Molecular and Regulatory Mechanisms of Desensitization and Resensitization of GABAA Receptors with a Special Reference to Propofol/Barbiturate



Abstract
:1. Introduction
2. PRIP-1/2 are Involved in Desensitization and Resensitization of GABAAR-Mediated Currents
3. [Ca2+]i Dependence of Desensitization and Resensitization of GABAAR-Mediated Currents and Their Abolishment by a Calcineurin Inhibitor
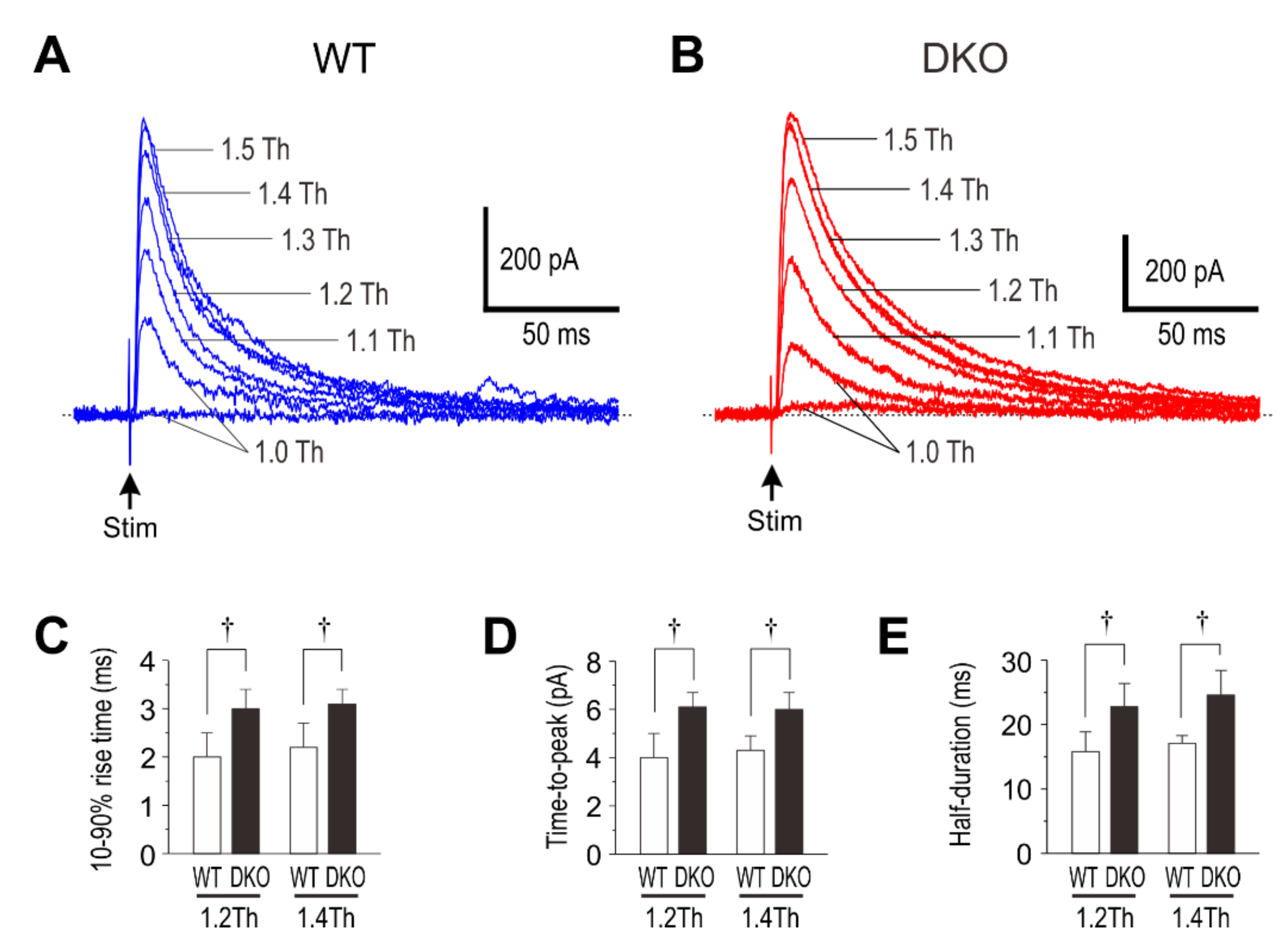
4. Deletion of PRIP-1/2 Prolongs eIPSCs in Layer II/III Pyramidal Cells
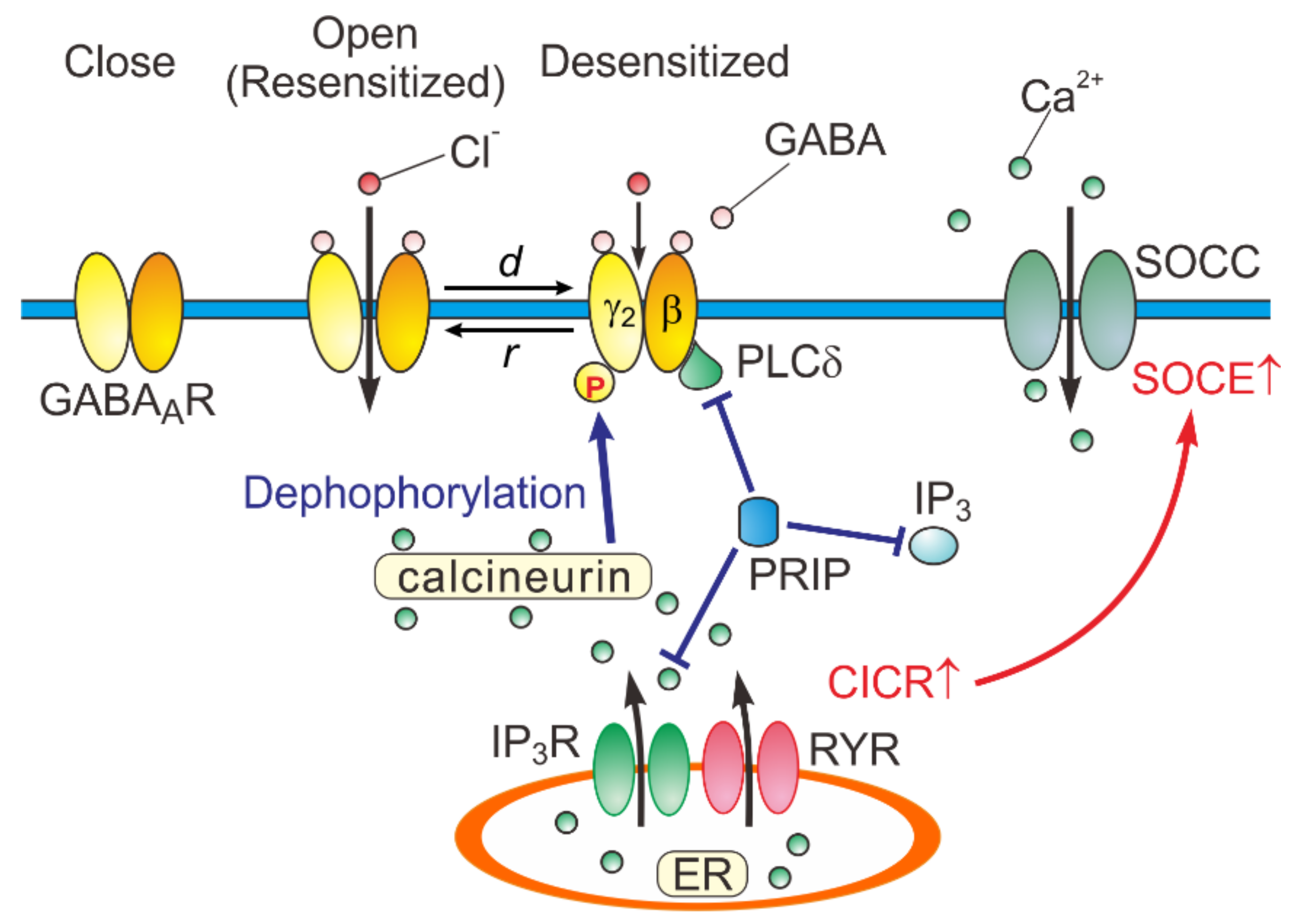
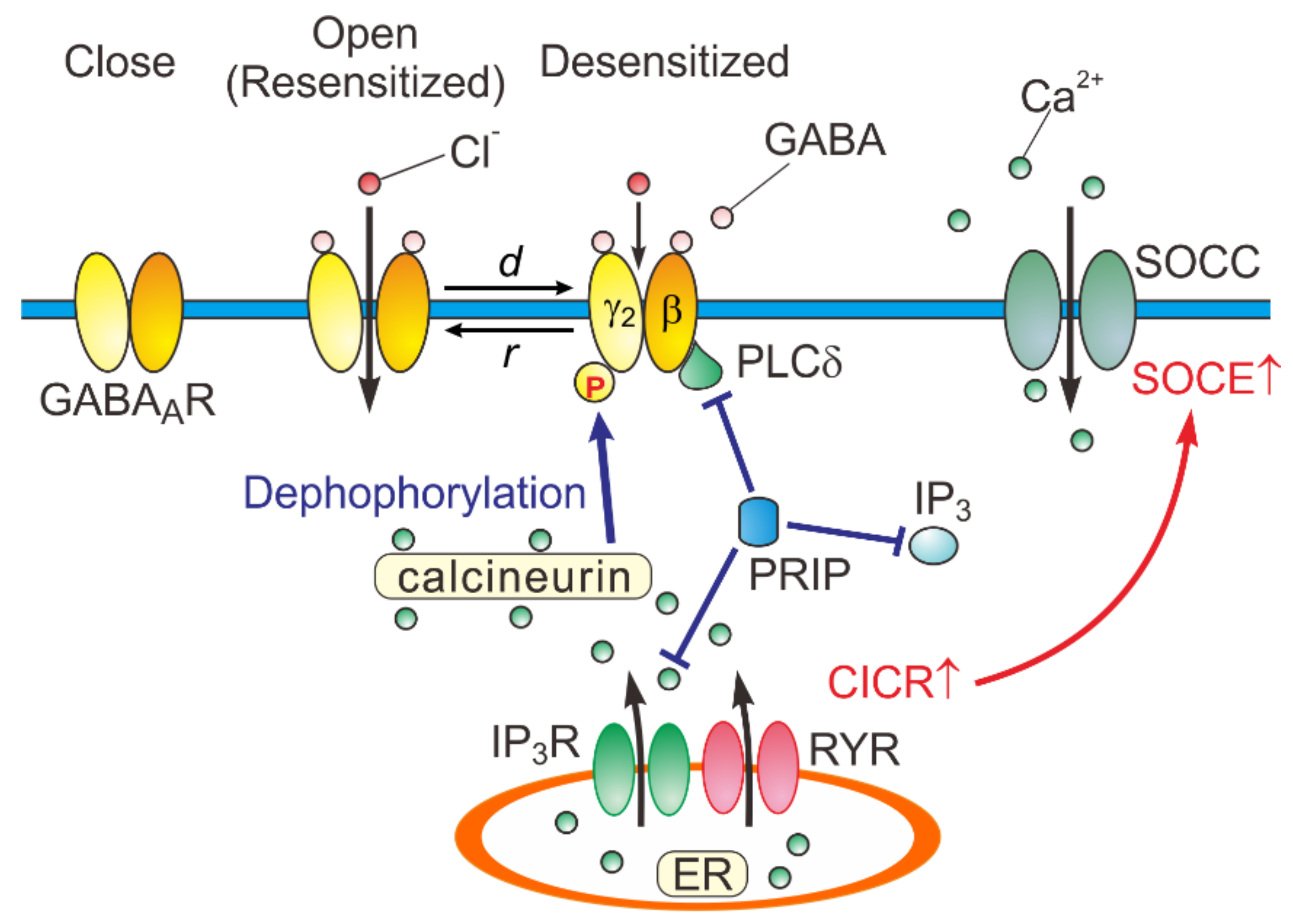
5. A Possible Kinetic Mechanism Underlying the Generation of the Hump-Like Tail-Currents and the Prolongation of eIPSCs
6. Physiological Significance of Desensitization and Resensitization of GABAAR-Mediated Currents
7. Clinical Significance of Desensitization and Resensitization of GABAAR-Mediated Currents
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CICR | Ca2+-induced Ca2+ release |
| DKO | double-knockout |
| GABAAR | GABAA receptor |
| GABARAP | GABAAR-associated protein |
| IPSC | inhibitory postsynaptic current |
| IPSP | inhibitory postsynaptic potential |
| NMDA | N-methyl-D-aspartate |
| PLC | phospholipase C |
| PRIP | phospholipase C-related catalytically inactive protein |
| RYR | ryanodine receptor |
| SOCC | store-operated Ca2+ channel |
| SOCE | store-operated Ca2+ entry |
References
- Jones, M.V.; Westbrook, G.L. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996, 19, 96–101. [Google Scholar] [CrossRef]
- Keramidas, A.; Lynch, J.W. An outline of desensitization in pentameric ligand-gated ion channel receptors. Cell Mol. Life Sci. 2013, 70, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.V.; Westbrook, G.L. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 1995, 15, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.V.; Westbrook, G.L. Shaping of IPSCs by endogenous calcineurin activity. J. Neurosci. 1997, 17, 7626–7633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overstreet, L.S.; Jones, M.V.; Westbrook, G.L. Slow desensitization regulates the availability of synaptic GABAA receptors. J. Neurosci. 2000, 20, 7914–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qazi, S.; Caberlin, M.; Nigam, N. Mechanism of psychoactive drug action in the brain: Simulation modeling of GABAA receptor interactions at non-equilibrium conditions. Curr. Pharm. Des. 2007, 13, 1437–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehrani, M.H.; Hablitz, J.J.; Barnes, E.M., Jr. cAMP increases the rate of GABAA receptor desensitization in chick cortical neurons. Synapse 1989, 4, 126–131. [Google Scholar] [CrossRef]
- Moss, S.J.; Smart, T.G.; Blackstone, C.D.; Huganir, R.L. Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science 1992, 257, 661–665. [Google Scholar] [CrossRef]
- Robello, M.; Amico, C.; Cupello, A. Evidence of two populations of GABAA receptors in cerebellar granule cells in culture: Different desensitization kinetics, pharmacology, serine/threonine kinase sensitivity, and localization. Biochem. Biophys. Res. Commun. 1999, 266, 603–608. [Google Scholar] [CrossRef]
- Hinkle, D.J.; Macdonald, R.L. Beta subunit phosphorylation selectively increases fast desensitization and prolongs deactivation of α1β1γ2L and α1β3γ2L GABAA receptor currents. J. Neurosci. 2003, 23, 11698–11710. [Google Scholar] [CrossRef] [Green Version]
- Krishek, B.J.; Xie, X.; Blackstone, C.; Huganir, R.L.; Moss, S.J.; Smart, T.G. Regulation of GABAA receptor function by protein kinase C phosphorylation. Neuron 1994, 12, 1081–1095. [Google Scholar] [CrossRef]
- Wang, R.A.; Cheng, G.; Kolaj, M.; Randic, M. Alpha-subunit of calcium/calmodulin-dependent protein kinase II enhances gamma-aminobutyric acid and inhibitory synaptic responses of rat neurons in vitro. J. Neurophysiol. 1995, 73, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- Martina, M.; Mozrzymas, J.W.; Boddeke, H.W.; Cherubini, E. The calcineurin inhibitor cyclosporin A-cyclophilin A complex reduces desensitization of GABAA-mediated responses in acutely dissociated rat hippocampal neurons. Neurosci. Lett. 1996, 215, 95–98. [Google Scholar] [CrossRef]
- Muir, J.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Smith, K.R.; Griffin, L.D.; Kittler, J.T. NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the 2 subunit. Proc. Natl. Acad. Sci. USA 2010, 107, 16679–16684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, H.; Saito, M.; Sato, H.; Tanaka, T.; Ogawa, T.; Yatani, H.; Kanematsu, T.; Hirata, M.; Kang, Y. Deletion of phospholipase C-related inactive protein-1/2 enhances desensitization and resensitization of GABAA receptors in pyramidal cells of the barrel cortex. Pflugers Arch. 2015, 467, 267–284. [Google Scholar] [CrossRef]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Luscher, B.; Fuchs, T.; Kilpatrick, C.L. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 2011, 70, 385–409. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, H.; Vicini, S.; Olsen, R.W. The γ-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc. Natl. Acad. Sci. USA 2000, 97, 11557–11562. [Google Scholar] [CrossRef] [Green Version]
- Orser, B.A.; Wang, L.Y.; Pennefather, P.S.; MacDonald, J.F. Propofol modulates activation and desensitization of GABAA receptors in cultured murine hippocampal neurons. J. Neurosci. 1994, 14, 7747–7760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.X.; Lu, H.; Dong, X.P.; Liu, J.; Xu, T.L. Kinetics of etomidate actions on GABAA receptors in the rat spinal dorsal horn neurons. Brain Res. 2002, 953, 93–100. [Google Scholar] [CrossRef]
- Akaike, N.; Hattori, K.; Inomata, N.; Oomura, Y. γ-Aminobutyric-acid- and pentobarbitone-gated chloride currents in internally perfused frog sensory neurones. J. Physiol. 1985, 360, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Akaike, N.; Maruyama, T.; Tokutomi, N. Kinetic properties of the pentobarbitone-gated chloride current in frog sensory neurones. J. Physiol. 1987, 394, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M.; Donevan, S.D.; Rogawski, M.A. Direct activation of GABAA receptors by barbiturates in cultured rat hippocampal neurons. J. Physiol. 1996, 497, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Gingrich, K.J.; Burkat, P.M.; Roberts, W.A. Pentobarbital produces activation and block of α1β2γ2S GABAA receptors in rapidly perfused whole cells and membrane patches: Divergent results can be explained by pharmacokinetics. J. Gen. Physiol. 2009, 133, 171–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.J.; Bianchi, M.T.; Macdonald, R.L. Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABAA receptor currents. Mol. Pharmacol. 2004, 66, 988–1003. [Google Scholar] [CrossRef]
- Krampfl, K.; Wolfes, H.; Dengler, R.; Bufler, J. Kinetic analysis of the agonistic and blocking properties of pentobarbital on recombinant rat α1β2γ2S GABAA receptor channels. Eur. J. Pharmacol. 2002, 435, 1–8. [Google Scholar] [CrossRef]
- Thompson, S.A.; Whiting, P.J.; Wafford, K.A. Barbiturate interactions at the human GABAA receptor: Dependence on receptor subunit combination. Br. J. Pharmacol. 1996, 117, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Wooltorton, J.R.; Moss, S.J.; Smart, T.G. Pharmacological and physiological characterization of murine homomeric β3 GABAA receptors. Eur. J. Neurosci. 1997, 9, 2225–2235. [Google Scholar] [CrossRef]
- Ziemba, A.M.; Forman, S.A. Correction for inhibition leads to an allosteric co-agonist model for pentobarbital modulation and activation of α1β3γ2L GABAA receptors. PLoS ONE 2016, 11, e0154031. [Google Scholar] [CrossRef] [Green Version]
- Brotherton, A.L.; Hamilton, E.P.; Kloss, H.G.; Hammond, D.A. Propofol for treatment of refractory alcohol withdrawal syndrome: A review of the literature. Pharmacotherapy 2016, 36, 433–442. [Google Scholar] [CrossRef]
- MacKinnon, G.L.; Parker, W.A. Benzodiazepine withdrawal syndrome: A literature review and evaluation. Am. J. Drug Alcohol Abuse 1982, 9, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Katz, A. The role of barbiturates for alcohol withdrawal syndrome. Psychosomatics 2016, 57, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Oomura, Y.; Yakushiji, T.; Akaike, N. Intracellular calcium ions decrease the affinity of the GABA receptor. Nature 1986, 324, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Mozrzymas, J.W.; Cherubini, E. Changes in intracellular calcium concentration affect desensitization of GABAA receptors in acutely dissociated P2–P6 rat hippocampal neurons. J. Neurophysiol. 1998, 79, 1321–1328. [Google Scholar] [CrossRef]
- Toyoda, H.; Saito, M.; Sato, H.; Kawano, T.; Kawakami, S.; Yatani, H.; Kanematsu, T.; Hirata, M.; Kang, Y. Enhanced lateral inhibition in the barrel cortex by deletion of phospholipase C-related catalytically inactive protein-1/2 in mice. Pflugers Arch. 2015, 467, 1445–1456. [Google Scholar] [CrossRef]
- Mizokami, A.; Tanaka, H.; Ishibashi, H.; Umebayashi, H.; Fukami, K.; Takenawa, T.; Nakayama, K.I.; Yokoyama, T.; Nabekura, J.; Kanematsu, T.; et al. GABAA receptor subunit alteration-dependent diazepam insensitivity in the cerebellum of phospholipase C-related inactive protein knockout mice. J. Neurochem. 2010, 114, 302–310. [Google Scholar] [CrossRef]
- Mizokami, A.; Kanematsu, T.; Ishibashi, H.; Yamaguchi, T.; Tanida, I.; Takenaka, K.; Nakayama, K.I.; Fukami, K.; Takenawa, T.; Kominami, E.; et al. Phospholipase C-related inactive protein is involved in trafficking of γ2 subunit-containing GABAA receptors to the cell surface. J. Neurosci. 2007, 27, 1692–1701. [Google Scholar] [CrossRef] [Green Version]
- Kanematsu, T.; Jang, I.S.; Yamaguchi, T.; Nagahama, H.; Yoshimura, K.; Hidaka, K.; Matsuda, M.; Takeuchi, H.; Misumi, Y.; Nakayama, K.; et al. Role of the PLC-related, catalytically inactive protein p130 in GABAA receptor function. EMBO J. 2002, 21, 1004–1011. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Ghansah, E.; Chen, Y.; Ye, J.; Weiss, D.S. Desensitization mechanism of GABA receptors revealed by single oocyte binding and receptor function. J. Neurosci. 2002, 22, 7982–7990. [Google Scholar] [CrossRef]
- Nicholson, M.W.; Sweeney, A.; Pekle, E.; Alam, S.; Ali, A.B.; Duchen, M.; Jovanovic, J.N. Diazepam-induced loss of inhibitory synapses mediated by PLCδ/Ca2+/calcineurin signalling downstream of GABAA receptors. Mol. Psychiatry 2018, 23, 1851–1867. [Google Scholar] [CrossRef]
- Armstrong-James, M.; Fox, K.; Das-Gupta, A. Flow of excitation within rat barrel cortex on striking a single vibrissa. J. Neurophysiol. 1992, 68, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.C.; Sakmann, B. Functionally independent columns of rat somatosensory barrel cortex revealed with voltage-sensitive dye imaging. J. Neurosci. 2001, 21, 8435–8446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Shimanuki, Y.; Saito, M.; Toyoda, H.; Nokubi, T.; Maeda, Y.; Yamamoto, T.; Kang, Y. Differential columnar processing in local circuits of barrel and insular cortices. J. Neurosci. 2008, 28, 3076–3089. [Google Scholar] [CrossRef] [PubMed]
- Laaris, N.; Carlson, G.C.; Keller, A. Thalamic-evoked synaptic interactions in barrel cortex revealed by optical imaging. J. Neurosci. 2000, 20, 1529–1537. [Google Scholar] [CrossRef] [Green Version]
- Laaris, N.; Keller, A. Functional independence of layer IV barrels. J. Neurophysiol. 2002, 87, 1028–1034. [Google Scholar] [CrossRef] [Green Version]
- Anstrom, K.K.; Schallert, T.; Woodlee, M.T.; Shattuck, A.; Roberts, D.C. Repetitive vibrissae-elicited forelimb placing before and immediately after unilateral 6-hydroxydopamine improves outcome in a model of Parkinson’s disease. Behav. Brain Res. 2007, 179, 183–191. [Google Scholar] [CrossRef]
- Bayard, M.; McIntyre, J.; Hill, K.R.; Woodside, J., Jr. Alcohol withdrawal syndrome. Am. Fam. Physician 2004, 69, 1443–1450. [Google Scholar]
- McKeon, A.; Frye, M.A.; Delanty, N. The alcohol withdrawal syndrome. J. Neurol. Neurosurg. Psychiatry 2008, 79, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Onyett, S.R. The benzodiazepine withdrawal syndrome and its management. J. R. Coll. Gen. Pract. 1989, 39, 160–163. [Google Scholar]
- Petursson, H. The benzodiazepine withdrawal syndrome. Addiction 1994, 89, 1455–1459. [Google Scholar] [CrossRef]
- Allison, C.; Pratt, J.A. Neuroadaptive processes in GABAergic and glutamatergic systems in benzodiazepine dependence. Pharmacol. Ther. 2003, 98, 171–195. [Google Scholar] [CrossRef]
- Chandler, L.J.; Newsom, H.; Sumners, C.; Crews, F. Chronic ethanol exposure potentiates NMDA excitotoxicity in cerebral cortical neurons. J. Neurochem. 1993, 60, 1578–1581. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.A.; Valverius, P.; Hudspith, M.; Tabakoff, B. Ethanol withdrawal seizures and the NMDA receptor complex. Eur. J. Pharmacol. 1990, 176, 289–296. [Google Scholar] [CrossRef]
- Koff, J.M.; Pritchard, G.A.; Greenblatt, D.J.; Miller, L.G. The NMDA receptor competitive antagonist CPP modulates benzodiazepine tolerance and discontinuation. Pharmacology 1997, 55, 217–227. [Google Scholar] [CrossRef]
- Tsuda, M.; Shimizu, N.; Yajima, Y.; Suzuki, T.; Misawa, M. Hypersusceptibility to DMCM-induced seizures during diazepam withdrawal in mice: Evidence for upregulation of NMDA receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1998, 357, 309–315. [Google Scholar] [CrossRef]
- Olsen, R.W.; Liang, J.; Cagetti, E.; Spigelman, I. Plasticity of GABAA receptors in brains of rats treated with chronic intermittent ethanol. Neurochem. Res. 2005, 30, 1579–1588. [Google Scholar] [CrossRef]
- Olsen, R.W.; Spigelman, I. GABAA Receptor Plasticity in Alcohol Withdrawal. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012. [Google Scholar]
- Tan, K.R.; Rudolph, U.; Luscher, C. Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends Neurosci. 2011, 34, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Wafford, K.A. GABAA receptor subtypes: Any clues to the mechanism of benzodiazepine dependence? Curr. Opin. Pharmacol. 2005, 5, 47–52. [Google Scholar] [CrossRef]
- Preskorn, S.H.; Denner, L.J. Benzodiazepines and withdrawal psychosis. Report of three cases. JAMA 1977, 237, 36–38. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinases/ Molecules | Neuron/Recombinant GABAARs | Effects | References |
|---|---|---|---|
| PKA | Chick cortical neurons | increases desensitization | [7] |
| Rat sympathetic ganglion neurons | decreases peak amplitude and increases fast desensitization | [8] | |
| Rat cerebellar granule cells | decreases fast desensitization | [9] | |
| α1β1γ2S, α1β3γ2LS | increases desensitization and slows deactivation | [10] | |
| PKC | α1β1 | decreases fast desensitization | [11] |
| PKG | Rat cerebellar granule cells | decreases fast desensitization | [9] |
| CaMKII | Rat spinal dorsal horn neurons | decreases desensitization | [12] |
| Calcineurin | Rat hippocampal neurons | increases desensitization and slows deactivation | [4] |
| PRIP | Mouse cortical pyramidal neurons | PRIP deletion increases desensitization and generates hump-like currents through increased calcineurin activity | [15] |
| GABARAP | α1β2γ2L | promotes clustering of GABAARs, facilitates deactivation, and slows desensitization | [18] |
| Drugs | Neurons/ Recombinant GABAARs | Effects | Refs. |
|---|---|---|---|
| Anesthetics | |||
| Propofol | Mouse hippocampal neurons | slows deactivation and increases apparent desensitization of GABA responses at low concentrations and directly elicits after-responses upon washout at high concentrations | [19] |
| Etomidate | Rat spinal dorsal horn neurons | slows deactivation of GABA responses at low concentrations while directly eliciting tail currents upon washout at high concentrations | [20] |
| Barbiturate | |||
| Pentobarbital | Frog sensory neurons | slows deactivation and increases apparent desensitization of GABA responses at low concentrations and directly elicits hump currents upon washout at high concentrations | [21,22] |
| Rat hippocampal neurons | slows deactivation and increases apparent desensitization of GABA responses at low concentrations and directly elicits rebound currents upon washout at high concentrations | [23] | |
| α1β2γ2L | directly elicits tail currents upon washout at high concentrations | [24,26] | |
| α1β3γ2L | slows deactivation and increases apparent desensitization of GABA responses at low concentrations and directly elicits rebound currents upon washout at high concentrations | [25] | |
| α1β2γ2S, α6β2γ2S | directly elicits hump currents upon washout at high concentrations | [27] | |
| β3 | increases apparent desensitization of GABA responses and directly elicits rebound currents upon washout at high concentrations | [28] | |
| α1β3γ2L | directly elicits tail currents upon washout at high concentrations | [29] | |
| Phenobarbital | Rat hippocampal neurons | slows deactivation and increases apparent desensitization of GABA responses at low concentrations and directly elicits rebound currents upon washout at high concentrations | [23] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Y.; Saito, M.; Toyoda, H. Molecular and Regulatory Mechanisms of Desensitization and Resensitization of GABAA Receptors with a Special Reference to Propofol/Barbiturate. Int. J. Mol. Sci. 2020, 21, 563. https://doi.org/10.3390/ijms21020563
Kang Y, Saito M, Toyoda H. Molecular and Regulatory Mechanisms of Desensitization and Resensitization of GABAA Receptors with a Special Reference to Propofol/Barbiturate. International Journal of Molecular Sciences. 2020; 21(2):563. https://doi.org/10.3390/ijms21020563
Chicago/Turabian StyleKang, Youngnam, Mitsuru Saito, and Hiroki Toyoda. 2020. "Molecular and Regulatory Mechanisms of Desensitization and Resensitization of GABAA Receptors with a Special Reference to Propofol/Barbiturate" International Journal of Molecular Sciences 21, no. 2: 563. https://doi.org/10.3390/ijms21020563