Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Dose IBA-Induced Embryogenic Redifferentiation in Cotton



Abstract
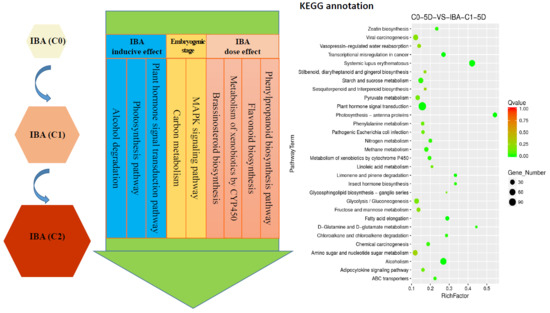
1. Introduction
2. Results
2.1. Callus Differentiation Rate of Cotton Induced by Different IBA Concentration and KT Combinations
2.2. Density Distribution of Reads on Chromosomes
2.3. Screening of Differentially Expressed Genes
2.4. Overall Transcriptome Sequencing Analysis
2.5. Screening of Differentially Expressed Genes from Venn Diagram between Different Comparison Groups
2.6. Type and Number of Alternative Splicing
2.7. Functional Annotation of Differentially Expressed Genes
2.8. Differential Metabolic Pathway Analysis
2.8.1. Enriched Metabolic Pathway in Comparison Groups with and without IBA
2.8.2. Enriched Differential Metabolic Pathway in Different IBA Doses
2.8.3. Enriched Differential Metabolic Pathway with Different Culture Times
2.9. Expression Profiles of Major Genes Associated with SE Regulation
2.10. Candidate Gene Expression Pattern Validation
3. Discussion
3.1. IBA Embryogenic Inductive Effect and Dose Effect During Differentiation of Cotton SE
3.2. Plant Hormone Signal Transduction and Alcohol Degradation Pathway in Embryogenic Induction Responsive Stage
3.3. Photosynthesis, Cell Cycle and SERK Pathway During Somatic Embryogenesis in Cotton
4. Materials and Methods
4.1. Plant Materials and Culture Conditions
4.2. RNA Extraction, cDNA Library Preparation, and RNA-Seq
4.3. Mapping and Differential Expression Analysis
4.4. GO Functional Annotation and KEGG Enrichment Analysis
4.5. Alternative Splicing Analysis
4.6. Validation of RNA-Seq Data by Real-Time RT-PCR
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| DEG | Differentially expressed gene |
| DGE | Digital gene expression |
| EC | Embryogenic callus |
| ERF | Ethylene response factor |
| GO | Gene Ontology |
| IAA | Indole acetic acid |
| IBA | Indole-3-butyric acid |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KT | Kinetin |
| MAPK | Mitogen-activated protein kinase |
| MS | Murashige and Skoog |
| NAA | Naphthalene acetic acid |
| NR | Non-redundant Protein Sequence Database in GenBank |
| RPKM | Reads Per Kilo bases per Million reads |
| RT-PCR | Reverse-transcription-polymerase chain reaction |
| SE | Somatic embryogenesis |
| SERK | Somatic embryogenesis receptor kinase |
| 2, 4-D | 2, 4-dichlorophenoxyacetic acid |
Appendix A





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene ID | Full Name | Primer Sequence (Forward/Reverse) | Tm (°C) | Product (bp) |
|---|---|---|---|---|---|
| CHIT1B | Gh_D06G0479 | Basic endochitinase | AGCAGGAGAAGCAATAAAGG/ATGACACCGTATCCAGGGAC | 56.6 | 201 |
| CBL4 | Gh_D11G0140 | Calcineurin B-like protein 4 | TCTTGCTGCTGAAACACCT/TCTAACGAACTCACCGAAC | 53.4 | 235 |
| PER73 | Gh_D03G0246 | Peroxidase 73 | GCAACTATCCGCCTCTTCT/GCTTAACCCATCCAACCTC | 54.3 | 291 |
| CYP82A3 | Gh_D05G2983 | Cytochrome P450 82A3 | CTTTGGTAGTGGTAGGAGGAG/GAGCAAGACGAGGTGAGAC | 51.9 | 194 |
| ARF4 | Gh_D05G0755 | Auxin response factor 4 | CAACATGAATCAAGCACCCAA/GTTTCCTGATGTCGTCCAC | 53 | 194 |
| AUX22B | Gh_D13G1819 | Auxin-induced protein 22B | GGCAACTCTTCGGAGCAAC/TATCCCACTAATTTCACCC | 49.8 | 193 |
| GhUB7 | DQ116441 | Ubiquitin 7 | GAAGGCATTCCACCTGACCAAC/CTTGACCTTCTTCTTCTTGTGCTT | 59 | 198 |
References
- Steward, F.C.; Mapes, M.O.; Smith, J. Growth and organized development of cultured cells I. Growth and division of freely suspended cells. Am. J. Bot. 1958, 45, 693–703. [Google Scholar] [CrossRef]
- Zimmerman, J.L. Somatic embryogenesis: A model for early development in higher plants. Plant Cell 1993, 5, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Chugh, A.; Khurana, P. Gene expression during somatic embryogenesis: Recent advances. Curr. Sci. 2002, 83, 715–730. [Google Scholar]
- Zeng, F.; Zhang, X.; Zhu, L. Isolation and characterization of genes associated to cotton somatic embryogenesis by suppression subtractive hybridization and macroarray. Plant Mol. Biol. 2006, 60, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Jin, S.; Guo, X.; Zhang, X. Somatic Embryogenesis and Plant Regeneration in Cotton Cultivars from Yellow and Yangtze River Planting Areas. Acta Agron. Sin. 2007, 33, 394–400. [Google Scholar]
- Feng, R.; Wang, Q.; Zhang, B.; Yu, X. Genotype analysis in cotton tissue culture and plant regeneration. Acta Agriculturae Boreali Occidentalis Sinica 1997, 6, 27–30. [Google Scholar]
- Zhang, C.; Yu, S.; Fan, S.; Zhang, J.; Li, F. Inheritance of somatic embryogenesis using leaf petioles as explants in upland cotton. Euphytica 2011, 181, 55–63. [Google Scholar] [CrossRef]
- Zhang, X.L.; Sun, J.Z.; Liu, J.L. Somatic embryogenesis and plant regeneration in upland cotton. Acta Genet. Sin. 1991, 18, 461–467. [Google Scholar]
- Mishra, R.; Wang, H.Y.; Yadav, N.R.; Wilkins, T.A. Development of a highly regenerable elite Acala cotton (Gossypium hirsutum cv. Maxxa): A step towards genotype-independent regeneration. Plant Cell Tissue Organ Cult. 2003, 73, 21–35. [Google Scholar] [CrossRef]
- Ganesan, M.; Jayabalan, N. Evaluation of haemoglobin (erythrogen): For improved somatic embryogenesis and plant regeneration in cotton (Gossypium hirsutum L. cv. SVPR 2). Plant Cell Rep. 2004, 23, 181–187. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X.L. Effects of brassinolide on somatic embryogenesis of Gossypium hirsutum. Plant Physiol. Commun. 1992, 28, 15–18. [Google Scholar]
- Wu, J.; Zhang, X.; Nie, Y. Factors affecting somatic embryogenesis and plant regeneration from a range of recalcitrant genotypes of Chinese cottons (Gossypium hirsutum L.). In Vitro Cell. Dev. Biol. Plant 2004, 40, 371–375. [Google Scholar] [CrossRef]
- Chen, Z.X.; Li, S.J.; Trolinder, N.L.; Goodin, J.R. Studies on some characteristics of cotton cell suspension culture embryogenesis and plant regeneration. Chin. Agric. Sci. 1987, 5, 6–11. [Google Scholar]
- Price, H.J.; Smith, R.H. Somatic embryogenesis in suspension cultures of Gossypium klotzschianum anderss. Planta 1979, 145, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.W.; Sun, J.S. Method of rapid plant formation by somatic regeneration of upland cotton. Chin. Sci. Bull. 2000, 45, 1771–1774. [Google Scholar] [CrossRef]
- Li, G.D.; Xiao, J.L.; Luo, X.L.; Zhang, A.H.; Wu, J.H. Somatic embryogenesis and plant regeneration of calli derived from cotton (Gossypium hirsutum L.) depending on their quality. J. Shanxi Agric. Sci. 2006, 34, 29–31. [Google Scholar]
- Sun, J.Y.; Li, W.M.; Zhang, H.S.; Zhao, J.L.; Yin, X.L.; Wang, L.A. Direct somatic embryogenesis and plant regeneration in glandless upland cotton (Gossypium hirsutum). Acta Agriculture Boreali-Sinica 2009, 24, 81–84. [Google Scholar]
- Chen, M.Y.; Zhang, X.L.; Nie, Y.C.; Wu, J.H. Study on improvement of plant regeneration technology of upland cotton. Acta Cotton Sinica 2002, 14, 344–347. [Google Scholar]
- Li, S.W.; Shi, R.F.; Leng, Y.; Zhou, Y. Transcriptomic analysis reveals the gene expression profile that specifically responds to IBA during adventitious rooting in mung bean seedlings. BMC Genom. 2016, 17, 1–23. [Google Scholar] [CrossRef]
- Zeng, F.; Zhang, X.; Jin, S.; Cheng, L.; Liang, S.; Hu, L.; Guo, X.; Nie, Y.; Cao, J. Chromatin reorganization and endogenous auxin/cytokinin dynamic activity during somatic embryogenesis of cultured cotton cell. Plant Cell Tissue Organ Cult. 2007, 90, 63–70. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Yuan, D.; Jin, F.; Zhang, Y.; Xu, J. Transcript profiling reveals complex auxin signalling pathway and transcription regulation involved in dedifferentiation and redifferentiation during SE in cotton. BMC Plant Biol. 2012, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, C.; Zhang, X.; Liu, C.; Wu, Z.; Yang, Z.; Zhou, K.; Yang, X.; Li, F. Transcriptome profiling reveals auxin and cytokinin regulating somatic embryogenesis in different sister lines of cotton cultivar CCRI24. J. Integr. Plant Biol. 2013, 55, 631–642. [Google Scholar] [CrossRef]
- Min, L.; Hu, Q.; Li, Y.; Xu, J.; Ma, Y.; Zhu, L.; Yang, X.; Zhang, X. LEAFY COTYLEDON1-CASEIN KINASE I-TCP15-PHYTOCHROME INTERACTING FACTOR4 network regulates somatic embryogenesis by regulating auxin homeostasis. Plant Physiol. 2015, 169, 2805–2821. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, X.; Huang, C.; Guo, X.; Nie, Y. Somatic embryogenesis and plant regeneration from different wild diploid cotton (Gossypium) species. Plant Cell Rep. 2006, 25, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Miao, Z.; Wang, Z.; Yu, L.; Cai, X.; Xiang, C.L. Arabidopsis ERF1 Mediates Cross-Talk between Ethylene and Auxin Biosynthesis during Primary Root Elongation by Regulating ASA1 Expression. PloS Genet. 2016, 12, e1005760. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, L.; Yuan, D.; Lindsey, K.; Zhang, X. Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J. Exp. Bot. 2013, 64, 1521–1536. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Passardi, F.; Theiler, G.; Zamocky, M.; Cosioa, C.; Rouhier, N.; Teixera, F.; Margis-Pinheiroe, M.; Ioannidis, V.; Penel, C.; Falquet, L.; et al. PeroxiBase: The peroxidase database. Phytochemistry 2007, 68, 1605–1611. [Google Scholar] [CrossRef]
- Guo, H.X.; Guo, H.H.; Zhang, L.; Fan, Y.J.; Fan, Y.P.; Tang, Z.; Zeng, F. Dynamic TMT-based quantitative proteomics analysis of critical initiation process of totipotency during cotton somatic embryogenesis transdifferentiation. Int. J. Mol. Sci. 2019, 20, 1691–1724. [Google Scholar]
- Guo, H.H.; Guo, H.X.; Zhang, L.; Tang, Z.; Yu, X.; Wu, J.; Zeng, F. Metabolome and transcriptome association analysis reveals dynamic regulation of purine metabolism and flavonoid synthesis in transdifferentiation during somatic embryogenesis in cotton. Int. J. Mol. Sci. 2019, 20, 2070. [Google Scholar] [CrossRef]
- Zhai, L.; Xu, L.; Wang, Y.; Zhu, X.; Feng, H.; Li, C.; Luo, X.; Everlyne, M.; Liu, L. Transcriptional identification and characterization of differentially expressed genes associated with embryogenesis in radish (Raphanus sativus L.). Sci. Rep. 2016, 6, 21652–21664. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lai, Z.; Tian, Q.; Lin, L.; Lai, R.; Yang, M.; Zhang, D.; Chen, Y.; Zhang, Z. Endogenous target mimics down-regulate miR160 mediation of ARF10, -16, and -17 cleavage during somatic embryogenesis in Dimocarpus longan Lour. Front. Plant Sci. 2015, 6, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Liu, Y.; Zhou, C.; Li, X.; Zhang, X. The microRNA167 controls somatic embryogenesis in Arabidopsis through regulating its target genes ARF6 and ARF8. Plant Cell Tissue Organ Cult. 2016, 124, 405–417. [Google Scholar] [CrossRef]
- Cao, A.; Zheng, Y.; Yu, Y.; Wang, X.; Shao, D.; Sun, J.; Cui, B. Comparative Transcriptome Analysis of SE initial dedifferentiation in cotton of different SE capability. Sci Rep. 2017, 7, 8583. [Google Scholar] [CrossRef]
- Xiao-Yi, Z.; Li, S.; Hong-Wei, X. Brassinosteroids Regulate the Differential Growth of Arabidopsis Hypocotyls through Auxin Signaling Components IAA19 and ARF7. Mol. Plant 2013, 6, 887–904. [Google Scholar]
- Kumar, G.P.; Subiramani, S.; Govindarajan, S.; Sadasivam, V.; Manickam, V.; Mogilicherla, K.; Thiruppathi, S.K.; Narayanasamy, J. Evaluation of different carbon sources for high frequency callus culture with reduced phenolic secretion in cotton (Gossypium hirsutum L.) cv. SVPR-2. Biotechnol. Rep. 2015, 7, 72–80. [Google Scholar] [CrossRef]
- Cheng, W.; Zhu, H.; Tian, W.; Zhu, S.; Xiong, X.; Sun, Y.; Zhu, Q.; Sun, J. De novo transcriptome analysis reveals insights into dynamic homeostasis regulation of somatic embryogenesis in upland cotton (G. hirsutum L.). Plant Mol. Biol. 2016, 92, 279–292. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, M.; Shi, H. Induction and Regulation of Plant Hormone on Somatic Embryogenesis in Cotton. Biotechnol. Commun. 2004, 15, 577–579. [Google Scholar]
- Wolters, H.; Jurgens, G. Survival of the flexible: Hormonal growth control and adaptation in plant development. Nat. Rev. Genet. 2009, 10, 305–317. [Google Scholar] [CrossRef]
- Perata, P.; Alpi, A. Ethanol-induced injuries to carrot cells: The role of acetaldehyde. Plant Physiol. 1991, 95, 748–752. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, Y.; Chu, D.; Aizawa, M.; Kim, M.; Yamomoto, T. Ethanol metabolism by tobacco callus in vitro. Plant Sci. 1995, 104, 147–151. [Google Scholar] [CrossRef]
- Zeng, F.; Zhang, X.; Cheng, L.; Hu, L.; Zhu, L.; Cao, J.; Guo, X. A draft gene regulatory network for cellular totipotency reprogramming during plant somatic embryogenesis. Genomic 2007, 90, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.H.; Guo, H.X.; Zhang, L.; Fan, Y.J.; Fan, Y.P.; Zeng, F. SELTP assembled battery drives totipotency of somatic plant cell. Plant Biotechnol. J. 2019, 17, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.D.; Guzzo, F.; Toonen, M.A. A leucine-rich repeat containing receptor-like kinase marks somatic plant cells competent to form embryos. Development 1997, 124, 2049–2062. [Google Scholar]
- Guo, H.H.; Wu, J.; Chen, C.; Wang, H.; Zhao, Y.; Zhang, C.; Jia, Y.; Liu, F.; Ning, T.; Chu, Z.; et al. Identification and characterization of cell cultures with various embryogenic/regenerative potential in cotton based on morphological, cytochemical, and cytogenetical assessment. J. Integr. Agric. 2019, 18, 1–8. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]










| IBA Concentration (mg dm−3) | KT Concentration (mg dm−3) | Callus Differentiation Rate (%) |
|---|---|---|
| 0.05 (C2) | 0.1 | 52.63 |
| 0.025 (C1) | 0.1 | 72.22 |
| 0 (C0) | 0.1 | 7.81 |
| Pairwise Comparisons | NR | GO | KEGG |
|---|---|---|---|
| 24D-C3-0D VS IBA-C1-5D | 7264 | 5007 | 1789 |
| 24D-C3-0D VS IBA-C1-20D | 5141 | 3507 | 1300 |
| 24D-C3-0D VS IBA-C2-5D | 9486 | 6444 | 2524 |
| 24D-C3-0D VS IBA-C2-20D | 4314 | 2930 | 1080 |
| 24D-C3-0D VS C0-5D | 2135 | 1434 | 550 |
| 24D-C3-0D VS C0-20D | 5349 | 3731 | 1415 |
| IBA-C1-5D VS IBA-C1-20D | 2924 | 2036 | 723 |
| IBA-C2-5D VS IBA-C2-20D | 11,235 | 7564 | 2828 |
| C0-5D VS C0-20D | 4118 | 2785 | 1191 |
| IBA-C1-5D VS IBA-C2-5D | 8889 | 6068 | 2318 |
| IBA-C1-20D VS IBA-C2-20D | 1542 | 1038 | 333 |
| C0-5D VS IBA-C1-5D | 6181 | 4269 | 1518 |
| C0-5D VS IBA-C2-5D | 10,227 | 6926 | 2685 |
| C0-20D VS IBA-C1-20D | 4404 | 2994 | 1244 |
| C0-20D VS IBA-C2-20D | 2363 | 1626 | 668 |
| No. | Gene ID | Gene Description | Pathway Annotation | p-Value | Regulation | Compare Combination |
|---|---|---|---|---|---|---|
| 1 | Gh_A05G0085 | EIN3-binding F-box protein 1-like isoform X2 | plant hormone signal transduction | 4.06 × 10−84 | Down | C0-5D VS IBA-C1-5D |
| 2 | Gh_D05G0148 | EIN3-binding F-box protein 1-like isoform X2 | plant hormone signal transduction | 1.08 × 10−67 | Down | C0-5D VS IBA-C1-5D |
| 3 | Gh_D09G0971 | pathogenesis-related protein 1-like | plant hormone signal transduction | 1.87 × 10−55 | Down | C0-5D VS IBA-C1-5D |
| 4 | Gh_D06G1764 | abscisic acid receptor PYL4-like | plant hormone signal transduction | 4.05 × 10−55 | Down | C0-5D VS IBA-C1-5D |
| 5 | Gh_A06G1418 | abscisic acid receptor PYL4-like | plant hormone signal transduction | 6.63 × 10−41 | Down | C0-5D VS IBA-C1-5D |
| 6 | Gh_D12G0182 | probable indole-3-acetic acid-amido synthetase GH3.1 | plant hormone signal transduction | 1.15 × 10−38 | Ups | C0-5D VS IBA-C1-5D |
| 7 | Gh_D08G0034 | histone H2A-like | alcohol degradation | 4.07 × 10−24 | Ups | C0-5D VS IBA-C1-5D |
| 8 | Gh_D09G0854 | histone H2B-like | alcohol degradation | 4.91 × 10−18 | Ups | C0-5D VS IBA-C1-5D |
| 9 | Gh_D02G1958 | histone deacetylase 19 isoform X1 | alcohol degradation | 5.05 × 10−16 | Ups | C0-5D VS IBA-C1-5D |
| 10 | Gh_A09G1018 | G2/mitotic-specific cyclin S13-7 | cell cycle | 1.47 × 10−15 | Ups | C0-20D VS IBA-C1-20D |
| 11 | Gh_D09G1039 | G2/mitotic-specific cyclin S13-7 | cell cycle | 2.00 × 10−6 | Ups | C0-20D VS IBA-C1-20D |
| 12 | Gh_D11G0102 | cell division cycle 20.2, cofactor of APC complex-like | cell cycle | 2.08 × 10−6 | Ups | C0-20D VS IBA-C1-20D |
| 13 | Gh_A05G2161 | cyclin-B2-4-like isoform X2 | cell cycle | 3.36 × 10−6 | Ups | C0-20D VS IBA-C1-20D |
| 14 | Gh_A08G1487 | probable serine/threonine-protein kinase cdc7 isoform X1 | cell cycle | 5.93 × 10−6 | Ups | C0-20D VS IBA-C1-20D |
| 15 | Gh_D08G0847 | putative cyclin-B3-1 | cell cycle | 1.65 × 10−5 | Ups | C0-20D VS IBA-C1-20D |
| 16 | Gh_A08G2544 | histone H2A | alcohol degradation | 1.48 × 10−23 | Ups | C0-20D VS IBA-C1-20D |
| 17 | Gh_A09G0836 | probable histone H2B.1 | alcohol degradation | 6.06 × 10−8 | Ups | C0-20D VS IBA-C1-20D |
| 18 | Gh_D05G0853 | histone H2B-like | alcohol degradation | 4.01 × 10−7 | Ups | C0-20D VS IBA-C1-20D |
| 19 | Gh_A11G1738 | oxygen-evolving enhancer protein 1, chloroplastic | photosynthesis | 4.66 × 10−54 | Down | C0-20D VS IBA-C1-20D |
| 20 | Gh_D11G1768 | ferredoxin--NADP reductase, leaf isozyme, chloroplastic | photosynthesis | 4.87 × 10−54 | Down | C0-20D VS IBA-C1-20D |
| 21 | Gh_D07G1624 | photosystem I reaction center subunit XI, chloroplastic-like | photosynthesis | 6.18 × 10−54 | Down | C0-20D VS IBA-C1-20D |
| 22 | Gh_D05G1742 | plastocyanin | photosynthesis | 6.16 × 10−50 | Down | C0-20D VS IBA-C1-20D |
| 23 | Gh_A02G1466 | peroxidase 73-like | Phenylpropanoid biosynthesis | 1.25 × 10−283 | Down | IBA-C1-5D VS IBA-C2-5D |
| 24 | Gh_A09G1415 | peroxidase 21 | Phenylpropanoid biosynthesis | 4.58 × 10−248 | Down | IBA-C1-5D VS IBA-C2-5D |
| 25 | Gh_A08G0747 | peroxidase 16-like | Phenylpropanoid biosynthesis | 3.63 × 10−226 | Down | IBA-C1-5D VS IBA-C2-5D |
| 26 | Gh_D08G0829 | peroxidase 59 | Phenylpropanoid biosynthesis | 4.52 × 10−207 | Ups | IBA-C1-5D VS IBA-C2-5D |
| 27 | Gh_D02G2386 | cytochrome P450 CYP749A22-like isoform X1 | Brassinosteroid biosynthesis | 5.39 × 10−19 | Down | IBA-C1-5D VS IBA-C1-20D |
| 28 | Gh_D08G2495 | cytochrome P450 734A1-like | Brassinosteroid biosynthesis | 3.35 × 10−17 | Ups | IBA-C1-5D VS IBA-C1-20D |
| 29 | Gh_A06G1446 | cytochrome P450 90A1 | Brassinosteroid biosynthesis | 7.63 × 10−5 | Down | IBA-C1-5D VS IBA-C1-20D |
| 30 | Gh_D02G0227 | crocetin glucosyltransferase, chloroplastic-like | Anthocyanin biosynthesis | 2.70 × 10−63 | Down | IBA-C1-5D VS IBA-C1-20D |
| 31 | Gh_D02G0225 | crocetin glucosyltransferase, chloroplastic-like | Anthocyanin biosynthesis | 1.09 × 10−60 | Down | IBA-C1-5D VS IBA-C1-20D |
| 32 | Gh_D02G0230 | crocetin glucosyltransferase, chloroplastic-like | Anthocyanin biosynthesis | 1.33 × 10−28 | Down | IBA-C1-5D VS IBA-C1-20D |
| 33 | Gh_A09G1509 | glutathione S-transferase U8-like | Metabolism of xenobiotics by cytochrome P450 | 2.76 × 10−118 | Ups | IBA-C1-5D VS IBA-C2-5D |
| 34 | Gh_D01G0907 | alcohol dehydrogenase-like 7 | Metabolism of xenobiotics by cytochrome P450 | 2.01 × 10−44 | Down | IBA-C1-5D VS IBA-C2-5D |
| 35 | Gh_D02G2184 | squalene synthase-like | Steroid biosynthesis | 1.43 × 10−63 | Down | IBA-C1-5D VS IBA-C2-5D |
| 36 | Gh_D04G2024 | squalene monooxygenase-like | Steroid biosynthesis | 9.41 × 10−58 | Ups | IBA-C1-5D VS IBA-C2-5D |
| 37 | Gh_D06G0846 | 3beta-hydroxysteroid-dehydrogenase/decarboxylase isoform 2 isoform X1 | Steroid biosynthesis | 1.22 × 10−23 | Down | IBA-C1-5D VS IBA-C2-5D |
| 38 | Gh_A05G1647 | dihydroflavonol-4-reductase-like | Flavonoid biosynthesis | 2.65 × 10−37 | Down | IBA-C1-5D VS IBA-C2-5D |
| 39 | Gh_D12G1798 | Flavonoid 3’-monooxygenase | Flavonoid biosynthesis | 3.84 × 10−34 | Down | IBA-C1-5D VS IBA-C2-5D |
| 40 | Gh_D03G0311 | phosphoenolpyruvate carboxykinase [ATP] | Carbon metabolism | 8.18 × 10−125 | Down | 24D-C3-0D VS IBA-C2-5D |
| 41 | Gh_D07G0203 | ATP-dependent 6-phosphofructokinase 6 isoform X1 | Carbon metabolism | 3.85 × 10−90 | Ups | 24D-C3-0D VS IBA-C2-5D |
| 42 | Gh_D05G3328 | malate dehydrogenase, mitochondrial | Carbon metabolism | 2.09 × 10−57 | Down | 24D-C3-0D VS IBA-C2-5D |
| 43 | Gh_D02G1553 | glyceraldehyde-3-phosphate dehydrogenase, cytosolic | Carbon metabolism | 1.02 × 10−259 | Ups | IBA-C2-5D VS IBA-C2-20D |
| 44 | Gh_A02G0386 | malate dehydrogenase | Carbon metabolism | 1.12 × 10−177 | Ups | IBA-C2-5D VS IBA-C2-20D |
| No. | Gene ID | Gene Description | p-Value | Regulation | Compare Combination |
|---|---|---|---|---|---|
| 1 | Gh_D05G1962 | WUSCHEL-related homeobox 4-like | 1.37 × 10−12 | Ups | C0-20D-VS-IBA-C1-20D |
| 2 | Gh_D11G3261 | WUSCHEL-related homeobox 8-like | 4.35 × 10−3 | Ups | C0-5D-VS-IBA-C1-5D |
| 3 | Gh_D05G1503 | WUSCHEL-related homeobox 9-like | 1.76 × 10−31 | Down | C0-5D-VS-IBA-C1-5D |
| 4 | Gh_D10G0270 | WUSCHEL-related homeobox 9-like isoform X1 | 2.31 × 10−6 | Down | C0-5D-VS-IBA-C1-5D |
| 5 | Gh_D05G1962 | WUSCHEL-related homeobox 4-like | 1.37 × 10−12 | Ups | C0-5D-VS-IBA-C1-5D |
| 6 | Gh_A07G1368 | zinc finger protein ZAT9-like | 4.07 × 10−6 | Down | C0-20D-VS-IBA-C1-20D |
| 7 | Gh_A09G0899 | zinc finger protein WIP3-like | 1.76 × 10−5 | Ups | C0-20D-VS-IBA-C1-20D |
| 8 | Gh_D11G1518 | zinc finger protein CONSTANS-LIKE 9-like isoform X1 | 6.71 × 10−6 | Ups | C0-20D-VS-IBA-C1-20D |
| 9 | Gh_D12G0712 | zinc finger protein AZF1-like | 2.55 × 10−5 | Ups | C0-5D-VS-IBA-C1-5D |
| 10 | Gh_A01G0135 | zinc finger CCCH domain-containing protein 2-like | 3.94 × 10−6 | Down | C0-5D-VS-IBA-C1-5D/C0-20D-VS-IBA-C1-20D |
| 11 | Gh_D01G0181 | zinc finger CCCH domain-containing protein 2-like | 1.17 × 10−5 | Down | C0-20D-VS-IBA-C1-20D |
| 12 | Gh_D11G0511 | transcription factor MYB44-like | 2.63 × 10−47 | Down | C0-5D-VS-IBA-C1-5D |
| 13 | Gh_A06G0188 | transcription factor MYB44-like | 2.21 × 10−66 | Down | C0-5D-VS-IBA-C1-5D/ C0-20D-VS-IBA-C1-20D |
| 14 | Gh_A09G0939 | transcription factor LHW | 1.88 × 10−47 | Ups | C0-5D-VS-IBA-C1-5D/ C0-20D-VS-IBA-C1-20D |
| 15 | Gh_D02G0841 | transcription factor bHLH96-like | 9.80 × 10−56 | Down | C0-5D-VS-IBA-C1-5D |
| 16 | Gh_D07G0897 | transcription factor bHLH62-like | 2.12 × 10−69 | Down | C0-20D-VS-IBA-C1-20D |
| 17 | Gh_A07G2268 | transcription factor bHLH62-like | 3.76 × 10−46 | Down | C0-5D-VS-IBA-C1-5D |
| 18 | Gh_D07G0897 | transcription factor bHLH62-like | 2.12 × 10−69 | Down | C0-5D-VS-IBA-C1-5D |
| 19 | Gh_D11G3340 | transcription factor bHLH106 | 4.72 × 10−71 | Ups | C0-5D-VS-IBA-C1-5D |
| 20 | Gh_A09G0433 | somatic embryogenesis receptor kinase 5 | 3.30 × 10−4 | Ups | IBA-C1-5D-VS-IBA-C2-5D |
| 21 | Gh_D09G0447 | somatic embryogenesis receptor kinase 5 | 1.28 × 10−4 | Ups | C0-20D-VS-IBA-C1-20D |
| 22 | Gh_A01G0158 | somatic embryogenesis receptor kinase 2-like isoform X1 | 3.31 × 10−4 | Ups | C0-20D-VS-IBA-C1-20D |
| 23 | Gh_D07G1063 | serine/threonine-protein kinase D6PKL2-like | 1.04 × 10−88 | Down | C0-5D-VS-IBA-C1-5D |
| 24 | Gh_A10G1074 | receptor-like protein kinase 2 | 1.77 × 10−54 | Down | C0-5D-VS-IBA-C1-5D |
| 25 | Gh_D10G1439 | receptor-like protein kinase 2 | 4.76 × 10−86 | Down | C0-20D-VS-IBA-C1-20D |
| 26 | Gh_A09G1432 | putative receptor protein kinase ZmPK1 isoform X1 | 1.58 × 10−53 | Ups | C0-5D-VS-IBA-C1-5D |
| 27 | |||||
| 28 | Gh_A06G0601 | probable nuclear hormone receptor HR38 isoform X2 | 3.92 × 10−3 | Down | C0-5D-VS-IBA-C1-5D |
| 29 | Gh_D06G0681 | probable nuclear hormone receptor HR38 isoform X2 | 2.00 × 10−8 | Down | C0-5D-VS-IBA-C1-5D |
| 30 | Gh_A06G1176 | ethylene-responsive transcription factor ERF091-like | 1.78 × 10−54 | Down | C0-20D-VS-IBA-C1-20D |
| 31 | Gh_D08G1225 | ethylene-responsive transcription factor ERF061-like | 8.50 × 10−49 | Ups | C0-5D-VS-IBA-C1-5D |
| 32 | Gh_A08G1503 | ethylene-responsive transcription factor 5-like | 8.27 × 10−50 | Down | C0-20D-VS-IBA-C1-20D |
| 33 | Gh_A12G1021 | dof zinc finger protein DOF3.7 | 5.10 × 10−6 | Ups | C0-20D-VS-IBA-C1-20D |
| 34 | Gh_A06G1830 | B-box zinc finger protein 21-like, partial | 3.86 × 10−6 | Down | C0-20D-VS-IBA-C1-20D |
| 35 | Gh_D11G1798 | B-box zinc finger protein 18-like isoform X1 | 4.60 × 10−6 | Ups | C0-5D-VS-IBA-C1-5D |
| 36 | Gh_A11G1640 | B-box zinc finger protein 18-like isoform X1 | 4.61 × 10−6 | Ups | C0-5D-VS-IBA-C1-5D |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Yu, X.; Guo, H.; Wei, J.; Guo, H.; Zhang, L.; Zeng, F. Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Dose IBA-Induced Embryogenic Redifferentiation in Cotton. Int. J. Mol. Sci. 2020, 21, 426. https://doi.org/10.3390/ijms21020426
Fan Y, Yu X, Guo H, Wei J, Guo H, Zhang L, Zeng F. Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Dose IBA-Induced Embryogenic Redifferentiation in Cotton. International Journal of Molecular Sciences. 2020; 21(2):426. https://doi.org/10.3390/ijms21020426
Chicago/Turabian StyleFan, Yupeng, Xiaoman Yu, Huihui Guo, Junmei Wei, Haixia Guo, Li Zhang, and Fanchang Zeng. 2020. "Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Dose IBA-Induced Embryogenic Redifferentiation in Cotton" International Journal of Molecular Sciences 21, no. 2: 426. https://doi.org/10.3390/ijms21020426
APA StyleFan, Y., Yu, X., Guo, H., Wei, J., Guo, H., Zhang, L., & Zeng, F. (2020). Dynamic Transcriptome Analysis Reveals Uncharacterized Complex Regulatory Pathway Underlying Dose IBA-Induced Embryogenic Redifferentiation in Cotton. International Journal of Molecular Sciences, 21(2), 426. https://doi.org/10.3390/ijms21020426