Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies

Abstract
:
1. Introduction
2. Classification of Target Cancer Antigens
2.1. The Ideal Therapeutic Cancer Antigen
2.2. Tumour Specific Antigens (TSA)
2.3. Tumour Associated Antigens (TAA)
2.4. Cancer Germline Antigens
3. Target Antigen Identification: From Screening to Validation
3.1. Cell Surface Molecule Identification
3.2. Refinement of Target Antigen Pool
3.3. Generation of Antibodies for Therapeutic Screening
3.4. Validation of Tools and Target
4. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Chimeric antigen receptor |
| BiTE | Bispecific T cell engager |
| scFv | Single chain variable fragment |
| HER2 | Human epidermal growth factor receptor 2 |
| EGFRvIII | Human epidermal growth factor receptor variant III |
| MHC | Major Histocompatibility Complex |
| MS | Mass spectrometry |
| CDR | Complementarity determining region |
| CGA | Cancer germline antigen |
| TAA | Tumour associated antigen |
| TCR | T cell receptor |
| TSA | Tumour specific antigen |
References
- Suryadevara, C.M.; Gedeon, P.C.; Sanchez-Perez, L.; Verla, T.; Alvarez-Breckenridge, C.; Choi, B.D.; Fecci, P.E.; Sampson, J.H. Are BiTEs the “missing link” in cancer therapy? OncoImmunology 2015, 4, e1008339. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Vallina, L.; Hawkins, R.E. Antigen-specific targeting of CD28-mediated T cell co-stimulation using chimeric single-chain antibody variable fragment-CD28 receptors. Eur J. Immunol. 1996, 26, 2304–2309. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Maude, S.L.; Barrett, D.M.; Rheingold, S.R.; Aplenc, R.; Teachey, D.T.; Callahan, C.; Baniewicz, D.; White, C.; Talekar, M.K.; Shaw, P.A.; et al. Efficacy of Humanized CD19-Targeted Chimeric Antigen Receptor (CAR)-Modified T Cells in Children and Young Adults with Relapsed/Refractory Acute Lymphoblastic Leukemia. Blood 2016, 128, 217. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1 specific TCR engineered T-cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; L, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Oncol. 2018, 11, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowrishankar, K.; Birtwistle, L.; Micklethwaite, K. Manipulating the tumor microenvironment by adoptive cell transfer of CAR T-cells. Mamm Genome 2018, 29, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.-T.; Song, H.; et al. Recognition of Glioma Stem Cells by Genetically Modified T Cells Targeting EGFRvIII and Development of Adoptive Cell Therapy for Glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-Specific T Cells Target Primary Glioblastoma Stem Cells and Induce Regression of Autologous Experimental Tumors. Clin. Cancer Res. 2010, 16, 474. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Garrett, T.P.J.; Burgess, A.W.; Gan, H.K.; Luwor, R.B.; Cartwright, G.; Walker, F.; Orchard, S.G.; Clayton, A.H.A.; Nice, E.C.; Rothacker, J.; et al. Antibodies specifically targeting a locally misfolded region of tumor associated EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 5082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Leyland-Jones, B. Trastuzumab: hopes and realities. Lancet Oncol. 2002, 3, 137–144. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.-T. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef]
- Kim, R.; Kulkarni, P.; Hannenhalli, S. Derepression of Cancer/Testis Antigens in cancer is associated with distinct patterns of DNA Hypomethylation. BMC Cancer 2013, 13, 144. [Google Scholar] [CrossRef] [Green Version]
- Debinski, W.; Gibo, D.M.; Hulet, S.W.; Connor, J.R.; Gillespie, G.Y. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. 1999, 5, 985–990. [Google Scholar] [PubMed]
- Kuhlmann, L.; Cummins, E.; Samudio, I.; Kislinger, T. Cell-surface proteomics for the identification of novel therapeutic targets in cancer. Expert Rev. Proteom. 2018, 15, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Mao, J.; Yao, Y.; Wang, K.; Qiao, Q.; Fang, Z.; Ye, M. Sensitive profiling of cell surface proteome by using an optimized biotinylation method. J Proteom. 2019, 196, 33–41. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361. [Google Scholar] [CrossRef] [Green Version]
- Koehler Leman, J.; Ulmschneider, M.B.; Gray, J.J. Computational modeling of membrane proteins. Proteins: Struct. Funct. Bioinform. 2015, 83, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Funk, C.C.; Caballero, J.; Shah, N.; Rouleau, K.; Earls, J.C.; Soroceanu, L.; Foltz, G.; Cobbs, C.S.; Price, N.D.; et al. A Cell-Surface Membrane Protein Signature for Glioblastoma. Cell Syst. 2017, 4, 516–529.e7. [Google Scholar] [CrossRef] [Green Version]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell 2017, 32, 506–519.e5. [Google Scholar] [CrossRef] [Green Version]
- Coscia, F.; Lengyel, E.; Duraiswamy, J.; Ashcroft, B.; Bassani-Sternberg, M.; Wierer, M.; Johnson, A.; Wroblewski, K.; Montag, A.; Yamada, S.D.; et al. Multi-level Proteomics Identifies CT45 as a Chemosensitivity Mediator and Immunotherapy Target in Ovarian Cancer. Cell 2018, 175, 159–170.e16. [Google Scholar] [CrossRef] [Green Version]
- Everest-Dass, A.V.; Moh, E.S.X.; Ashwood, C.; Shathili, A.M.M.; Packer, N.H. Human disease glycomics: technology advances enabling protein glycosylation analysis—part 1. Expert Rev. Proteom. 2018, 15, 165–182. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, J.-L. Mass-spectrometry-based lipidomics. J. Sep. Sci. 2018, 41, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Bausch-Fluck, D.; Goldmann, U.; Muller, S.; van Oostrum, M.; Muller, M.; Schubert, O.T.; Wollscheid, B. The in silico human surfaceome. Proc. Natl. Acad. Sci. USA 2018, 115, E10988–E10997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudet, P.; Michel, P.A.; Zahn-Zabal, M.; Britan, A.; Cusin, I.; Domagalski, M.; Duek, P.D.; Gateau, A.; Gleizes, A.; Hinard, V.; et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2017, 45, D177–D182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, K.; Keerthikumar, S.; Goel, R.; Mathivanan, S.; Patankar, N.; Shafreen, B.; Renuse, S.; Pawar, H.; Ramachandra, Y.L.; Acharya, P.K.; et al. Human Proteinpedia: a unified discovery resource for proteomics research. Nucleic Acids Res. 2009, 37, D773–D781. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.P.; Hayes, C.A.; Struwe, W.B.; Wilkins, M.R.; Aoki-Kinoshita, K.F.; Harvey, D.J.; Rudd, P.M.; Kolarich, D.; Lisacek, F.; Karlsson, N.G.; et al. UniCarbKB: Putting the pieces together for glycomics research. PROTEOMICS 2011, 11, 4117–4121. [Google Scholar] [CrossRef]
- Foster, J.M.; Moreno, P.; Fabregat, A.; Hermjakob, H.; Steinbeck, C.; Apweiler, R.; Wakelam, M.J.O.; Vizcaíno, J.A. LipidHome: A Database of Theoretical Lipids Optimized for High Throughput Mass Spectrometry Lipidomics. PLoS ONE 2013, 8, e61951. [Google Scholar] [CrossRef] [Green Version]
- Nuñez-Prado, N.; Compte, M.; Harwood, S.; Álvarez-Méndez, A.; Lykkemark, S.; Sanz, L.; Álvarez-Vallina, L. The coming of age of engineered multivalent antibodies. Drug Discov. Today 2015, 20, 588–594. [Google Scholar] [CrossRef]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423. [Google Scholar] [CrossRef]
- Ansar, W.; Ghosh, S. Monoclonal Antibodies: A Tool in Clinical Research. Indian J. Clin. Med. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- KÖHler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Nair, R.R.; Fisher, E.M.C.; Cunningham, T.J. Humanising the mouse genome piece by piece. Nat. Commun. 2019, 10, 1845. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.C.; Liang, Q.; Ali, H.; Bayliss, L.; Beasley, A.; Bloomfield-Gerdes, T.; Bonoli, L.; Brown, R.; Campbell, J.; Carpenter, A.; et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat. Biotechnol. 2014, 32, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, M.S.; Williams, G.T.; Fox, R.O. Recombinant antibodies possessing novel effector functions. Nature 1984, 312, 604–608. [Google Scholar] [CrossRef]
- Riechmann, L.; Clark, M.; Waldmann, H.; Winter, G. Reshaping human antibodies for therapy. Nature 1988, 332, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Parola, C.; Neumeier, D.; Reddy, S.T. Integrating high-throughput screening and sequencing for monoclonal antibody discovery and engineering. Immunology 2018, 153, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Kamat, K.; Johnson, B.P.; Yin, C.C.; Scholler, N.; Abbott, K.L. Generation of a Fully Human scFv that binds Tumor-Specific Glycoforms. Sci. Rep. 2019, 9, 5101. [Google Scholar] [CrossRef]
- Li, D.; Hu, S.; Fan, Q.; Bao, W.; Zhou, W.; Xu, T.; Ye, T.; Liu, H.; Song, L. Phage display screening of TIGIT-specific antibody for antitumor immunotherapy. Biosci. Biotechnol. Biochem. 2019, 83, 1683–1696. [Google Scholar] [CrossRef]
- Jalilzadeh-Razin, S.; Mantegi, M.; Tohidkia, M.R.; Pazhang, Y.; Pourseif, M.M.; Barar, J.; Omidi, Y. Phage antibody library screening for the selection of novel high-affinity human single-chain variable fragment against gastrin receptor: An in silico and in vitro study. Daru 2019, 27, 21–34. [Google Scholar] [CrossRef]
- Hua, C.K.; Gacerez, A.T.; Sentman, C.L.; Ackerman, M.E. Development of unique cytotoxic chimeric antigen receptors based on human scFv targeting B7H6. Protein Eng. Des. Sel. 2017, 30, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Staehr, M.; Masakayan, R.; Tatake, I.J.; Purdon, T.J.; Wang, X.; Wang, P.; Liu, H.; Xu, Y.; Garrett-Thomson, S.C.; et al. Development and Evaluation of an Optimal Human Single-Chain Variable Fragment-Derived BCMA-Targeted CAR T Cell Vector. Mol. Ther. 2018, 26, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassev, D.V.; Cheng, M.; Cheung, N.K. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene 2012, 19, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ahmed, M.; Tassev, D.V.; Hasan, A.; Kuo, T.Y.; Guo, H.F.; O’Reilly, R.J.; Cheung, N.K. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia 2015, 29, 2238–2247. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef] [Green Version]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [Green Version]
- Gohil, S.H.; Paredes-Moscosso, S.R.; Harrasser, M.; Vezzalini, M.; Scarpa, A.; Morris, E.; Davidoff, A.M.; Sorio, C.; Nathwani, A.C.; Della Peruta, M. An ROR1 bi-specific T-cell engager provides effective targeting and cytotoxicity against a range of solid tumors. Oncoimmunology 2017, 6, e1326437. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.S.; Reuter, V.E.; Slovin, S.F.; Scher, H.I.; Livingston, P.O. Expression of potential target antigens for immunotherapy on primary and metastatic prostate cancers. Clin. Cancer Res. 1998, 4, 295. [Google Scholar]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2017, 24, 20. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Qin, W.; Liu, T.; Jiang, D.; Cui, L.; Liu, X.; Fang, Y.; Tang, X.; Jin, H.; Qian, Q. PiggyBac-engineered T cells expressing a glypican-3-specific chimeric antigen receptor show potent activities against hepatocellular carcinoma. Immunobiology 2019. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.B.; Choudhari, N.; Perazzelli, J.; Storm, J.; Hofmann, T.J.; Jain, P.; Storm, P.B.; Pardi, N.; Weissman, D.; Waanders, A.J.; et al. Purification of mRNA Encoding Chimeric Antigen Receptor Is Critical for Generation of a Robust T-Cell Response. Hum. Gene Ther. 2019, 30, 168–178. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
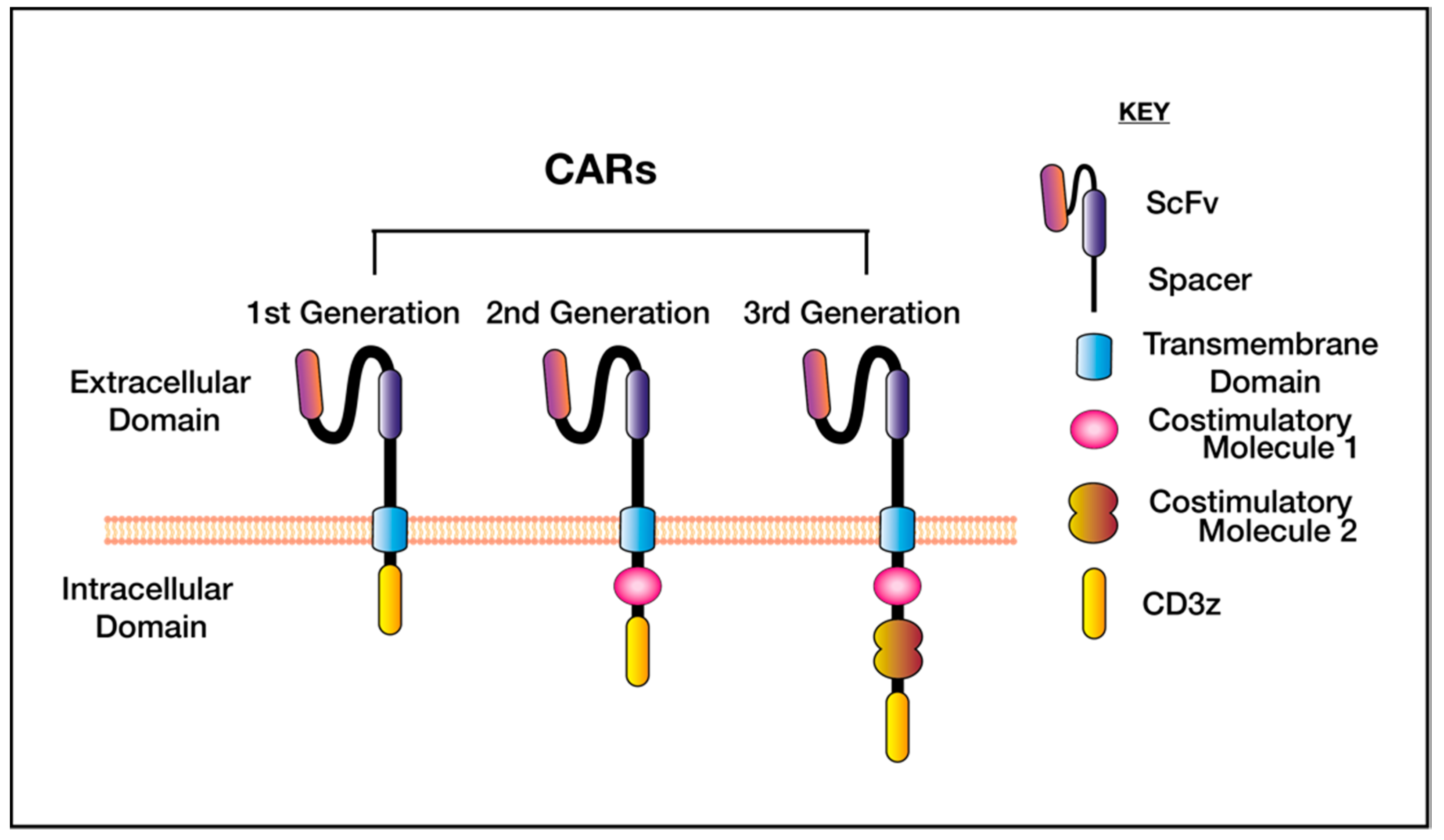{kind=link}
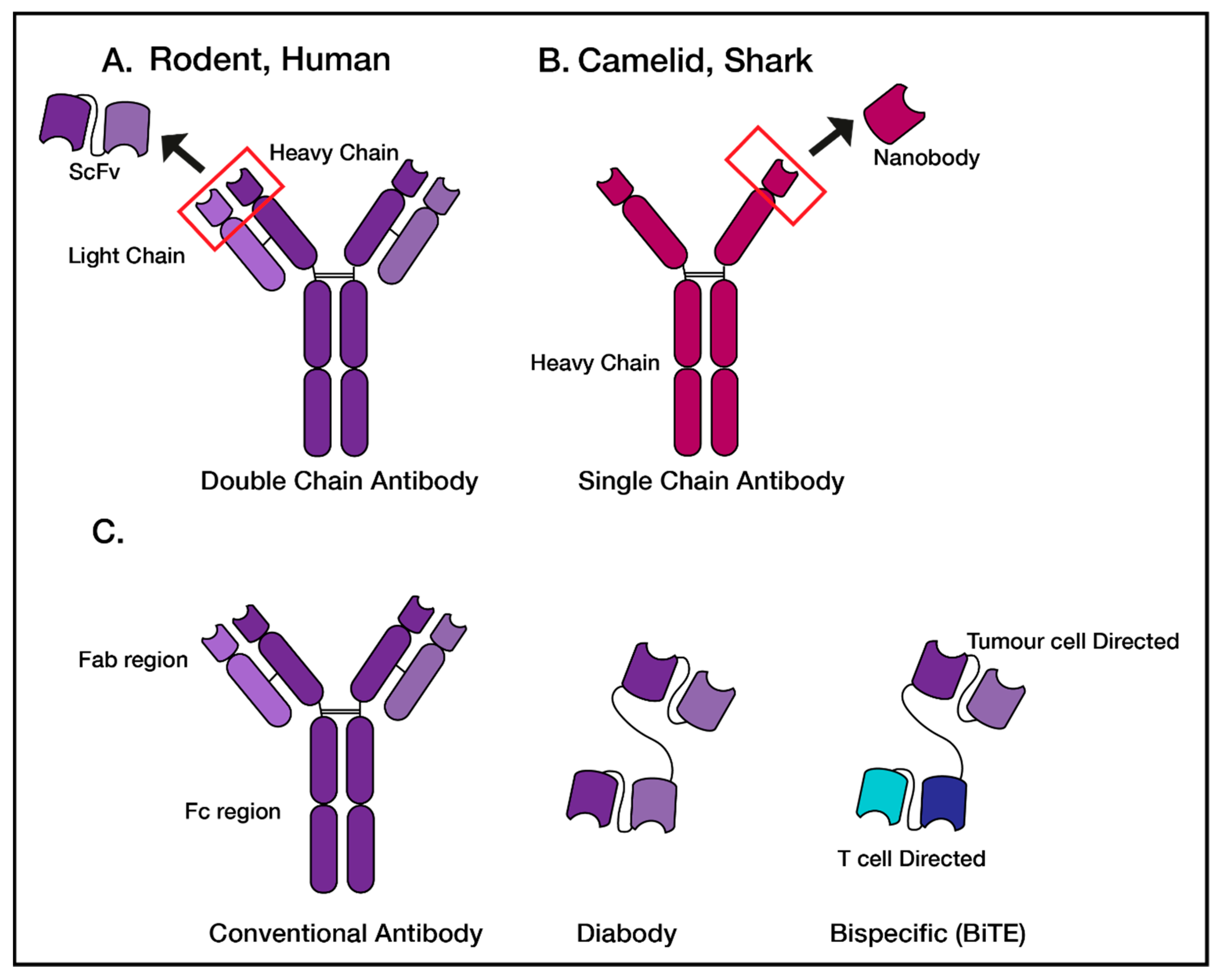{kind=link}
| Type of Antigen | Expression | Advantage | Disadvantage | Example |
|---|---|---|---|---|
| Tumour Specific | Expressed only on malignant tissue | Unlikely to cause on target off tumour effects | Difficult to identify tumour specific targets—limited pool of possible proteins. Tumour heterogeneity between patients may make pan marker identification difficult. | EGFRvIII [26,27] |
| Tumour Associated | Expressed at low levels on healthy tissue or organs, expressed at a higher level on malignant tissue. | Increased likelihood of target identification | Possibility to cause on target off tumour effects. Extensive preclinical validation required to determine safety. | HER2 [28,29] |
| Embryonic Cancer Germline Antigen | Frequent expression on malignant cells, expressed on embryonic tissues and minimal expression on healthy adult tissue. | Regulators of many important cellular function pathways. Minimal expression on healthy tissue indicates this class of antigens may be potentially safer to target therapeutically. | Application of CARs targeting these antigens could result in the destruction of healthy reproductive tissue. Limited pool of antigens. | IL-13Rα2 [14] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbott, R.C.; Cross, R.S.; Jenkins, M.R. Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies. Int. J. Mol. Sci. 2020, 21, 515. https://doi.org/10.3390/ijms21020515
Abbott RC, Cross RS, Jenkins MR. Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies. International Journal of Molecular Sciences. 2020; 21(2):515. https://doi.org/10.3390/ijms21020515
Chicago/Turabian StyleAbbott, Rebecca C., Ryan S. Cross, and Misty R. Jenkins. 2020. "Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies" International Journal of Molecular Sciences 21, no. 2: 515. https://doi.org/10.3390/ijms21020515
APA StyleAbbott, R. C., Cross, R. S., & Jenkins, M. R. (2020). Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies. International Journal of Molecular Sciences, 21(2), 515. https://doi.org/10.3390/ijms21020515