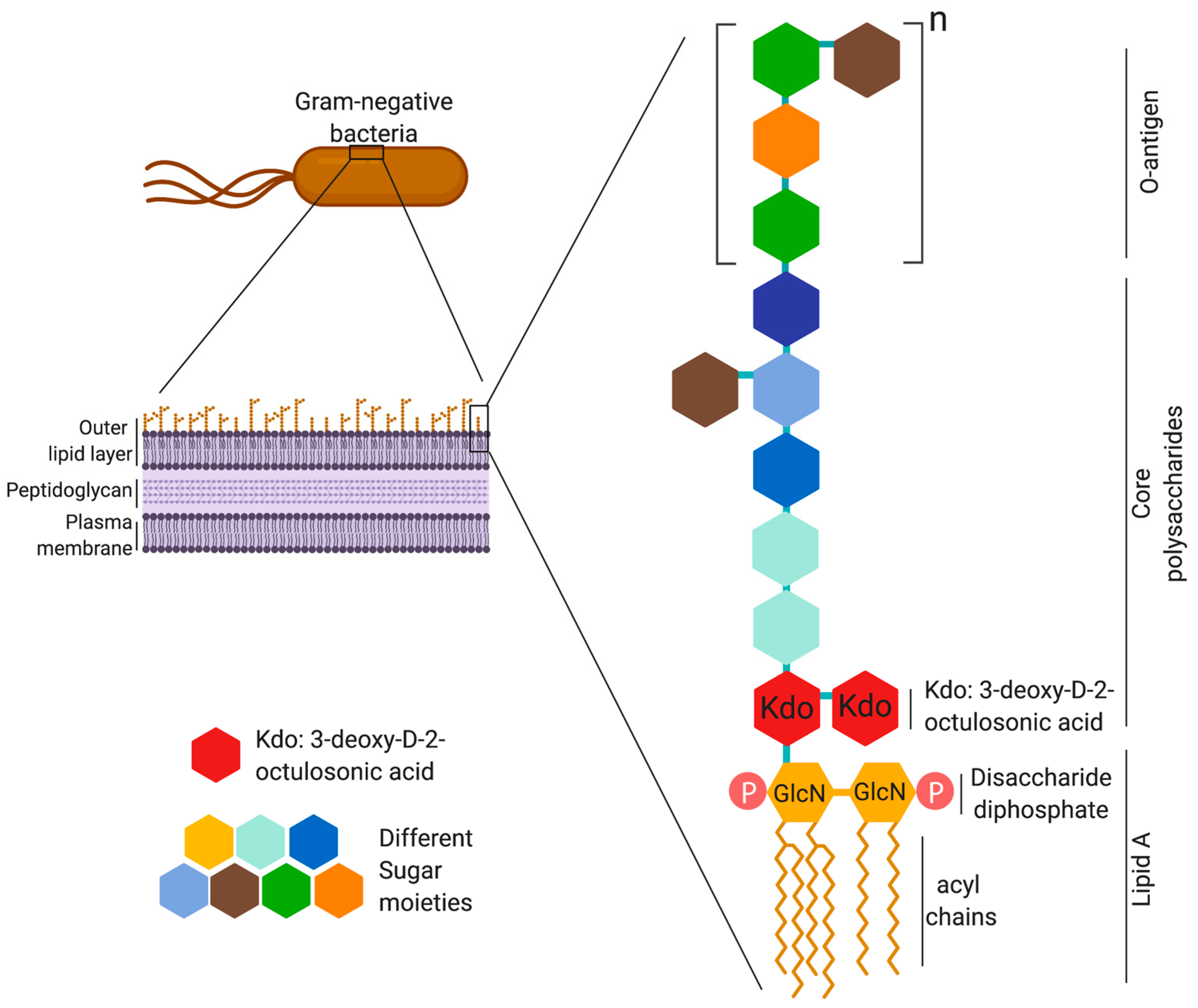
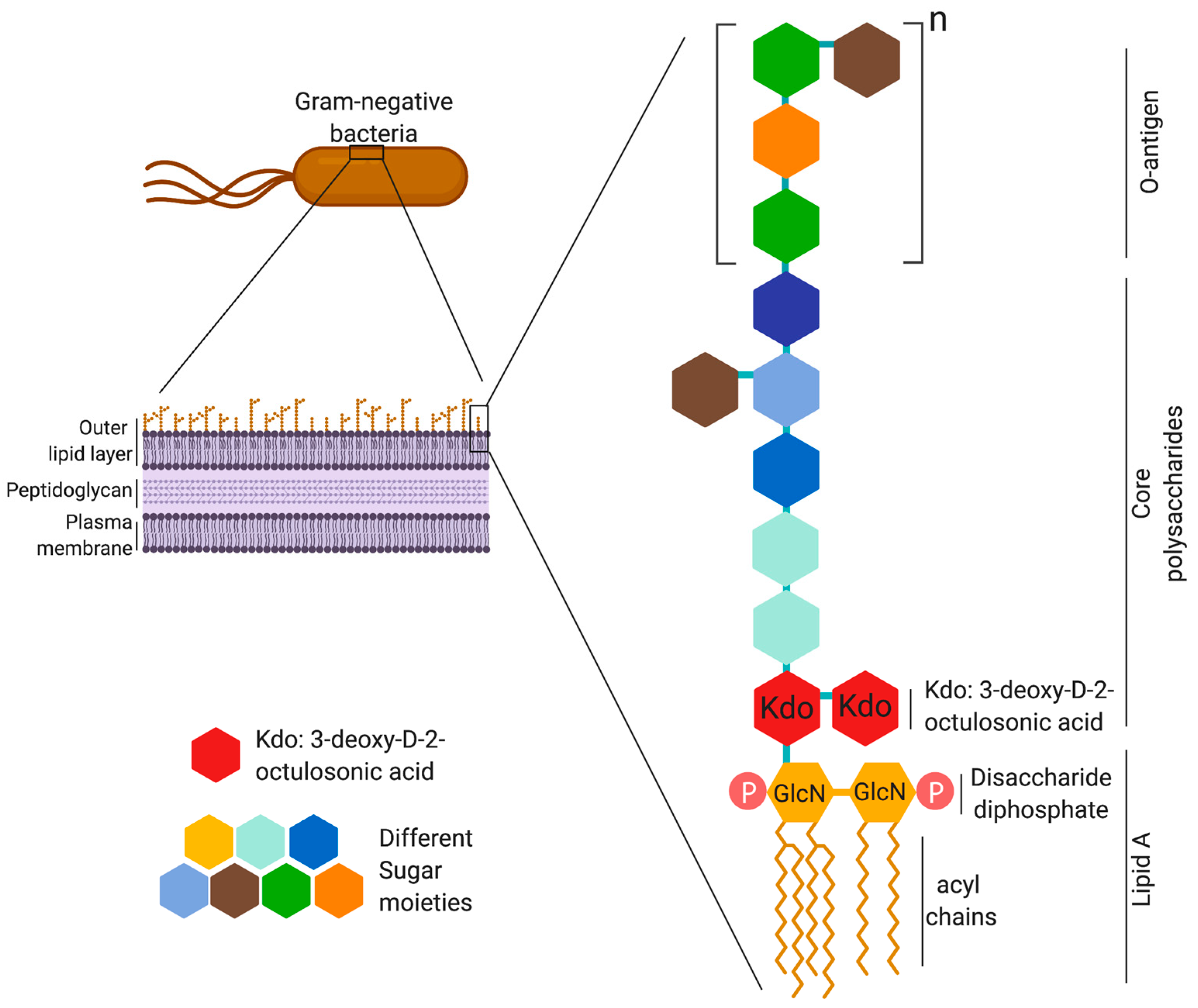
2. LPS Structure and Immunogenicity
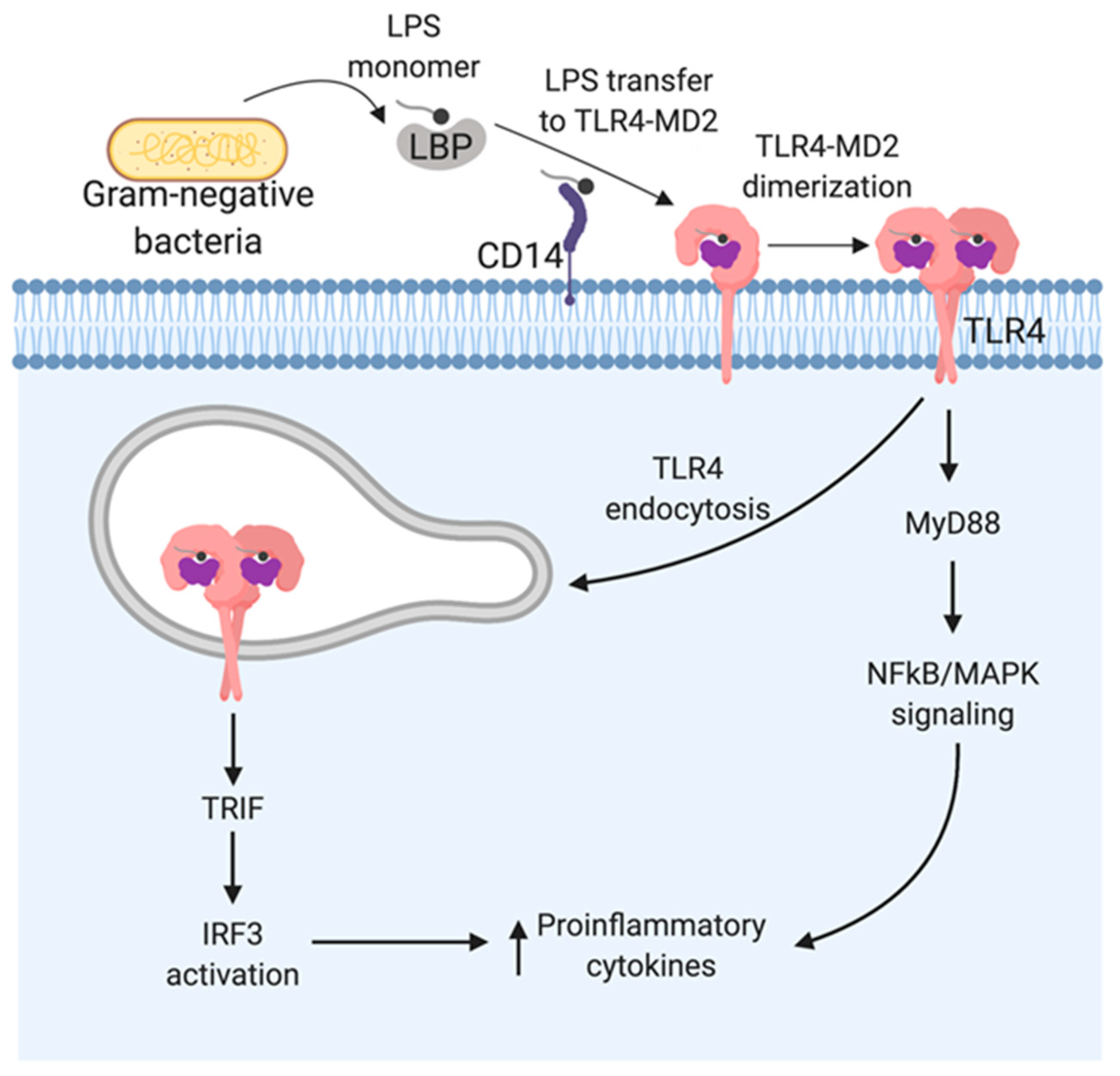
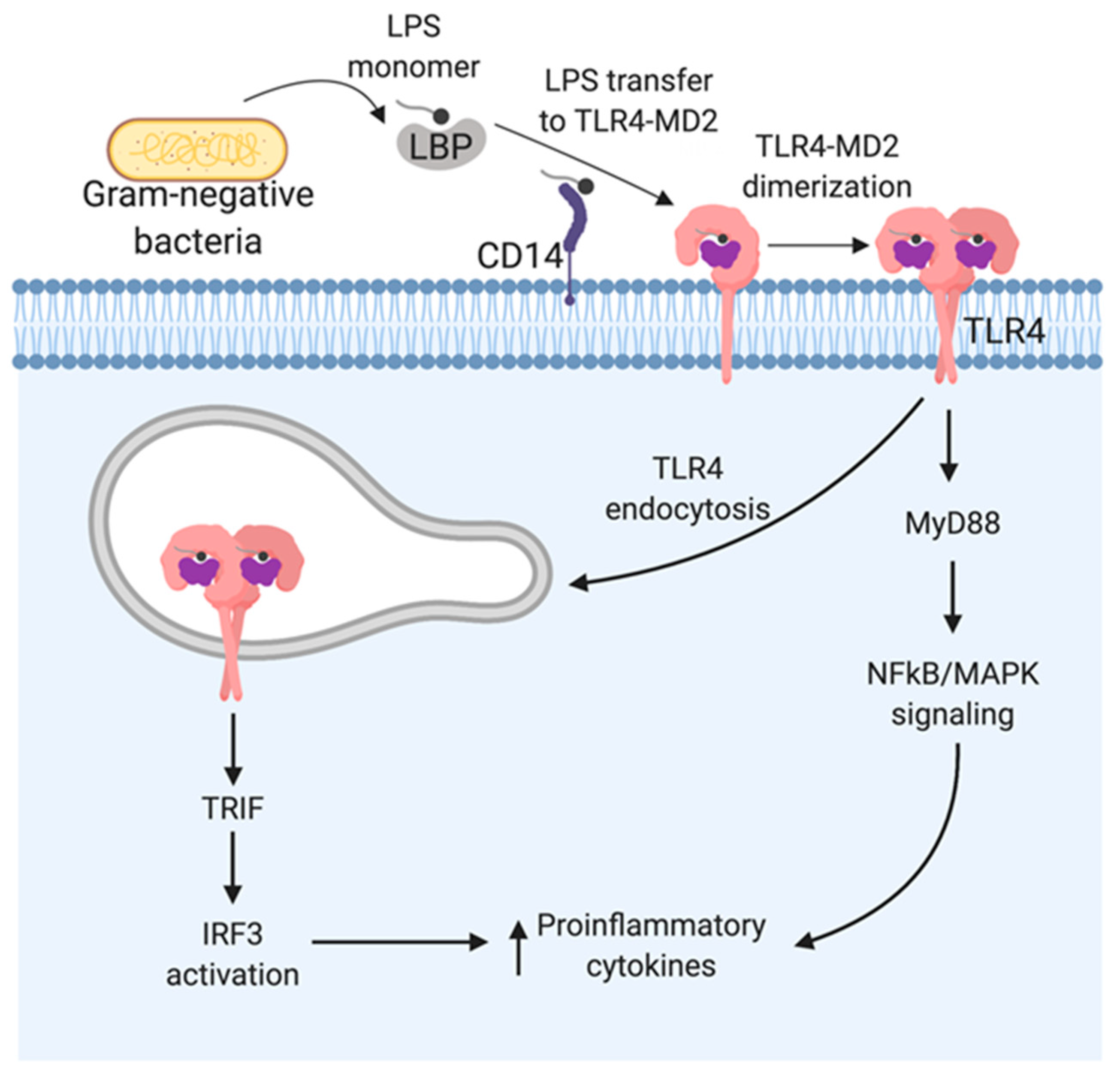
3. LPS Recognition by TLR4
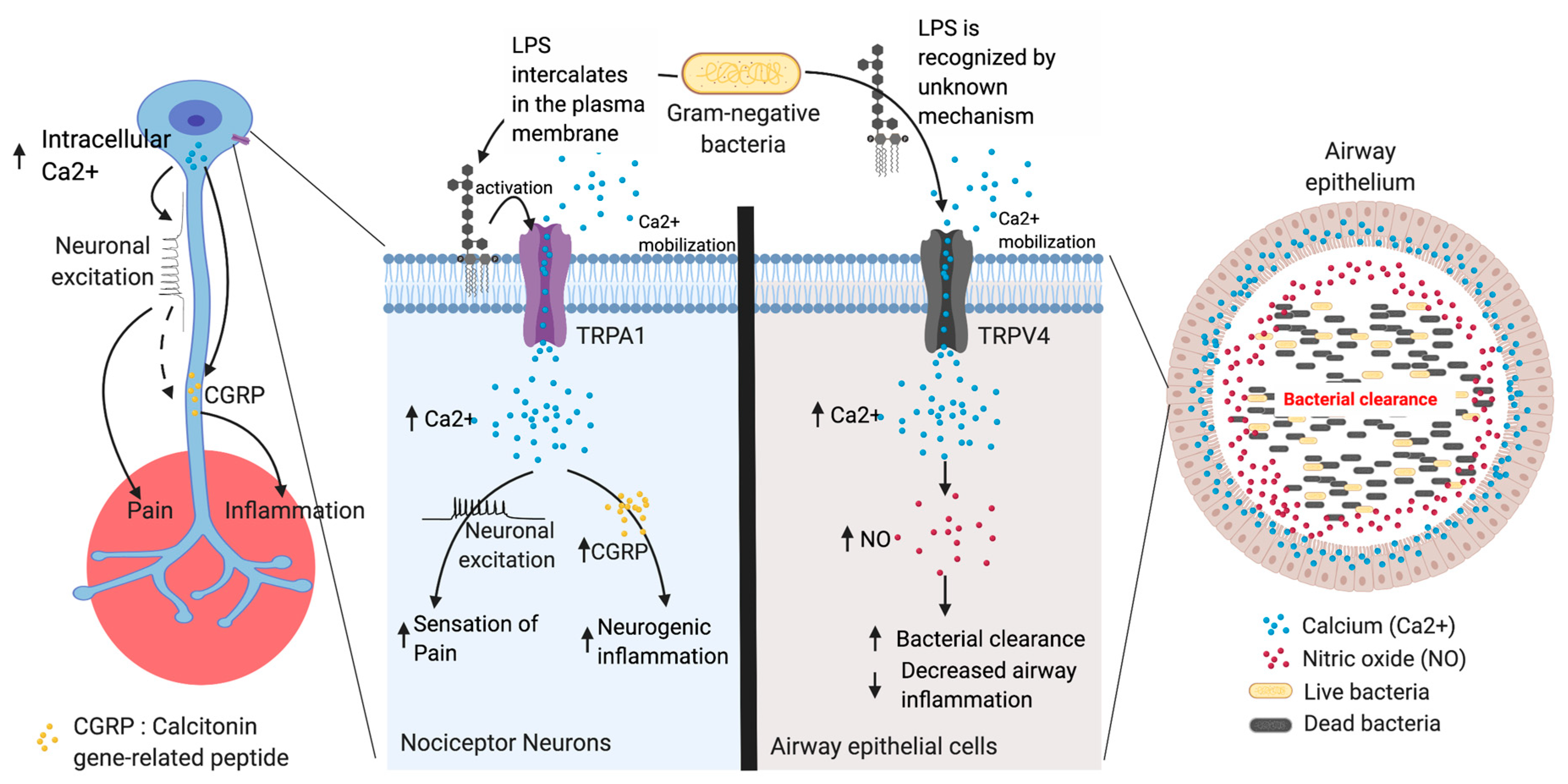
4. LPS Recognition by TRP Channels
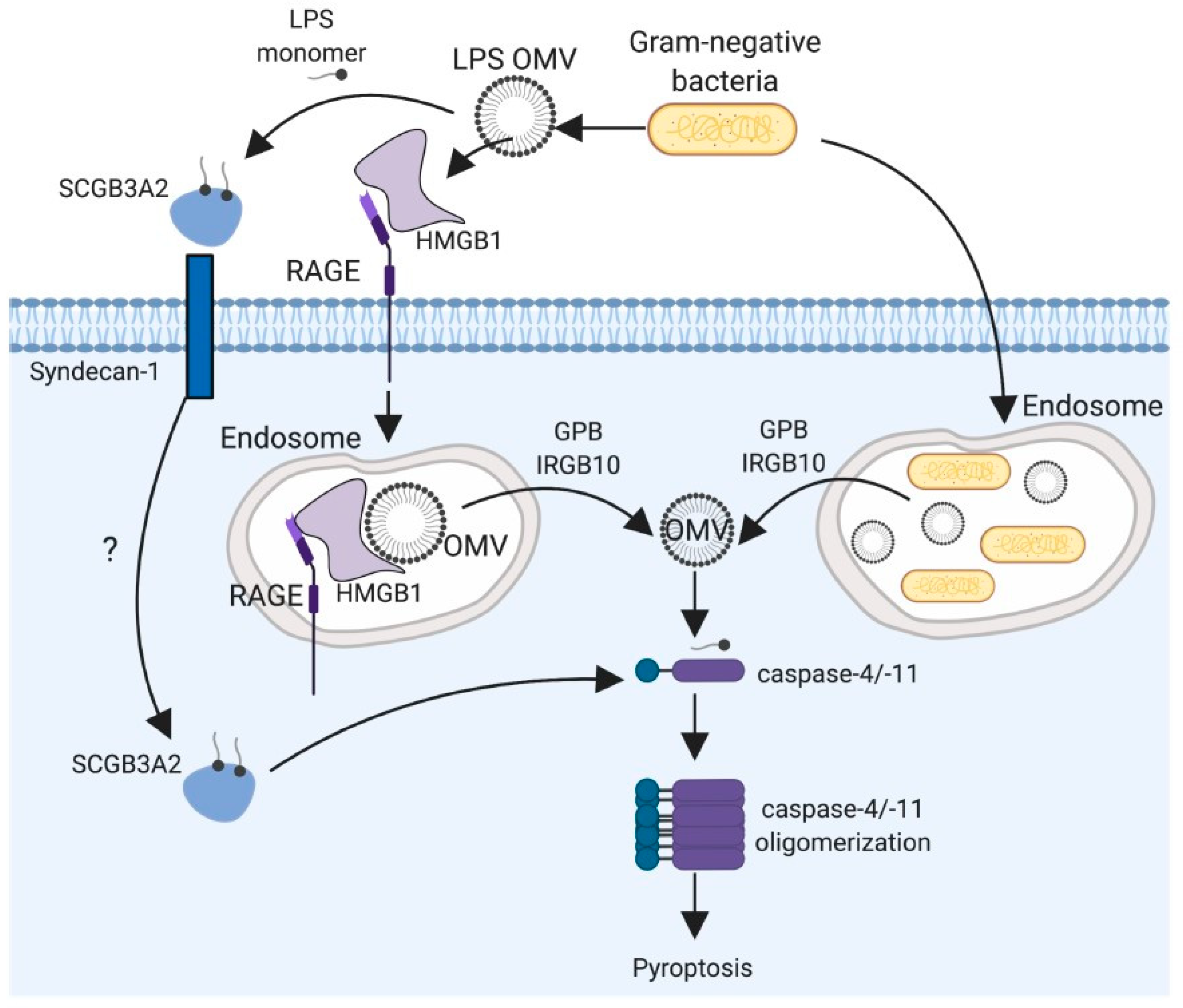
5. LPS Recognition by Caspase-11
6. Summary
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Heine, H.; Rietschel, E.T.; Ulmer, A.J. The Biology of Endotoxin. Mol. Biotechnol. 2001, 19, 279–296. [Google Scholar] [CrossRef]
- Uchiyama, T.; Jacobs, D.M. Modulation of immune response by bacterial lipopolysaccharide (LPS): Multifocal effects of LPS-induced suppression of the primary antibody response to a T-dependent antigen. J. Immunol. 1978, 121, 2340–2346. [Google Scholar]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A Modification Systems in Gram-Negative Bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [Green Version]
- Steimle, A.; Autenrieth, I.B.; Frick, J.S. Structure and function: Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, R.F.; Sá-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef]
- Imoto, M.; Yoshimura, H.; Shimamoto, T.; Sakaguchi, N.; Kusumoto, S.; Shiba, T. Total Synthesis of Escherichia coli Lipid A, the Endotoxically Active Principle of Cell-Surface Lipopolysaccharide. Bull. Chem. Soc. Jpn. 2006, 60, 2205–2214. [Google Scholar] [CrossRef] [Green Version]
- Rietschel, E.T.; Seydel, U.; Zähringer, U.; Schade, U.F.; Brade, L.; Loppnow, H.; Feist, W.; Wang, M.H.; Ulmer, A.J.; Flad, H.D.; et al. Bacterial endotoxin: Molecular relationships between structure and activity. Infect. Dis. Clin. N. Am. 1991, 5, 753–779. [Google Scholar]
- Tan, Y.; Kagan, J.C. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol. Cell 2014, 54, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [Green Version]
- Glauser, M.P.; Heumann, D.; Baumgartner, J.D.; Cohen, J. Pathogenesis and potential strategies for prevention and treatment of septic shock: An update. Clin. Infect. Dis. 1994, 18, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, V.; Alpizar, Y.A.; Luis, E.; Tajada, S.; Denlinger, B.; Fajardo, O.; Manenschijn, J.A.; Fernández-Peña, C.; Talavera, A.; Kichko, T.; et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat. Commun. 2014, 5, 3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauvliege, R.; Vanrobaeys, J.; Schotte, P.; Beyaert, R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-γ requires nuclear factor-κB and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem. 2002, 277, 41624–41630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurung, P.; Malireddi, R.K.; Anand, P.K.; Demon, D.; Vande Walle, L.; Liu, Z.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.D. TRIF-mediated caspase-11 production integrates TLR4- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J. Biol. Chem. 2012, 287, 34474–34483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerala, R. Structural biology of the LPS recognition. Int. J. Med. Microbiol. 2007, 297, 353–363. [Google Scholar] [CrossRef]
- Caroff, M.; Karibian, D. Structure of bacterial lipopolysaccharides: Bacterial antigens and vaccines. Carbohydr. Res. 2003, 338, 2431–2447. [Google Scholar] [CrossRef]
- Joiner, K.A.; Schmetz, M.A.; Goldman, R.C.; Leive, L.; Frank, M.M. Mechanism of bacterial resistance to complement-mediated killing: Inserted C5b-9 correlates with killing for Escherichia coli O111B4 varying in O-antigen capsule and O-polysaccharide coverage of lipid A core oligosaccharide. Infect. Immun. 1984, 45, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Ulmer, A.J.; Holst, O.; Brade, H.; Schmidt, G.; Mamat, U.; Grimmecke, H.D.; Kusumoto, S.; et al. The chemical structure of bacterial endotoxin in relation to bioactivity. Immunobiology 1993, 187, 169–190. [Google Scholar] [CrossRef]
- Luderitz, O.; Tanamoto, K.; Galanos, C.; McKenzie, G.R.; Brade, H.; Zähringer, U.; Rietschel, E.T.; Kusumoto, S.; Shiba, T. Lipopolysaccharides: Structural Principles and Biologic Activities. Clin. Infect. Dis. 1984, 6, 428–431. [Google Scholar] [CrossRef]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; Di Padova, F. Bacterial endotoxin: Molecular relationships of structure to activity and function. FASEB J. 1994, 8, 217–225. [Google Scholar] [CrossRef]
- Cochet, F.; Peri, F. The role of carbohydrates in the lipopolysaccharide (LPS)/toll-like receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 2017, 18, 2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, N.R.; Morrison, D.C. Binding and activation of the first component of human complement by the lipid A region of lipopolysaccharides. J. Immunol. 1978, 120, 1862–1868. [Google Scholar] [PubMed]
- Morrison, D.C.; Kline, L.F. Activation of the classical and properdin pathways of complement by bacterial lipopolysaccharides (LPS). J. Immunol. 1977, 118, 362–368. [Google Scholar] [PubMed]
- Galanos, C. Physical state and biological activity of lipopolysaccharides. Toxicity and immunogenicity of the lipid A component. Z. Immun. Exp. Klin. Immunol. 1975, 149, 214–229. [Google Scholar]
- Kusumoto, S.; Fukase, K.; Shiba, T. Erratum to “Key structures of bacterial peptidoglycan and lipopolysaccharide triggering the innate immune system of higher animals: Chemical synthesis and functional studies”. Proc. Jpn. Acad. Ser. B 2010, 86, 322–337. [Google Scholar] [CrossRef] [Green Version]
- Dixon, D.R.; Darveau, R.P. Lipopolysaccharide Heterogeneity: Innate Host Responses to Bacterial Modification of Lipid A Structure. J. Dent. Res. 2005, 84, 584–595. [Google Scholar] [CrossRef]
- Okan, N.A.; Kasper, D.L. The atypical lipopolysaccharide of Francisella. Carbohydr. Res. 2013, 378, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Needham, B.D.; Trent, M.S. Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481. [Google Scholar] [CrossRef]
- Valentin-Hansen, P.; Johansen, J.; Rasmussen, A.A. Small RNAs controlling outer membrane porins. Curr. Opin. Microbiol. 2007, 10, 152–155. [Google Scholar] [CrossRef]
- Barker, J.H.; Weiss, J.; Apicella, M.A.; Nauseef, W.M. Basis for the failure of Francisella tularensis lipopolysaccharide to prime human polymorphonuclear leukocytes. Infect. Immun. 2006, 74, 3277–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, E.; Perry, M.B.; Conlan, J.W. Structural analysis of Francisella tularensis lipopolysaccharide. Eur. J. Biochem. 2002, 269, 6112–6118. [Google Scholar] [CrossRef] [PubMed]
- Zughaier, S.; Agrawal, S.; Stephens, D.S.; Pulendran, B. Hexa-acylation and KDO2-glycosylation determine the specific immunostimulatory activity of Neisseria meningitidis lipid a for human monocyte derived dendritic cells. Vaccine 2006, 24, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Gaekwad, J.; Zhang, Y.; Zhang, W.; Reeves, J.; Wolfert, M.A.; Boons, G.J. Differential induction of innate immune responses by synthetic lipid a derivatives. J. Biol. Chem. 2010, 285, 29375–29386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroi, M.; Tanamoto, K. The polysaccharide portion plays an indispensable role in Salmonella lipopolysaccharide-induced activation of NF-κB through human toll-like receptor 4. Infect. Immun. 2002, 70, 6043–6047. [Google Scholar] [CrossRef] [Green Version]
- Munoz, C.; Carlet, J.; Fitting, C.; Misset, B.; Blériot, J.P.; Cavaillon, J.M. Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Investig. 1991, 88, 1747–1754. [Google Scholar] [CrossRef] [Green Version]
- Kitchens, R.L.; Thompson, P.A.; Viriyakosol, S.; O’Keefe, G.E.; Munford, R.S. Plasma CD14 decreases monocyte responses to LPS by transferring cell-bound LPS to plasma lipoproteins. J. Clin. Investig. 2001, 108, 485–493. [Google Scholar] [CrossRef]
- Ohno, N.; Morrison, D. Lipopolysaccharide interactions with lysozyme differentially affect lipopolysaccharide immunostimulatory activity. Eur. J. Biochem. 1989, 186, 629–636. [Google Scholar] [CrossRef]
- Weiss, J. Bactericidal/permeability-increasing protein (BPI) and lipopolysaccharide-binding protein (LBP): Structure, function and regulation in host defence against Gram-negative bacteria. Biochem. Soc. Trans. 2003, 31, 785–790. [Google Scholar] [CrossRef]
- Wang, D.; Pabst, K.M.; Aida, Y.; Pabst, M.J. Lipopolysaccharide-inactivating activity of neutrophils is due to lactoferrin. J. Leukoc. Biol. 1995, 57, 865–874. [Google Scholar] [CrossRef]
- Rana, S.V.; Reimers, H.J.; Pathikonda, M.S.; Bajaj, S.P. Expression of tissue factor and factor VIIa/tissue factor inhibitor activity in endotoxin or phorbol ester stimulated U937 monocyte-like cells. Blood 1988, 71, 259–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrick, J.W.; Hirata, M.; Balint, R.F.; Lee, J.; Zhong, J.; Wright, S.C. Human CAP18: A novel antimicrobial lipopolysaccharide-binding protein. Infect. Immun. 1995, 63, 1291–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaby, R. Lipopolysaccharide-binding molecules: Transporters, blockers and sensors. Cell. Mol. Life Sci. 2004, 61, 1697–1713. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.H. Lipopolysaccharide: Basic Biochemistry, Intracellular Signaling, and Physiological Impacts in the Gut. Intest. Res. 2014, 12, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrä, J.; Gutsmann, T.; Müller, M.; Schromm, A.B. Interactions between lipid A and serum proteins. Adv. Exp. Med. Biol. 2009, 667, 39–51. [Google Scholar]
- Tobias, P.; Ulevitch, R.; Wright, S.; Mathison, J.; Ramos, R. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 2006, 249, 1431–1433. [Google Scholar]
- Kobayashi, M.; Saitoh, S.; Tanimura, N.; Takahashi, K.; Kawasaki, K.; Nishijima, M.; Fujimoto, Y.; Fukase, K.; Akashi-Takamura, S.; Miyake, K. Regulatory Roles for MD-2 and TLR4 in Ligand-Induced Receptor Clustering. J. Immunol. 2014, 176, 6211–6218. [Google Scholar] [CrossRef] [Green Version]
- Esparza, G.A.; Teghanemt, A.; Zhang, D.; Gioannini, T.L.; Weiss, J.P. Endotoxin·albumin complexes transfer endotoxin monomers to MD-2 resulting in activation of TLR4. Innate Immun. 2012, 18, 478–491. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.J.; Lee, H.; Park, J.D.; Hyun, H.C.; Sohn, H.O.; Lee, D.W.; Kim, Y.S. Kinetics of binding of LPS to recombinant CD14, TLR4, and MD-2 proteins. Mol. Cells 2007, 24, 119–124. [Google Scholar]
- Beveridge, T.J. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 1999, 181, 4725–4733. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.; Barker, J. Diverse pro-inflammatory endotoxin recognition systems of mammalian innate immunity. F1000Research 2018, 7, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailman, E.; Lichenstein, H.S.; Wurfel, M.M.; Miller, D.S.; Johnson, D.A.; Kelley, M.; Busse, L.A.; Zukowski, M.M.; Wright, S.D. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J. Exp. Med. 1994, 179, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Kim, S.J.; Rah, S.H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.O.; Park, B.S.; Yoon, T.Y.; et al. Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS Recognition and Transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamping, N.; Hoess, A.; Yu, B.; Park, T.C.; Kirschning, C.J.; Pfeil, D.; Reuter, D.; Wright, S.D.; Herrmann, F.; Schumann, R.R. Effects of site-directed mutagenesis of basic residues (Arg 94, Lys 95, Lys 99) of lipopolysaccharide (LPS)-binding protein on binding and transfer of LPS and subsequent immune cell activation. J. Immunol. 1996, 157, 4648–4656. [Google Scholar] [PubMed]
- Han, J.; Mathison, J.C.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide (LPS) binding protein, truncated at Ile-197, binds LPS but does not transfer LPS to CD14. J. Biol. Chem. 1994, 269, 8172–8175. [Google Scholar] [PubMed]
- Akashi, S.; Saitoh, S.I.; Wakabayashi, Y.; Kikuchi, T.; Takamura, N.; Nagai, Y.; Kusumoto, Y.; Fukase, K.; Kusumoto, S.; Adachi, Y.; et al. Lipopolysaccharide interaction with cell surface toll-like receptor 4-MD-2: Higher affinity than that with MD-2 or CD14. J. Exp. Med. 2003, 198, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Golenbock, D.T. Endotoxin recognition and signal transduction by the TLR4/MD-2 complex. Microbes Infect. 2004, 6, 1361–1367. [Google Scholar] [CrossRef]
- Kruger, C.L.; Zeuner, M.T.; Cottrell, G.S.; Widera, D.; Heilemann, M. Quantitative single-molecule imaging of TLR4 reveals ligand-specific receptor dimerization. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Paramo, T.; Piggot, T.J.; Bryant, C.E.; Bond, P.J. The structural basis for endotoxin-induced allosteric regulation of the toll-like receptor 4 (tlr4) innate immune receptor. J. Biol. Chem. 2013, 288, 36215–36225. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.G.; Berglund, N.A.; Kargas, V.; Marzinek, J.K.; Holdbrook, D.A.; Khalid, S.; Piggot, T.J.; Schmidtchen, A.; Bond, P.J. A Thermodynamic Funnel Drives Bacterial Lipopolysaccharide Transfer in the TLR4 Pathway. Structure 2018, 26, 1151–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horng, T.; Barton, G.M.; Flavell, R.A.; Medzhitov, R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002, 420, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Nijland, R.; Hofland, T.; Van Strijp, J.A.G. Recognition of LPS by TLR4: Potential for anti-inflammatory therapies. Mar. Drugs 2014, 12, 4260–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-induced Cytokine Production in Human Monocytes and Macrophages. Crit. Rev. Immunol. 2012, 31, 379–446. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Akira, S.; Kawai, T. TLR signaling. Semin. Immunol. 2007, 19, 24–32. [Google Scholar]
- Tanimura, N.; Saitoh, S.; Matsumoto, F.; Akashi-Takamura, S.; Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368, 94–99. [Google Scholar] [CrossRef]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of TLR4 to the induction of interferon beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Nagaoka, K.; Takahara, K.; Tanaka, K.; Yoshida, H.; Steinman, R.M.; Saitoh, S.; Akashi-Takamura, S.; Miyake, K.; Kang, Y.S.; Park, C.G.; et al. Association of SIGNR1 with TLR4-MD-2 enhances signal transduction by recognition of LPS in gram-negative bacteria. Int. Immunol. 2005, 17, 827–836. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, S.J.; Steeghs, L.; Bruijns, S.C.; Vaezirad, M.M.; Snijders Blok, C.; Arenas Busto, J.A.; Deken, M.; van Putten, J.P.; van Kooyk, Y. Variation of Neisseria gonorrhoeae lipooligosaccharide directs dendritic cell-induced T helper responses. PLoS Pathog. 2009, 5, e1000625. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Innate signaling by the C-type lectin DC-SIGN dictates immune responses. Cancer Immunol. Immunother. 2009, 58, 1149–1157. [Google Scholar] [CrossRef] [Green Version]
- Saunders, S.P.; Walsh, C.M.; Smith, P.; Fallon, P.G.; Barlow, J.L.; Bellsoi, A.; McKenzie, A.N.J. C-type lectin SIGN-R1 has a role in experimental colitis and responsiveness to lipopolysaccharide. J. Immunol. 2010, 184, 2627–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, A.D.; Rosenstreich, D.L.; Scher, I.; Campbell, G.H.; MacDermott, R.P.; Formal, S.B. Genetic control of susceptibility to Salmonella typhimurium in mice: Role of the LPS gene. J. Immunol. 1980, 124, 20–24. [Google Scholar]
- O’Brien, G.C.; Wang, J.H.; Redmond, H.P. Bacterial Lipoprotein Induces Resistance to Gram-Negative Sepsis in TLR4-Deficient Mice via Enhanced Bacterial Clearance. J. Immunol. 2005, 174, 1020–1026. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Owsianik, G.; D’hoedt, D.; Voets, T.; Nilius, B. Structure–function relationship of the TRP channel superfamily. Rev. Physiol. Biochem. Pharmacol. 2006, 156, 61–90. [Google Scholar] [PubMed]
- Cheng, W.; Sun, C.; Zheng, J. Heteromerization of TRP channel subunits: Extending functional diversity. Protein Cell 2010, 1, 802–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa-Valente, J.; Brain, S.D. A historical perspective on the role of sensory nerves in neurogenic inflammation. Semin. Immunopathol. 2018, 40, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, F.Q.; Poole, S.; Lorenzetti, B.B.; Ferreira, S.H. The pivotal role of tumour necrosis factor α in the development of inflammatory hyperalgesia. Br. J. Pharmacol. 1992, 107, 660–664. [Google Scholar] [CrossRef]
- Kichko, T.I.; Reeh, P.W. TRPV1 controls acid- and heat-induced calcitonin gene-related peptide release and sensitization by bradykinin in the isolated mouse trachea. Eur. J. Neurosci. 2009, 29, 1896–1904. [Google Scholar] [CrossRef]
- Richardson, J.D.; Vasko, M.R. Cellular mechanisms of neurogenic inflammation. J. Pharmacol. Exp. Ther. 2002, 302, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.; Pulver, S.R.; Panzano, V.C.; Chang, E.C.; Griffith, L.C.; Theobald, D.L.; Garrity, P.A. Analysis of Drosophila TRPA1 reveals an ancient origin for human chemical nociception. Nature 2010, 464, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Soldano, A.; Alpizar, Y.A.; Boonen, B.; Franco, L.; López-Requena, A.; Liu, G.; Mora, N.; Yaksi, E.; Voets, T.; Vennekens, R.; et al. Gustatory-mediated avoidance of bacterial lipopolysaccharides via TRPA1 activation in Drosophila. eLife 2016, 5, e13133. [Google Scholar] [CrossRef] [PubMed]
- Startek, J.B.; Talavera, K.; Voets, T.; Alpizar, Y.A. Differential interactions of bacterial lipopolysaccharides with lipid membranes: Implications for TRPA1-mediated chemosensation. Sci. Rep. 2018, 8, 12010. [Google Scholar] [CrossRef]
- Boonen, B.; Alpizar, Y.A.; Sanchez, A.; López-Requena, A.; Voets, T.; Talavera, K. Differential effects of lipopolysaccharide on mouse sensory TRP channels. Cell Calcium 2018, 73, 72–81. [Google Scholar] [CrossRef]
- Alenmyr, L.; Uller, L.; Greiff, L.; Högestätt, E.D.; Zygmunt, P.M. TRPV4-Mediated Calcium Influx and Ciliary Activity in Human Native Airway Epithelial Cells. Basic Clin. Pharmacol. Toxicol. 2014, 114, 210–216. [Google Scholar] [CrossRef]
- Diogenes, A.; Ferraz, C.C.R.; Akopian, A.N.; Henry, M.A.; Hargreaves, K.M. LPS Sensitizes TRPV1 via Activation of TLR4 in Trigeminal Sensory Neurons. J. Dent. Res. 2011, 90, 759–764. [Google Scholar] [CrossRef]
- Ferraz, C.C.R.; Henry, M.A.; Hargreaves, K.M.; Diogenes, A. Lipopolysaccharide from porphyromonas gingivalis sensitizes capsaicin-sensitive nociceptors. J. Endod. 2011, 37, 45–48. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Tschopp, J. Inflammatory Caspases: Linking an Intracellular Innate Immune System to Autoinflammatory Diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, I.M.; Liedtke, W.; Sanderson, M.J.; Valverde, M.A. TRPV4 channel participates in receptor-operated calcium entry and ciliary beat frequency regulation in mouse airway epithelial cells. Proc. Natl. Acad. Sci. USA 2008, 105, 12611–12616. [Google Scholar] [CrossRef] [Green Version]
- Everaerts, W.; Zhen, X.; Ghosh, D.; Vriens, J.; Gevaert, T.; Gilbert, J.P.; Hayward, N.J.; McNamara, C.R.; Xue, F.; Moran, M.M.; et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc. Natl. Acad. Sci. USA 2010, 107, 19084–19089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorneloe, K.S.; Sulpizio, A.C.; Lin, Z.; Figueroa, D.J.; Clouse, A.K.; McCafferty, G.P.; Chendrimada, T.P.; Lashinger, E.S.; Gordon, E.; Evans, L.C.; et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-Dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a Novel and Potent Transient Receptor Potential Vanilloid 4 Channel Agonist Induces Urinary. J. Pharmacol. Exp. Ther. 2008, 326, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, N.M.; Ravindran, K.; Kuebler, W.M. TRPV4: Physiological role and therapeutic potential in respiratory diseases. Naunyn-Schmiedeberg’s. Arch. Pharmacol. 2015, 388, 421–436. [Google Scholar] [CrossRef]
- Thornberry, N. Caspases: A decade of death research. Cell Death Differ. 1999, 6, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Jurg, T.; Martinon, F.; Burns, K. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J.; et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Faucheu, C.; Diu, A.; Chan, A.W.; Blanchet, A.M.; Miossec, C.; Hervé, F.; Collard-Dutilleul, V.; Gu, Y.; Aldape, R.A.; Lippke, J.A.; et al. A novel human protease similar to the interleukin-1 beta converting enzyme induces apoptosis in transfected cells. EMBO J. 1995, 14, 1914–1922. [Google Scholar] [CrossRef]
- Kamens, J.; Paskind, M.; Hugunin, M.; Talanian, R.V.; Allen, H.; Banach, D.; Bump, N.; Hackett, M.; Johnston, C.G.; Li, P.; et al. Identification and characterization of ICH-2, a novel member of the interleukin-1 beta-converting enzyme family of cysteine proteases. J. Biol. Chem. 1995, 270, 15250–15256. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Feng, Y.; Xiong, G.; Whyte, S.; Duan, J.; Yang, Y.; Wang, K.; Yang, S.; Geng, Y.; Ou, Y.; et al. Caspase-11, a specific sensor for intracellular lipopolysaccharide recognition, mediates the non-canonical inflammatory pathway of pyroptosis. Cell Biosci. 2019, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, I.; Tharakan, B.; Bhat, G.K. Caspases—An update. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 151, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Miura, M.; Jung Yk Zhu, H.; Gagliardini, V.; Shi, L.; Greenberg, A.H.; Yuan, J. Identification and characterization of ich-3, a member of the interleukin-1β converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J. Biol. Chem. 1996, 271, 20580–20587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.F.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Kang, S.J.; Wang, S.; Hara, H.; Peterson, E.P.; Namura, S.; Amin-Hanjani, S.; Huang, Z.; Srinivasan, A.; Tomaselli, K.J.; Thornberry, N.A.; et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J. Cell Biol. 2000, 149, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef]
- Napier, B.A.; Brubaker, S.W.; Sweeney, T.E.; Monette, P.; Rothmeier, G.H.; Gertsvolf, N.A.; Puschnik, A.; Carette, J.E.; Khatri, P.; Monack, D.M. Complement pathway amplifies caspase-11–dependent cell death and endotoxin-induced sepsis severity. J. Exp. Med. 2016, 213, 2365–2382. [Google Scholar] [CrossRef] [Green Version]
- Case, C.L.; Kohler, L.J.; Lima, J.B.; Strowig, T.; de Zoete, M.R.; Flavell, R.A.; Zamboni, D.S.; Roy, C.R. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl. Acad. Sci. USA 2013, 110, 1851–1856. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4 Nobuhiko Kayagaki. Science 2013, 1246, 1246–1250. [Google Scholar] [CrossRef]
- Aachoui, Y.; Leaf, I.A.; Hagar, J.A.; Fontana, M.F.; Campos, C.G.; Zak, D.E.; Tan, M.H.; Cotter, P.A.; Vance, R.E.; Aderem, A.; et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science 2013, 339, 975–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Kanneganti, T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Zhao, Y.; Shao, F. Innate immunity to intracellular LPS. Nat. Immunol. 2019, 20, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.H.; Oh, Y.J.; Kim, E.S.; Choi, J.E.; Shin, J.S. High Mobility Group Box 1 Protein Binding to Lipopolysaccharide Facilitates Transfer of Lipopolysaccharide to CD14 and Enhances Lipopolysaccharide-Mediated TNF-α Production in Human Monocytes. J. Immunol. 2008, 180, 5067–5074. [Google Scholar] [CrossRef] [Green Version]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef]
- Yokoyama, S.; Cai, Y.; Murata, M.; Tomita, T.; Yoneda, M.; Xu, L.; Pilon, A.L.; Cachau, R.E.; Kimura, S. A novel pathway of LPS uptake through syndecan-1 leading to pyroptotic cell death. eLife 2018, 7, e37854. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A.K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef] [Green Version]
- Finethy, R.; Luoma, S.; Orench-Rivera, N.; Feeley, E.M.; Haldar, A.K.; Yamamoto, M.; Kanneganti, T.D.; Kuehn, M.J.; Coers, J. Inflammasome activation by bacterial outer membrane vesicles requires guanylate binding proteins. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.C.; Dick, M.S.; Lagrange, B.; Degrandi, D.; Pfeffer, K.; Yamamoto, M.; Meunier, E.; Pelczar, P.; Henry, T.; Broz, P. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. 2018, 37, e98089. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Dick, M.S.; Dreier, R.F.; Schürmann, N.; Kenzelmann Broz, D.; Warming, S.; Roose-Girma, M.; Bumann, D.; Kayagaki, N.; Takeda, K.; et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 2014, 509, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Pilla, D.M.; Hagar, J.A.; Haldar, A.K.; Mason, A.K.; Degrandi, D.; Pfeffer, K.; Ernst, R.K.; Yamamoto, M.; Miao, E.A.; Coers, J. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc. Natl. Acad. Sci. USA 2014, 111, 6046–6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazgaeen, L.; Gurung, P. Recent Advances in Lipopolysaccharide Recognition Systems. Int. J. Mol. Sci. 2020, 21, 379. https://doi.org/10.3390/ijms21020379
Mazgaeen L, Gurung P. Recent Advances in Lipopolysaccharide Recognition Systems. International Journal of Molecular Sciences. 2020; 21(2):379. https://doi.org/10.3390/ijms21020379
Chicago/Turabian StyleMazgaeen, Lalita, and Prajwal Gurung. 2020. "Recent Advances in Lipopolysaccharide Recognition Systems" International Journal of Molecular Sciences 21, no. 2: 379. https://doi.org/10.3390/ijms21020379
APA StyleMazgaeen, L., & Gurung, P. (2020). Recent Advances in Lipopolysaccharide Recognition Systems. International Journal of Molecular Sciences, 21(2), 379. https://doi.org/10.3390/ijms21020379