4.2. Synthesis
4.2.1. General Method for Photoinduced Addition of Thiols to Exoglycals or Sugar Exomethylene Derivatives
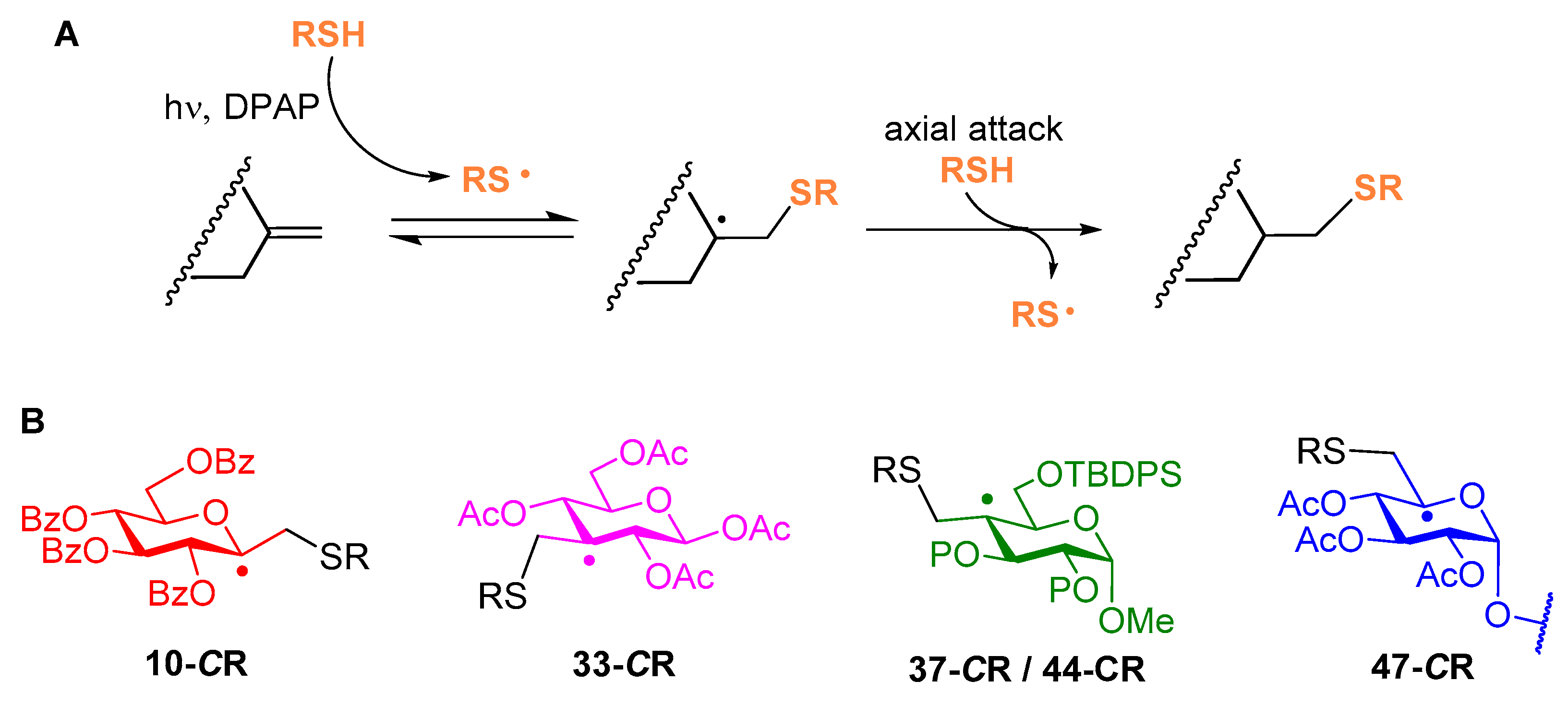
Sugar thiol (1.2–1.5 equiv.) and 2,2-dimethoxy-2-phenylacetophenone (DPAP, 0.10 equiv/alkene) were added to a solution of the starting unsaturated monosaccharide in dry toluene (it is indicated when some other solvent was used) (7–8 mL/1 mmol alkene). The solution was irradiated at room temperature (it is indicated when the reaction was performed at lower temperature) for 15 min. The progress of the reaction was monitored by TLC after this reaction period and irradiation and addition of DPAP were repeated if necessary, once or twice more. In these cases, no additional thiol was added to the reaction mixture. Subsequently, the solution was concentrated and the residue was purified by column chromatography or flash column chromatography.
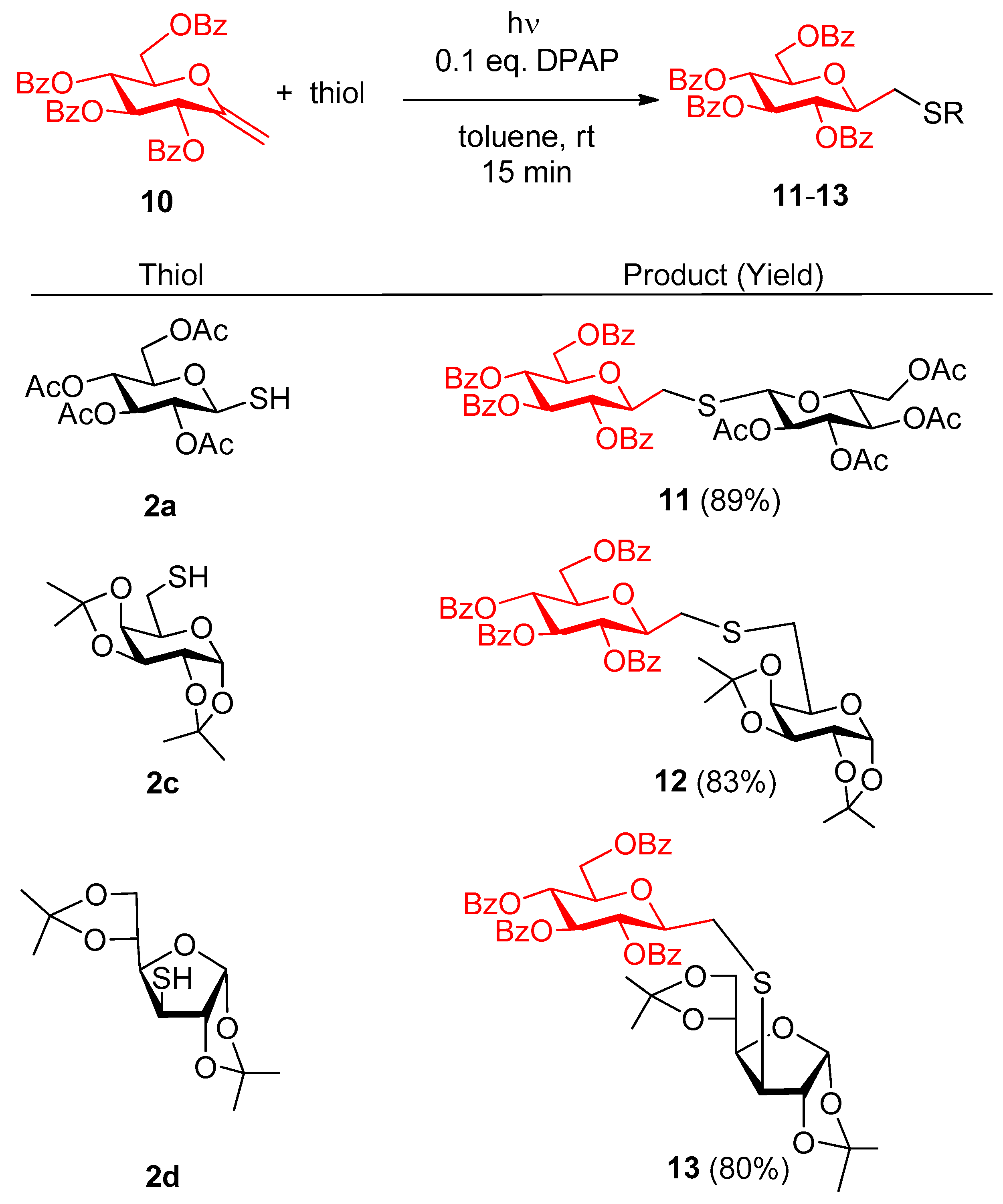
4.2.2. 2,6-Anhydro-3,4,5,7-tetra-O-benzoyl-1-deoxy-1-S-(2′,3′,4′,6′-tetra-O-acetyl-β-d-glucopyranosyl)-1-thio-d-glycero-d-gulo-heptitol (11)
Exo-glucal 10 (50 mg, 0.084 mmol) and thiol 2a (44 mg, 0.12 mmol) were reacted according to the general method. The crude product was purified by silica gel chromatography in hexane/ethyl acetate 6/4 to give compound 11 (72 mg).
Yield: 89%, white foam. [α]D20 = −14.6 (c 0.2, CHCl3). Rf 0.45 (CH2Cl2/acetone 95/5 ). 1H NMR (360 MHz, CDCl3): δ = 8.10–7.79 (m, 8H, arom), 7.59-7.24 (m, 12H, arom), 5.89 (t, J = 9.6 Hz, 1H), 5.70 (t, J = 9.8 Hz, 1H), 5.54 (t, J = 9.7 Hz, 1H), 5.25 (t, J = 9.6 Hz, 1H), 5.09 (t, J = 9.7 Hz, 1H), 4.48 (t, J = 9.8 Hz, 1H), 4.73 (d, J = 10 Hz, 1H, H-1′), 4.72 (dd, J = 9.6, 2.1 Hz, 2H), 4.50 (dd, J = 12.2, 5.2 Hz, 1H), 4.24–4.02 (m, 4H), 3.70–3.65 (m, 1H), 3.13 (dd, J = 14.6, 9.0 Hz, 1H, SCH2a), 2.81 (dd, J = 14.6, 2.5 Hz, 1H, SCH2b), 2.11 (s, 3H, COCH3), 2.05 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 1.98 (s, 3H, COCH3). 13C NMR (91 MHz, CDCl3): δ = 170.6, 170.0, 169.3, 169.3, 166.0, 165.8, 165.1, 165.1 (8C, 8×CO), 133.4-128.2 (24 C, arom), 82.9, 78.5, 76.3, 75.7, 74.3, 73.8, 71.4, 70.2, 69.4, 68.1 (10C, skeleton carbons), 62.8, 61.7 (C-7, C-6’), 30.5 (SCH2), 20.7, 20.5, 20.5, 20.5 (4C, 4×COCH3). MS (ESI-TOF) m/z: [M + Na]+ Calcd for C49H48O18NaS 979.2439; Found 979.2459; [M + Na]+.
4.2.3. 2,6-Anhydro-3,4,5,7-tetra-O-benzoyl-1-deoxy-1-S-(1′,2′:3′,4′-di-O-isopropylidene-α-d-galactopyranose-6′-yl)-1-thio-d-glycero-d-gulo-heptitol (12)
Exo-glucal 10 (59 mg, 0.10 mmol) and thiol 2c (42 mg, 0.15 mmol) were reacted according to the general method. The crude product was purified by silica gel chromatography in CH2Cl2/acetone 95/5 to give compound 12 (70 mg).
Yield: 83%, colorless syrup. [α]D20 = −11.4 (c 0.9, CHCl3). Rf 0.45, (CH2Cl2/acetone 9/1). 1H NMR (360 MHz, CDCl3): δ = 8.19–7.73 (m, 8H, arom.), 7.64–7.15 (m, 12H, arom.), 5.89 (t, J = 9.6 Hz, 1H), 5.63 (t, J = 9.6 Hz, 1H), 5.50 (t, J = 9.6 Hz, 1H), 5.45 (d, J = 5.1 Hz, 1H, H-1′), 4.64 (dd, J = 12.2, 2.8 Hz, 1H), 4.52 (dd, J = 7.9, 2.3 Hz, 1H), 4.44 (dd, J = 12.2, 5.2 Hz, 1H), 4.25 (dd, J = 5.1, 2.4 Hz, 1H), 4.21 (dd, J = 7.9, 1.8 Hz, 1H), 4.18–4.09 (m, 1H), 4.10–3.98 (m, 1H), 3.85 (t, J = 6.2 Hz, 1H), 3.00–2.73 (m, 4H, 2×SCH2); 1.50 (s, 3H ,CH3), 1.38 (s, 3H ,CH3), 1.27 (s, 3H ,CH3), 1.26 (s, 3H, CH3); 13C NMR (91 MHz, CDCl3): δ =166.3, 166.0, 165.5 and 165.4 (4C, 4×COPh), 133.5, 133.3, 133.2, 130.0, 129.8, 129.0, 128.5 and 128.4 (24C, arom.), 109.3 and 108.6 (2C, 2×Cq, i-propylidene), 96.7 (C-1), 79.8, 76.4, 74.4, 72.2, 71.9, 71.1, 70.6, 69.9 and 67.3 (9C, skeleton carbons), 63.5 (C-7), 33.3 (C-6’), 32.7 (SCH2), 26.2, 26.0, 25.0 and 24.5 (4C, 4×CCH3). MS (ESI-TOF) m/z: [M + Na]+ Calcd for C47H48O14NaS 891.2662; Found 891.2657 [M + Na]+.
4.2.4. 2,6-Anhydro-3,4,5,7-tetra-O-benzoyl-1-deoxy-1-S-(1′,2′:5′,6′-di-O-isopropylidene-α-d-glucopyranose-3′-yl)-1-thio-d-glycero-d-gulo-heptitol (13)
Exo-glucal 10 (59 mg, 0.10 mmol) and thiol 2d (42 mg, 1.5 mmol) were reacted, according to the general method. The crude product was purified by silica gel chromatography in hexane/acetone 8/2, and then in CH2Cl2/acetone 95/5 to give compound 13 (69 mg).
Yield: 80%, colorless syrup. [α]D20 = +2.1 (c 0.5, CHCl3). Rf 0.67 (CH2Cl2/acetone 9/1). 1H NMR (360 MHz, CDCl3): δ 8.05–7.79 (m, 8H, arom), 7.55–7.23 (m, 12H, arom), 5.92 (t, 1H, J = 9.6 Hz), 5.78 (d, 1H, J = 3.4 Hz,H-1), 5.69 (t, 1H, J = 9.7 Hz), 5.61 (t, 1H, J = 9.6 Hz), 4.72 (d, 1H, J = 3.5 Hz), 4.63 (dd, 1H, J = 3.1 Hz, J = 12.2 Hz), 4.51 (dd, 1H, J = 5.1 Hz, J = 12.1 Hz), 4.39–4.34 (m, 1H), 4.19–4.12 (m, 2H), 4.11–4.03 (m, 2H), 3.97 (dd, 1H, J = 5.1 Hz, J = 8.6 Hz), 3.55 (d, 1H, J= 3.6 Hz), 3.04–2.92 (m, 2H, SCH2), 1.41 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.35 (s, 3H, CH3), 1.22 (s, 3H, CH3); 13C NMR (90 MHz, CDCl3): δ = 166.0, 165.8, 165.3 and 165.1 (4C, 4×COPh), 133.5–128.2 (24C, arom), 111.8 and 109.4 (2C, 2×Cq, i-propylidene), 104.7 (C-1), 86.0, 80.1, 78.7, 76.1, 74.2, 73.9, 71.8 and 69.7 (8C, skeleton carbons), 67.6 (C-7), 63.3 (C-6′), 53.2 (C-3′), 33.8 (SCH2), 26.8, 26.5, 26.1 and 25.2 (4C, 4×CCH3); MS (ESI-TOF) m/z: [M + Na]+ Calcd for C47H48O14NaS 891.2662; Found 891.2657 [M + Na]+.
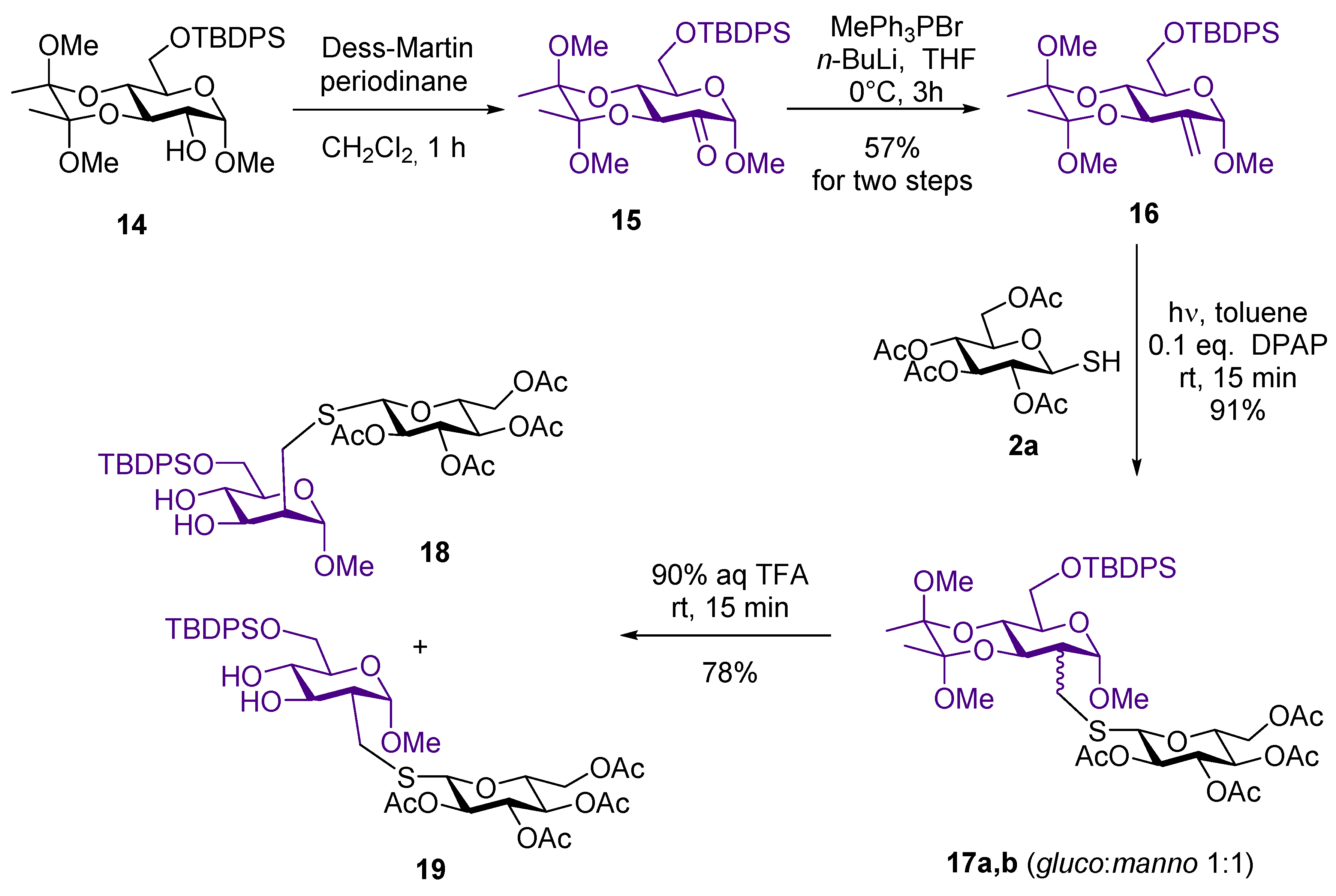
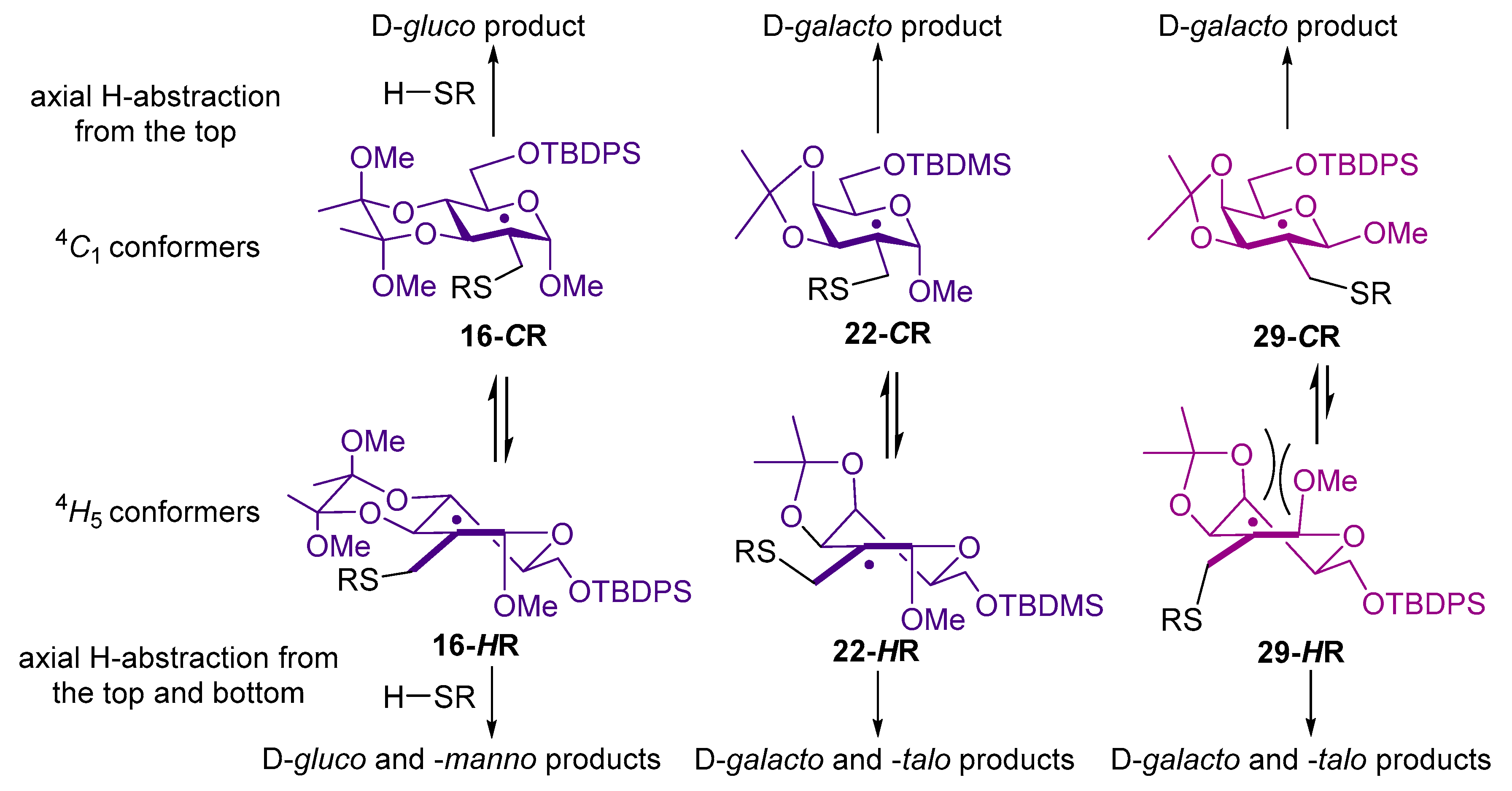
4.2.5. Methyl-6-O-tert-butyldiphenylsilyl-2,3-(2′,3′-dimethoxybutane-2′3′-diyl)-2-deoxy-2-C-methylene-α-d-arabino-hexopyranoside (16)
Compound 14 (1.989 g, 3.638 mmol) was dissolved in abs. CH2Cl2 (20 mL). Dess-Martin periodinane (1.855 g, 4.366 mmol, 1.2 equiv.) was added and the reaction was stirred for one hour. When TLC showed complete disappearance of the starting material, the reaction mixture was diluted with CH2Cl2, aq NaOH solution (28 mL, 1.3 M) was added and the mixture was vigorously stirred for 10 min. In the next step the organic layer was separated and washed with water, dried over MgSO4 and concentrated in vacuo to yield 15 (1.965 g, 99%). This compound was used in the next step without further purification. Dry tetrahydrofurane (20 mL) was stirred under argon and methyltriphenylphosphonium bromide (2.062 g, 5.772 mmol, 1.6 equiv.) was added. The suspension was cooled to 0 °C and n-butyllithium in hexane (2.309 mL, 5.772 mmol, c = 2.5 M, 1.6 equiv.) was added dropwise. After stirring the mixture for 30 min., 15 (1.965 g, 3.067 mmol) dissolved in dry tetrahydrofurane (10 mL) was added dropwise. The reaction was monitored by TLC. After three hours, ethyl acetate (200 mL) was added, and the organic layer was washed three times with satd aq NH4Cl solution and water, dried over MgSO4, and evaporated in vacuo. The crude product was purified by column chromatography to give 16 (1.130 g)
Yield: 57% from 14, colorless syrup. [α]D20 = +170.3 (c 0.1, CHCl3). Rf 0.72 (hexane/acetone 7/3). 1H NMR (400 MHz, CDCl3): δ = 7.74–7.70 (m, 4H, arom), 7.42–7.32 (m, 6H, arom), 5.27 (s, 1H, CH2a), 5.09 (s, 1H, CH2b), 5.00 (s, 1H, H-1), 4.60 (dt, J = 9.9, 2.2 Hz, 1H, H-3), 3.94 (ddd, J = 10.1, 6.8, 3.8 Hz, 1H, H-5), 3.90 (d, J = 3.3 Hz, 2H, H-6a and H-6b), 3.68 (t, J = 9.8 Hz, 1H, H-4), 3.36 (s, 3H, OCH3), 3.27 (s, 3H, OCH3), 3.17 (s, 3H, OCH3), 1.36 (s, 3H, CH3, butanedione), 1.29 (s, 3H, CH3 butanedione), 1.03 (s, 9H,3×CH3, t-Bu); 13C NMR (101 MHz, CDCl3): δ = 141.4 (C-2), 136.1, 135.7, 134.1, 133.6, 129.6, 127.7, 127.6 (arom), 109.0 (CH2), 102.2 (C-1), 100.1, 99.8 (2×Cq, butanedione), 71.5, 69.2 and 68.3 (C-5, C-4 and C-3), 62.4 (C-6), 54.3 (OCH3), 48.2, 48.2 (2×OCH3, butanedione), 26.9 (3×CH3, t-Bu), 19.5 (Cq, t-Bu), 18.0, 17.9 (2×CH3, butanedione). MS (ESI-TOF) m/z: [M + Na]+ Calcd for C30H42O7NaSi 565.2597; Found 565.2592 [M + Na]+.
4.2.6. Methyl 6-O-tert-butyldiphenylsilyl-2-deoxy-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-2,3-(2″,3″-dimethoxybutane-2″3″-diyl)-α-d-glucopyranoside (17a) and Methyl 6-O-tert-butyldiphenylsilyl-2-deoxy-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-2,3-(2″,3″-dimethoxybutane-2″3″-diyl)-α-d-mannopyranoside (17b)
Compound 16 (535 mg, 986 mmol) and 2a (431 mg, 1.183 mmol, 1.2 equiv.) were reacted, according to the general method, to give an inseparable 1:1 mixture of 17a and 17b (813 mg). The diastereoisomeric ratio was determined on the basis of 1H NMR spectrum.
Yield: 91%. Rf 0.40 (hexane/acetone 7/3). MS (ESI-TOF) m/z: [M + Na]+ Calcd for C44H62O16NaSSi 929.3426; Found 929.3421 [M + Na]+.
4.2.7. Methyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-α-d-mannopyranoside (18) and Methyl-6-O-tert-butyldiphenylsilyl-2-deoxy-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-α-d-glucopyranoside (19)
The mixture of 17a and 17b (295 mg, 0.325 mmol) was dissolved in CH2Cl2 (5 mL) and 2 mL 90 v/v% trifluoroacetic acid (1.8 mL trifluoroacetic acid + 0.2 mL water) was added dropwise. After 15 min. toluene (5 mL) was added and the mixture was evaporated in vacuo. The crude product was purified by flash chromatography to give 18 (45 mg), 19 (64 mg), and a mixture of 18 and 19 (94 mg).
18: Yield: 17%, colorless syrup. [α]D20 = +5.0 (c 0.06 CHCl3). Rf 0.50 (CH2Cl2/MeOH 95/5). 1H NMR (400 MHz, CDCl3): δ = 7.73–7.37 (m, 10H, arom), 5.21 (t, J = 9.3 Hz, 1H, H-3′), 5.14–5.08 (m, 1H, H-4′), 5.06 (t, J = 8.3 Hz, 1H, H-2’), 4.75 (s, 1H, H-1), 4.48 (d, J = 10.0 Hz, 1H, H-1’), 4.27 (dd, J = 12.4, 4.4 Hz, 1H, H-6’a), 4.15–4.10 (m, 1H, H-6′b), 4.04 (dd, J = 9.0, 5.4 Hz, 1H, H-3), 3.88 (s, 1H, H-6a), 3.87 (s, 1H, H-6b), 3.72–3.67 (m, 1H, H-5′), 3.62 (t, J = 9.3 Hz, 1H, H-4), 3.57–3.52 (m, 1H, H-5), 3.28 (s, 3H, OCH3), 3.23 (dd, J = 13.9, 2.1 Hz, 1H, SCH2a), 2.44 (dd, J = 13.6, 11.1 Hz, 1H, SCH2b), 2.32–2.25 (m, 1H, H-2), 2.07 (s, 3H, COCH3), 2.05 (s, 3H, COCH3), 2.02 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 1.07 (s, 9H, 3×CH3, t-Bu), 13C NMR (101 MHz, CDCl3): δ = 170.9, 170.3, 169.5 and 169.5 (4×COCH3), 135.7, 135.7, 133.0, 132.9, 130.1, 130.1, 128.0 and 128.0 (10C, arom), 100.3 (C-1), 83.8 (C-1′), 76.0, 74.0, 70.9, 70.7, 70.5, 69.9 and 68.3 (7C, skeleton carbons), 65.1 (C-6), 62.0 (C-6’), 55.0 (OCH3), 46.2 (C-2), 27.0 (3C, 3×CH3, t-Bu), 25.9 (SCH2), 20.8, 20.8, 20.7 and 20.7 (4C, 4×COCH3), 19.3 (Cq, t-Bu); MS (ESI-TOF) m/z: [M + Na]+ Calcd for C38H52O14SSiNa 815.274; Found 815.276.
19: Yield: 25%, colorless syrup. [α]D20 = +28.6 (c = 0.07 CHCl3). Rf 0.56 (CH2Cl2/MeOH 95/5). 1H NMR (400 MHz, CDCl3): δ = 7.73–7.34 (m, 10H, arom), 5.21 (t, J = 9.3 Hz, 1H, H-3’), 5.09–4.98 (m, 2H, H-2′, H-4′), 4.80 (d, J = 3.3 Hz, 1H, H-1), 4.52 (d, J = 10.1 Hz, 1H, H-1′), 4.21 (dd, J = 12.4, 4.9 Hz, 1H, H-6′a), 4.15 (dd, J = 12.3, 2.3 Hz, 1H, H-6′b), 3.88 (s, 1H, H-6a), 3.87 (s, 1H, H-6b), 3.74–3.58 (m, 3H, H-3, H-5 and H-5′), 3.48 (t, J = 9.1 Hz, 1H, H-4), 3.28 (s, 3H, OCH3), 3.13 (s, 1H, OH), 3.07 (dd, J = 13.6, 4.0 Hz, 1H, SCH2a), 2.84 (s, 1H, OH), 2.74 (dd, J = 13.5, 10.0 Hz, 1H, SCH2b), 2.06 (s, 3H, COCH3), 2.06 (s, 3H, COCH3), 2.02 (s, 3H, COCH3), 2.00 (s, 3H, COCH3), 1.98–1.92 (m, 1H, H-2), 1.06 (s, 9H, 3×CH3, t-Bu), 13C NMR (101 MHz, CDCl3): δ = 170.8, 170.3, 169.6 and 169.5 (4×COCH3), 135.7, 133.0, 132.9, 130.0, 130.0 and 127.9 (10C, arom), 99.4 (C-1), 85.0 (C-1’), 77.5, 77.2, 76.8, 76.0, 74.5, 73.9, 72.9, 70.2 and 68.4 (7C, skeleton carbons), 65.5 (C-6), 62.2 (C-6’), 54.9 (OCH3), 46.3 (C-2), 29.2 (SCH2), 26.9 (3C, 3×CH3, t-Bu), 20.9, 20.8, 20.7 and 20.7 (4C, 4×COCH3), 19.3 (Cq, t-Bu). Anal. Calcd for C38H52O14SSi: C, 55.96; H, 6.61; O, 28.25; S, 4.04; Si, 3.54. Found: C, 6.60, H, 6.60; S, 4.11.
Mixture of 18 and 19: Yield: 36%, colorless syrup.
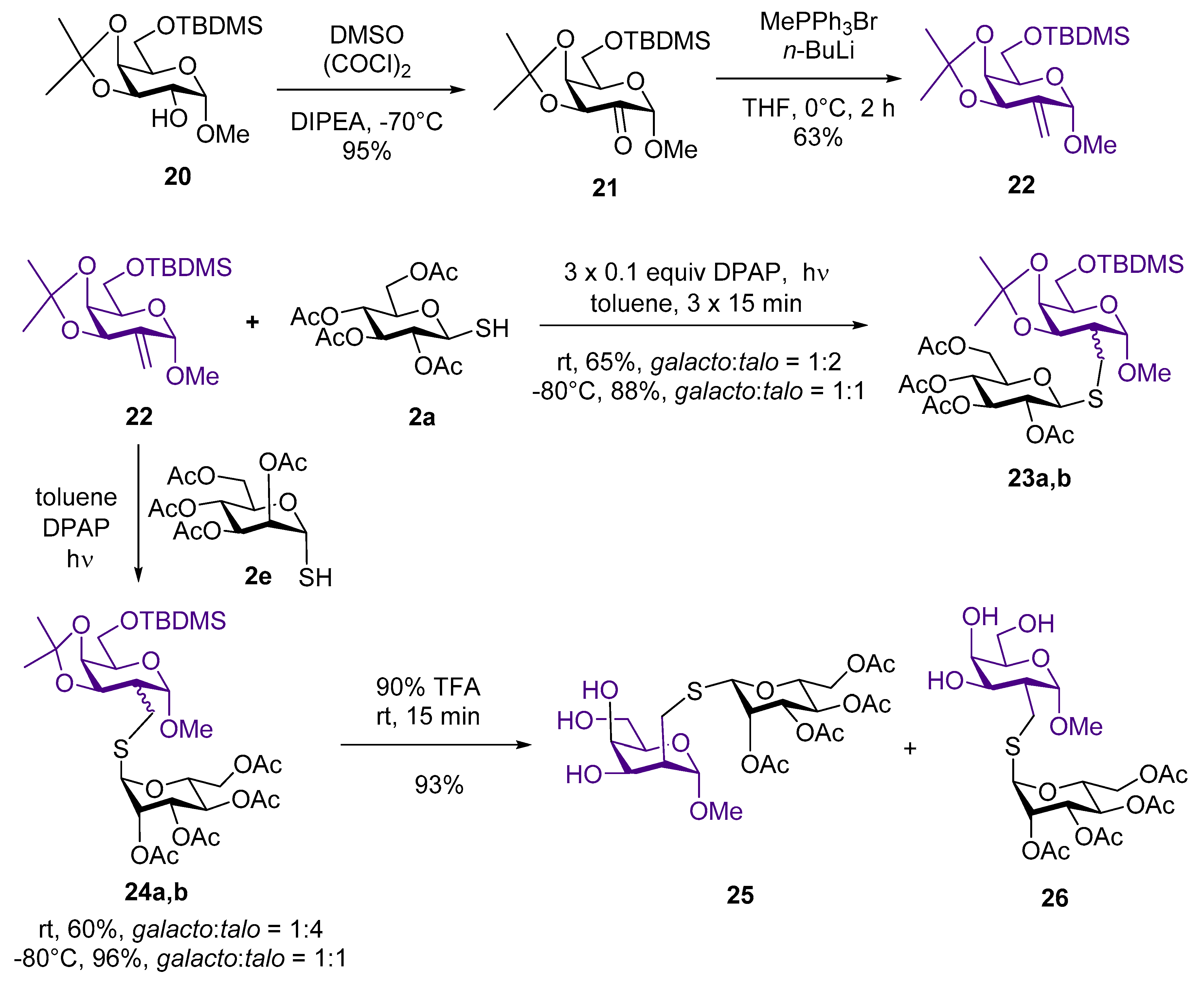
4.2.8. Methyl-6-O-tert-butyldimethylsilyl-3,4-di-O-isopropylidene-α-d-lyxo-hexopyranoside-2-ulose (21)
A mixture of dimethyl sulfoxide (163 μL, 179 mg, 2.30 mmol, 4 equiv.) and abs. CH2Cl2 (2 mL) was cooled to −80 °C and oxalyl chloride (97 μL, 144 mg, 1.15 mmol, 2 equiv.) was added. After 15 min., compound 20 (200 mg, 0.57 mmol) dissolved in CH2Cl2 (1 mL) was added. After 30 min., N,N-diisopropylethylamine (1.000 mL, 0.742 g, 5.741 mmol, 10 equiv.) was added and the mixture was then allowed to warm to room temperature. After 2 h, when TLC showed complete conversion, the mixture was diluted with CH2Cl2, extracted with 1 M HCl and water. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography (hexane/ethyl acetate 7/3) to give 21 (189 mg).
Yield: 95%, colorless syrup. [α]D20 = +58.6 (c 0.1, CHCl3). Rf 0.52 (hexane/ethyl acetate 7/3). 1H NMR (400 MHz, CDCl3): δ = 4.61 (s, 1H, H-1), 4.57 (d, J = 5.5 Hz, 1H, H-3), 4.48 (dd, J = 5.6, 1.9 Hz, 1H, H-4), 4.26 (td, J = 6.6, 1.9 Hz, 1H, H-5), 3.83 (dd, J = 10.0, 6.8 Hz, 1H, H-6a), 3.77 (dd, J = 10.0, 6.5 Hz, 1H, H-6b), 3.40 (s, 3H, OCH3), 1.35, 1.29 (2s, 6H, 2×i-Pr CH3), 0.84 (s, 9H, 3×t-Bu CH3), 0.03 (s, 6H, 2×Si-CH3); 13C NMR (101 MHz, CDCl3): δ = 199.2 (CO), 110.6 (i-Pr Cq), 100.5 (C-1), 77.1, 75.5, 68.2 (C-3, C-4, C-5), 61.9 (C-6), 55.3 (OCH3), 27.2, 26.1 (2C, 2×i-Pr CH3), 25.8 (3C, 3×t-Bu CH3), 18.2 (C-2), -5.4, -5.5 (2C, Si-CH3).
4.2.9. Methyl 6-O-tert-butyldimethylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-methylene-α-d-lyxo-hexopyranoside (22)
Methyltriphenylphosphonium bromide (332 mg, 0.929 mmol, 1.6 equiv.) was suspended in tetrahydrofurane (1.5 mL) under argon and cooled to 0 °C. n-Butyllithium (372 μL, 0.929 mmol, 1.6 equiv, 2.5 M solution in hexane) was added and the suspension was stirred. After 30 min. 21 (200 mg, 0.581 mmol) dissolved in tetrahydrofurane (1.5 mL) was added. When TLC showed the complete disappearance of 21, the mixture was diluted with ethyl acetate and washed with satd aq NH4Cl solution. The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography (hexane/ethyl acetate 9/1) to give 22 (125 mg)
Yield: 63%, colorless syrup. [α]D20 = +71.8 (c 0.4, CHCl3). Rf 0.48 (hexane/ethyl acetate 9/1). 1H NMR (400 MHz, CDCl3): δ = 5.52–5.48 (m, 2H, CH2), 5.23 (s, 1H, H-1), 4.83 (d, J = 6.9 Hz, 1H, H-3), 4.34 (dd, J = 6.9, 1.5 Hz, 1H, H-4), 3.92–3.80 (m, 3H, H-5, H-6a, H-6b), 3.52 (s, 3H, OCH3), 1.55, 1.42 (2s, 6H, 2 x i-Pr CH3), 0.97 (s, 9H, 3×t-Bu CH3), 0.15 (s, 6H, 2 x Si-CH3); 13C NMR (101 MHz, CDCl3): δ = 141.0 (C-2), 118.4 (CH2), 110.0 (i-Pr Cq), 99.4 (C-1), 75.5, 74.8, 70.4 (C-3, C-4, C-5), 62.3 (C-6), 54.8 (OCH3), 26.9, 26.0 (2C, 2×i-Pr CH3), 25.7 (3C, 3×t-Bu CH3), 18.4 (C-2), -5.2, -5.4 (2C, Si-CH3); MS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H32O5NaSi 367.1911; Found 367.1917.
4.2.10. Methyl 6-O-tert-butyldimethylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-α-d-galactopyranoside (23a) and Methyl 6-O-tert-butyldimethylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-D-glucopyranosyl)-α-d-talopyranoside (23b)
Exomethylene 22 (100 mg, 0.290 mmol), thiol 2a (127 mg, 0.348 mmol, 1.2 equiv.), and DPAP (7 mg, 0.29 mmol) were reacted with three irradiation cycles according to the general method. The crude product was purified by column chromatography (hexane/ethyl acetate 75/25) to give 23 (134 mg, 65%) as an inseparable diastereoisomeric mixture with a ratio of 23a galacto:23b talo ~ 1:2. The reaction was also carried out at −80 °C to give 23 (182 mg, 88%), with a ratio of 23a galacto:23b talo ~ 1:1. The diastereoisomeric ratios were determined on the basis of 1H NMR spectrum.
Yield (mixture of 23a and 23b): 88%, white foam. Rf 0.39 (hexane/ethyl acetate 75/25). 1H NMR (400 MHz, CDCl3): δ = 5.21 (t, J = 9.3 Hz, 1.59H), 5.11–4.94 (m, 2.98H), 4.89 (d, J = 3.2 Hz, 0.58H), 4.67 (dd, J = 7.9, 2.5 Hz, 1.05H), 4.63 (d, J = 10.1 Hz, 0.97H), 4.48 (d, J = 10.1 Hz, 0.60H), 4.42 (d, J = 7.4 Hz, 1.02H), 4.27–4.15 (m, 3.71H), 4.13–4.06 (m, 1.02H), 4.00–3.82 (m, 1.58H), 3.84–3.61 (m, 4.32H), 3.37 (s, 3.00H, OCH3), 3.34 (s, 1.70H, OCH3), 3.06–2.82 (m, 2.70H), 2.68–2.59 (m, 0.63H, COCH3), 2.14–1.97 (m, 17.42H, COCH3), 1.48, 1.43, 1.32 (3s, 11.03H, i-Pr CH3), 0.90, 0.89 (2s, 13.81H, t-Bu CH3), 0.11–0.03 (m, 7.81H, Si-CH3); 13C NMR (101 MHz, CDCl3): δ = 170.8, 170.7, 170.2, 170.2, 169.5, 169.4, 169.3, 169.3 (CO), 109.2, 109.0 (2×i-Pr Cq), 101.0, 98.7 (2×C-1), 85.7, 85.4 (2×C-1′), 76.0, 75.9, 74.9, 73.9, 73.6, 71.6, 71.5, 70.8, 70.5, 70.2, 68.4 67.9 (skeleton carbons), 62.7, 62.4, 62.2, 62.2 (4×C-6), 55.1, 54.9 (2×OCH3), 43.4, 41.1, 31.3, 31.0 (2×S-CH2), 28.6, 26.6, 26.2 (i-Pr CH3), 25.9, 25.9, 25.1 (t-Bu CH3), 20.8, 20.6 (COCH3), 18.3, 17.1 (2×C-2), -5.3, -5.4 (2C, Si-CH3).
4.2.11. Methyl 6-O-tert-butyldimethylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-α-d-mannopyranosyl)-α-d-galactopyranoside (24a) and Methyl 6-O-tert-butyldimethylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-α-d-mannopyranosyl)-α-d-talopyranoside (24b)
Exomethylene
22 (100 mg, 0.290 mmol), thiol
2e (127 mg, 0.348 mmol, 1.2 equiv.) and 2,2-dimethoxy-2-phenylacetophenone (7 mg, 0.029 mmol, 0.1 equiv.) were dissolved in toluene (2.3 mL) and then reacted according to the general method. The reaction mixture was concentrated in vacuo, the crude product was purified by flash chromatography (hexane/acetone 9/1 → 85/15) to give an inseparable mixture of
24a galacto and
24b talo (123 mg, 60%) as a colorless syrup. R
f 0.42 (hexane/acetone 75/25) in a 1:4 ratio of
24a galacto and
24b talo. The reaction was also carried out at −80 °C to give
37 (197 mg, 96%) with a ratio of
24a galacto:
24b talo ~ 1:1. The diastereoisomeric ratios were determined on the basis of
1H NMR spectrum (see
Supplementary Materials).
Yield (Mixture of 24a and 24b): 96%, white foam. Rf 0.42 (hexane/acetone 75/25).
4.2.12. Methyl 2-deoxy-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-α-d-mannopyranosyl)-α-d-galactopyranoside (25) and Methyl 2-deoxy-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-α-d-mannopyranosyl)-α-d-talopyranoside (26)
Compound 24 (161 mg, 0.227 mmol, 1:1 mixture of 24a galacto and 24b talo) was dissolved in CH2Cl2 (2 mL) and 500 μL 70 v/v% trifluoroacetic acid (350 μL trifluoroacetic acid + 150 μL water) was added dropwise. After 15 min. toluene (5 mL) was added and the mixture was evaporated in vacuo. The crude product was purified by flash chromatography to give 25 (37 mg, 29%) and a mixture of 25 and 26 (80 mg, 64%).
25 Yield: 29%, colorless syrup. [α]D20 = +91.8 (c 0.15, CHCl3), 1H NMR (400 MHz, CDCl3): δ = 5.35 (dd, J = 3.0, 1.4 Hz, 1H, H-2′), 5.32–5.27 (m, 2H, H-4′, H-1′), 5.24 (dd, J = 9.9, 3.1 Hz, 1H, H-3′), 4.91 (s, 1H, H-1), 4.34 (ddd, J = 9.2, 5.2, 2.3 Hz, 1H, H-5′), 4.28 (dd, J = 12.1, 5.3 Hz, 1H, H-6′a), 4.15 (dd, J = 12.0, 2.2 Hz, 1H, H-6′b), 4.06 (dd, J = 4.9, 3.8 Hz, 1H, H-3), 3.97–3.92 (m, 2H, H-4, H-6a), 3.88 (dd, J = 11.7, 4.3 Hz, 1H, H-6b), 3.77 (td, J = 4.6, 1.8 Hz, 1H, H-5), 3.46 (s, 1H, OH), 3.37 (s, 3H, OMe), 3.15 (dd, J = 13.7, 3.3 Hz, 1H, SCH2a), 2.93 (dd, J = 13.6, 11.1 Hz, 1H, SCH2b), 2.20–2.14 (m, 4H, COCH3 and H-2), 2.11 (s, 3H, COCH3), 2.06 (s, 3H, COCH3), 2.00 (s, 3H, COCH3); 13C NMR (101 MHz, CDCl3): δ = 171.1, 170.2, 170.1 and 169.9 (4C, 4×COCH3), 100.8 (C-1), 84.5 (C-1′), 71.2, 70.1, 69.5, 69.4, 69.2, 66.9 and 66.5 (7C, skeleton carbons), 63.6 (C-6), 62.6 (C-6’), 55.2 (OCH3), 44.3 (C-2), 30.3 (SCH2), 21.0, 20.8, 20.8 and 20.7 (4C, 4×COCH3). MS (ESI-TOF) m/z: [M + Na]+ Calcd for C22H34O14NaS 577.1567; Found 557.1560; [M + Na]+.
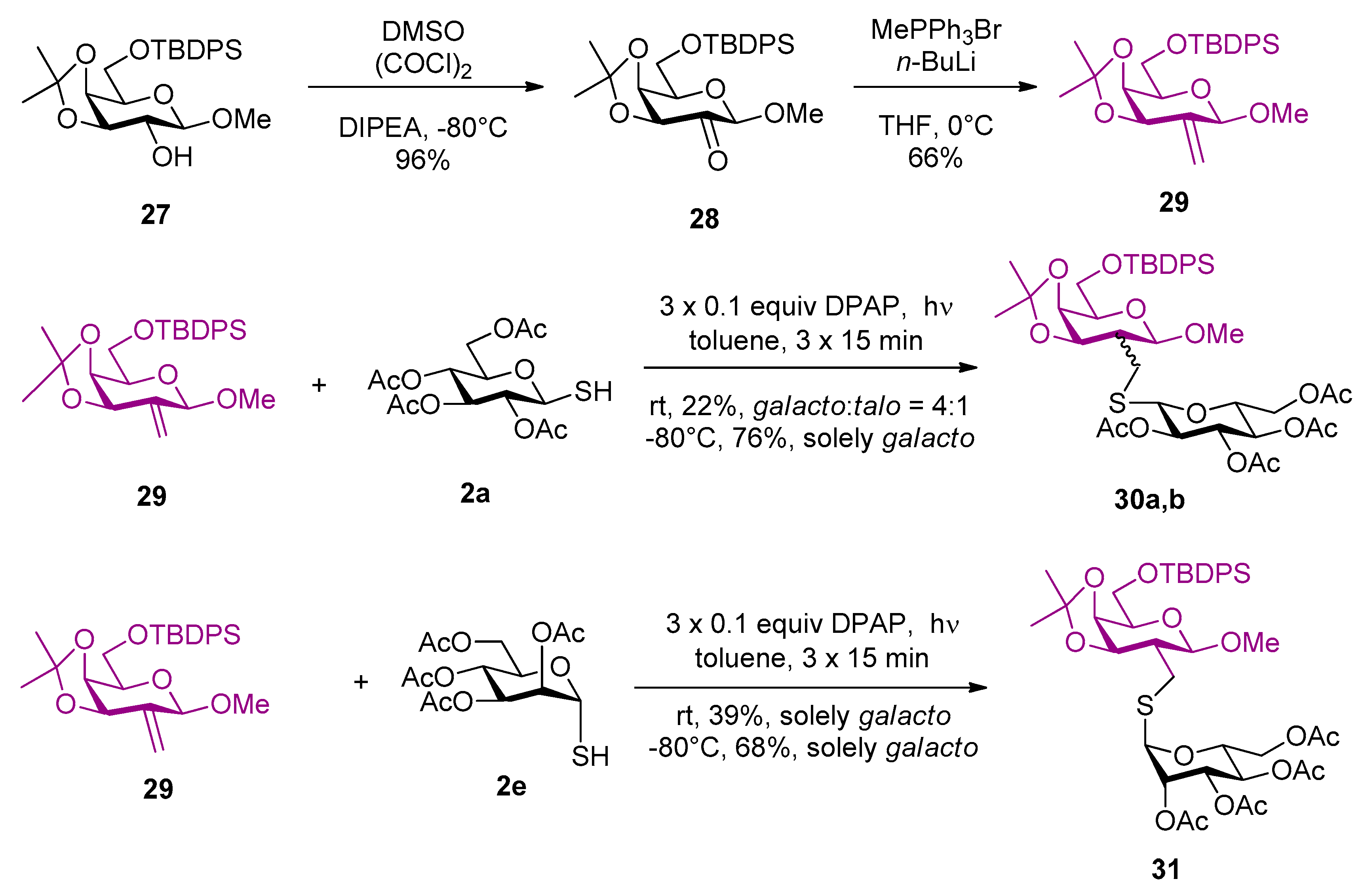
4.2.13. Methyl-6-O-tert-butyldiphenylsilyl-3,4-O-isopropylidene-β-d-lyxo-hexopyranoside-2-ulose (28)
Dimethyl sulfoxide (1.135 mL, 1.248 g, 15.978 mmol, 4 equiv.) was dissolved in abs. CH2Cl2 (19 mL), cooled to −80 °C and oxalyl chloride (676 μL, 1.014 g, 7.989 mmol, 2 equiv.) was added. After 15 min. 27 (1.888 g, 3.994 mmol) dissolved in CH2Cl2 (19 mL) was added. After 30 min, N,N-diisopropylethylamine (2.783 mL, 2.065 g, 15.978 mmol, 4 equiv.) was added and the mixture was then allowed to warm to room temperature. After 2 h, when TLC showed complete conversion, the mixture was diluted with CH2Cl2, extracted with 1 M HCl and water. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (hexane/acetone 75/25) to give 28 (1.798 g).
Yield: 96%, colorless syrup. [α]D20= −13.1 (c 0.2, CHCl3). Rf 0.27 (hexane/acetone 8/2). 1H NMR (400 MHz, CDCl3): δ = 7.74–7.68 (m, 4H, arom.), 7.46–7.36 (m, 6H, arom.), 4.75 (d, J = 0.5 Hz, 1H, H-1), 4.68 (dd, J = 5.6, 1.7 Hz, 1H, H-4), 4.50 (d, J = 5.6 Hz, 1H, H-3), 4.15 (td, J = 6.6, 1.6 Hz, 1H, H-5), 4.02–3.97 (m, 2H, H-6a, H-6b), 3.57 (s, 3H, OCH3), 1.42 (s, 3H, CH3, i-Pr), 1.38 (s, 3H, CH3, i-Pr), 1.08 (s, 9H, 3×CH3, t-Bu); 13C NMR (101 MHz, CDCl3): δ = 198.7 (C-2), 135.7, 135.6, 133.3, 133.2, 129.9, 127.9 and 127.8 (12C, arom.), 111.2 (Cq, i-Pr), 100.4 (C-1), 77.8, 77.7 and 73.3 (C-3, C-4 and C-5), 62.5 (C-6), 56.9 (OCH3), 27.3 (CH3, i-Pr), 26.8(3C, 3×CH3, t-Bu), 26.1 (CH3, i-Pr), 19.3 (Cq, t-Bu). Anal. Calcd for C26H34O6Si: C, 66.36; H, 7.28. Found: C, 66.71; H, 7.07.
4.2.14. Methyl 6-O-tert-butyldiphenylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-methylene-β-d-lyxo-hexopyranoside (29)
Methyltriphenylphosphonium bromide (2.119 g, 5.932 mmol, 1.6 equiv.) was suspended in tetrahydrofurane (20 mL) under argon and cooled to 0 °C. n-Butyllithium (2.373 mL, 5.932 mmol, 1.6 equiv, 2.5 M solution in hexane) was added and the suspension was stirred. After 30 min. 28 (1.745 g, 3.708 mmol) dissolved in tetrahydrofurane (17 mL) was added. When TLC showed complete disappearance of 28 the mixture was diluted with ethyl acetate and washed with satd aq NH4Cl solution. The organic layer was separated, dried over MgSO4, and then concentrated in vacuo. The crude product was purified by column chromatography to give 29 (1.153 g).
Yield: 66%, colorless syrup. [α]D20= +16.7 (c= 0.18, CHCl3). Rf 0.68 (hexane/acetone 8/2). 1H NMR (400 MHz, CDCl3): δ = 7.74–7.68 (m, 4H), 7.44–7.34 (m, 6H), 5.47–5.45 (m, 1H, CH2a), 5.44–5.42 (m, 1H, CH2b), 4.84 (s, 1H, H-1), 4.69 (dt, J = 6.3, 1.4 Hz, 1H, H-3), 4.30 (dd, J = 6.3, 1.9 Hz, 1H, H-4), 3.96 (dd, J = 10.0, 6.9 Hz, 1H, H-6a), 3.90 (dd, J = 10.1, 6.3 Hz, 1H, H-6b), 3.74–3.70 (m, 1H, H-5), 3.50 (s, 3H, OCH3), 1.46 (s, 3H, CH3, i-Pr), 1.36 (s, 3H, CH3, i-Pr), 1.06 (s, 9H, 3×CH3, t-Bu); 13C NMR (101 MHz, CDCl3): δ = 141.7 (C-2), 135.8, 135.7, 133.7, 133.6, 129.8, 127.8 and 127.7 (12C arom.), 115.8 (C-2), 110.3 (Cq, i-Pr), 101.0 (C-1), 75.3, 73.7 and 73.3 (C-5, C-4, C-3), 63.0 (C-6), 56.2 (OCH3), 27.4 (CH3, i-Pr), 26.9 (3C, 3×CH3, t-Bu), 26.2 (CH3, i-Pr), 19.4 (Cq, t-Bu); MS (MALDI-TOF) m/z: [M + Na]+ Calcd for C27H36O5NaSi 491.223; Found: 491.290.
4.2.15. Methyl 6-O-tert-butyldiphenylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-β-d-galactopyranoside (30a) and Methyl 6-O-tert-butyldiphenylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-β-d-talopyranoside (30b)
Compound 29 (100 mg, 0.213 mmol), 2a (93 mg, 0.256 mmol, 1.2 equiv.) and 2,2-dimethoxy-2-phenylacetophenone (5 mg, 0.021 mmol, 0.1 equiv.) were dissolved in toluene (1.9 mL) and reacted according to the general method with three irradiation cycles. The crude product was purified by column chromatography (hexane/acetone 8/2 → 75/25) to give 30 (39 mg, 22%) in 4:1 ratio of the d-galacto and d-talo isomers (the diastereoisomeric ratio was determined on the basis of 1H NMR spectrum). The reaction was also carried out at −80 °C to exclusively give the d-galacto isomer 30a (135 mg).
30a galacto Yield: 76%, colorless syrup. [α]D20= −37.7 (c 0.2, CHCl3). Rf 0.45 (hexane/acetone 7/3). 1H NMR (400 MHz, CDCl3): δ = 7.74–7.67 (m, 4H, arom.), 7.46–7.34 (m, 6H, arom.), 5.21 (t, J = 9.4 Hz, 1H, H-3’), 5.12–5.02 (m, 2H, H-4’, H-2’), 4.49 (d, J = 10.0 Hz, 1H, H-1’), 4.27–4.21 (m, 2H) and 4.18–4.12 (m, 3H) (H-6’a, H-6’b, H-4, H-3, H-1), 3.99 (dd, J = 9.8, 7.3 Hz, 1H, H-6a), 3.92 (dd, J = 9.8, 6.1 Hz, 1H, H-6b), 3.85–3.80 (m, 1H, H-5), 3.71 (ddd, J = 9.9, 5.0, 2.3 Hz, 1H, H-5’), 3.46 (s, 3H, OCH3), 3.02 (dd, J = 12.7, 5.1 Hz, 1H, SCH2a), 2.95 (dd, J = 12.7, 3.6 Hz, 1H, SCH2b), 2.07 (s, 3H, COCH3), 2.07 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 1.98–1.91 (m, 1H, H-2), 1.47 (s, 3H, CH3, i-Pr), 1.33 (s, 3H, CH3, i-Pr), 1.05 (s, 9H, 3×CH3, t-Bu); 13C NMR (101 MHz, CDCl3): δ = 170.7 (COCH3), 170.3 (COCH3), 169.5 (COCH3), 169.5 (COCH3), 135.7, 135.7, 133.6, 133.5, 129.8, 127.8 and 127.7 (12C, arom.), 109.7 (Cq, i-Pr), 102.4 (C-1), 84.2 (C-1’), 76.0, 75.2, 74.0, 73.4, 71.8, 70.3, 68.5, 63.0 (C-6), 62.4 (C-6’), 56.6 (OCH3), 44.8 (C-2), 28.5 (CH3, i-Pr), 28.4 (SCH2), 26.8 (3C, 3×CH3, t-Bu), 26.6 (CH3, i-Pr), 20.8 (COCH3), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3), 19.3 (Cq, t-Bu); MS (MALDI-TOF) m/z: [M + Na]+ Calcd for C41H56NaO14SSi 855.306; Found: 855.209.
4.2.16. Methyl 6-O-tert-butyldiphenylsilyl-2-deoxy-3,4-O-isopropylidene-2-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-α-d-mannopyranosyl)-β-d-galactopyranoside (31)
Compound 29 (100 mg, 0.213 mmol), 2e (93 mg, 0.256 mmol, 1.2 equiv.) and 2,2-dimethoxy-2-phenylacetophenone (5 mg, 0.021 mmol, 0.1 equiv.) were dissolved in toluene (1.9 mL) and reacted according to the general method with three irradiation cycles. The crude product was purified by column chromatography (hexane/acetone 8/2) to give 31 (69 mg, 39%) as a colorless syrup. The reaction was also carried out at −80°C to give 31 (120 mg).
Compound 31: Yield: 68%, [α]D20= +84.7 (c 0.2, CHCl3). Rf 0.41 (hexane/acetone 7/3). 1H NMR (400 MHz, Acetone-d6): δ = 7.81–7.75 (m, 4H, arom.), 7.51–7.41 (m, 6H, arom), 5.35–5.32 (m, 2H, H-1’, H-2’), 5.29 (t, J = 10.0 Hz, 1H, H-4’), 5.21 (dd, J = 10.1, 3.0 Hz, 1H, H-3’), 4.40 (ddd, J = 8.3, 5.6, 2.2 Hz, 1H, H-5’), 4.33 - 4.24 (m, 4H, H-6’a, H-4, H-3, H-1), 4.15 - 4.08 (m, 2H, H-6’b, H-5), 4.00 (dd, J = 9.8, 6.3 Hz, 1H, H-6a), 3.92 (dd, J = 9.8, 6.7 Hz, 1H, H-5), 3.44 (s, 3H, OCH3), 3.01 (dd, J = 12.9, 4.8 Hz, 1H, CH2a), 2.92 (dd, J = 12.9, 3.5 Hz, 1H, CH2b), 2.13 (s, 3H, COCH3), 2.05 (s, 6H, 2×COCH3), 1.96 (s, 3H, COCH3), 1.94–1.87 (m, 1H, H-2), 1.42 (s, 3H, CH3, i-Pr), 1.31 (s, 3H, CH3, i-Pr), 1.06 (s, 9H, CH3, t-Bu); 13C NMR (101 MHz, Acetone-d6): δ = 170.7, 170.4, 170.4 and 170.2 (4C, 4×COCH3), 136.4, 134.2, 130.7, 128.7 and 128.6 (12C, arom.), 110.0 (Cq, i-Pr), 103.0 (C-1), 83.6 (C-1’), 76.3, 74.2, 72.9, 71.5, 70.3, 70.1, 66.9, 64.2 and 63.0 (C-6’ and C-6), 56.4 (OCH3), 46.0 (C-2), 29.6 (SCH2) 28.7 (CH3, i-Pr), 27.1 (3C, 3×CH3, t-Bu), 26.7 (CH3, i-Pr), 20.7, 20.7, 20.6 and 20.5 (4C, 4×COCH3), 19.7 (t-Bu, Cq). MS (ESI-TOF) m/z: [M + Na]+ Calcd for C41H56O14SSiNa 855.3058; Found 855.3055; [M + Na]+.
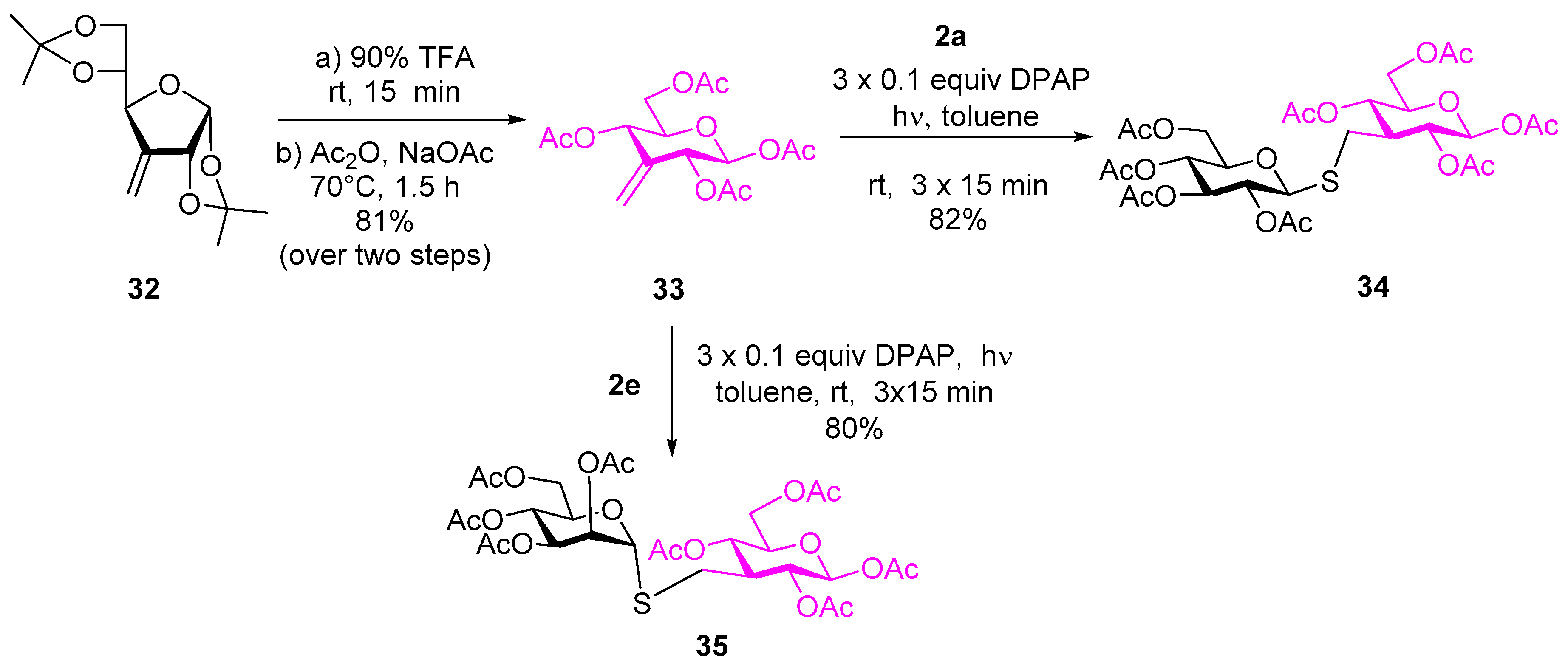
4.2.17. 1,2,4,6-Tetra-O-acetyl-3-deoxy-3-C-methylene-β-d-ribo-hexopyranose (33)
Compound 32 (1.613 g, 6.293 mmol) was dissolved in 5 mL 90 v/v% trifluoroacetic acid (4.5 mL trifluoroacetic acid + 0.5 mL water) and the mixture was stirred at room temperature. After 15 min., toluene (12 mL) was added and the mixture was concentrated in vacuo. Acetic anhydride (4.73 mL, 50.1 mmol, 8 equiv.) and sodium acetate (825 mg, 10.020 mmol, 1.6 equiv.) were added to the residue and the suspension was stirred at 70 °C. After 1.5 h the mixture was allowed to cool down, ethyl acetate was added, and the mixture was washed with water, satd aq NaHCO3, and water. The organic layer was dried over MgSO4, filtered, and evaporated in vacuo. The crude product was purified by column chromatography (hexane/ethyl acetate 7/3) to give 33 (1.755 g).
Yield: 81% over two steps, colorless syrup. [α]D20= +6.1 (c 0.2, CHCl3). Rf 0.47 (hexane/ethyl acetate 6/4). 1H NMR (400 MHz, CDCl3): δ = 5.62 (d, J = 7.5 Hz, 1H, H-1), 5.42–5.34 (m, 2H, H-2 and H-4), 5.14 (t, J = 1.7 Hz, 1H, CH2a), 5.11 (t, J = 1.6 Hz, 1H CH2b), 4.32 (dd, J = 12.3, 4.7 Hz, 1H, H-6a), 4.14 (dd, J = 12.2, 2.8 Hz, 1H, H-6b), 3.78 (ddd, J = 9.0, 4.7, 2.8 Hz, 1H, H-5), 2.16 (s, 3H, COCH3), 2.15 (s, 3H, COCH3), 2.13 (s, 3H, COCH3), 2.09 (s, 3H, COCH3); 13C NMR (101 MHz, CDCl3): δ = 169.1 and 169.0 (4×COCH3), 137.5 (CH2), 109.8 (C-3), 93.4 (C-1), 75.8, 70.6 and 68.0 (3C, C-5, C-4, C-2), 62.2 (C-6), 21.0 (COCH3), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3). Anal. Calcd for C15H20O9: C, 52.32; H, 5.85. Found: C, 57.06; H, 6.01.
4.2.18. 1,2,4,6-Tetra-O-acetyl-3-deoxy-3-C-(2′,3′,4′,6′-tetra-O-acetyl-1’-thiomethyl-β-d-glucopyranosyl)-β-d-glucopyranose (34)
Compound 33 (200 mg, 0.581 mmol) and thiol 2a (254 mg, 697 mmol, 1.2 equiv.) were reacted according to the general method with three irradiation cycles. The crude product was purified by flash chromatograpy to give 34 (332 mg)
Yield: 82%, white foam. [α]D20= −30.7 (c 0.1, CHCl3). Rf 0.23 (hexane/acetone 7/3). 1H NMR (400 MHz, CDCl3): δ = 5.67 (d, J = 7.9 Hz, 1H, H-1), 5.21 (t, J = 9.4 Hz, 1H) and 5.11–4.91 (m, 4H) (H-4, H-4′, H-3′, H-2, H-2′), 4.40 (d, J = 10.0 Hz, 1H, H-1′), 4.27 (dd, J = 12.4, 4.8 Hz, 1H), 4.18 (dd, J = 12.4, 4.9 Hz, 1H) and 4.12–4.04 (m, 2H) (H-6a, H-6′a, H-6b, H-6′b), 3.77 (ddd, J = 9.5, 4.7, 2.3 Hz, 1H, H-5), 3.72 (ddd, J = 10.0, 4.7, 2.1 Hz, 1H, H-5′), 2.86–2.76 (m, 2H, CH2), 2.38 - 2.30 (m, 1H, H-3), 2.12 (s, 3H), 2.10 (s, 6H), 2.08 (s, 6H), 2.06 (s, 3H), 2.03 (s, 3H) and 2.00 (s, 3H) (8×COCH3); 13C NMR (101 MHz, CDCl3): δ = 170.7, 170.5, 170.0, 169.6, 169.5, 169.4, 169.3 and 169.2 (8C, 8×COCH3), 93.3 (C-1), 82.5 (C-1′), 76.0 (C-5’), 75.4 (C-5), 73.7, 69.6, 69.2, 68.2 and 67.5 (C-4, C-4′, C-3′, C-2, C-2′), 62.0 (2C, C-6, C-6’), 43.8 (C-3), 26.3 (CH2), 20.9, 20.8, 20.7, 20.7 and 20.6 (8C, 8×COCH3). Anal. Calcd for C29H40O18S: C, 49.14; H, 5.69; S, 4.51. Found: C, 48.93; H, 5.65; S, 4.62.
4.2.19. 1,2,4,6-Tetra-O-acetyl-3-deoxy-3-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-α-d-mannopyranosyl)-β-d-glucopyranose (35)
Compound 33 (100 mg, 0.290 mmol) and thiol 2e (127 mg, 0.349 mmol, 1.2 equiv.) dissolved in toluene (2.3 mL) were reacted according to the general method with three irradiation cycles. The reaction mixture was concentrated in vacuo and the crude product was purified with column chromatography (hexane/acetone 65/35) to give 35 (162 mg).
Yield: 80%, colorless syrup. [α]D20= +77.8 (c 0.3, CHCl3). Rf 0.33 (hexane/acetone 65/35). 1H NMR (400 MHz, Acetone-d6): δ = 5.73 (d, J = 8.0 Hz, 1H, H-1), 5.31 (d, J = 1.4 Hz, 1H, H-1′), 5.29 (dd, J = 3.3, 1.4 Hz, 1H, H-2′), 5.29–5.24 (m, 1H, H-4′), 5.19 (dd, J = 10.1, 3.4 Hz, 1H, H-3′), 5.03 (t, J = 9.3 Hz, 1H, H-4), 4.98 (dd, J = 10.1, 7.2 Hz, 1H, H-2), 4.29 (ddd, J = 9.5, 5.5, 2.3 Hz, 1H, H-5’), 4.25 - 4.16 (m, 2H, H-6′a, H-6’b), 4.11 (dd, J = 12.0, 2.3 Hz, 1H, H-6a), 4.03 (dd, J = 12.3, 2.5 Hz, 1H, H-6b), 3.97 (ddd, J = 9.6, 5.0, 2.5 Hz, 1H, H-5), 2.84 (dd, J = 13.9, 3.8 Hz, 1H, SCH2a), 2.75 (dd, J = 13.9, 5.5 Hz, 1H, SCH2b), 2.53 (tdd, J = 10.8, 5.4, 3.9 Hz, 1H, H-3), 2.12 (s, 3H, COCH3), 2.11 (s, 3H, COCH3), 2.07 (s, 3H, COCH3), 2.05 (s, 3H, COCH3), 2.05 (s, 3H, COCH3), 2.04 (s, 3H, COCH3), 2.00 (s, 3H, COCH3), 1.94 (s, 3H, COCH3); 13C NMR (101 MHz, Acetone-d6): δ = 170.8, 170.8, 170.4, 170.3, 170.2 and 169.6 (8C, 8×COCH3), 93.9 (C-1), 82.6 (C-1′), 75.8, 71.2, 70.7, 70.3, 70.2, 69.3 and 66.8 (7C, skeleton carbons), 63.1 and 62.9 (2C, C-6 and C-6′), 45.0 (C-3), 29.2 (SCH2), 21.1, 20.8, 20.7, 20.7 and 20.6 (8C, 8×COCH3); MALDI-TOF m/z: [M + Na]+ Calcd for C29H40O18SNa 731.183; Found 731.135.
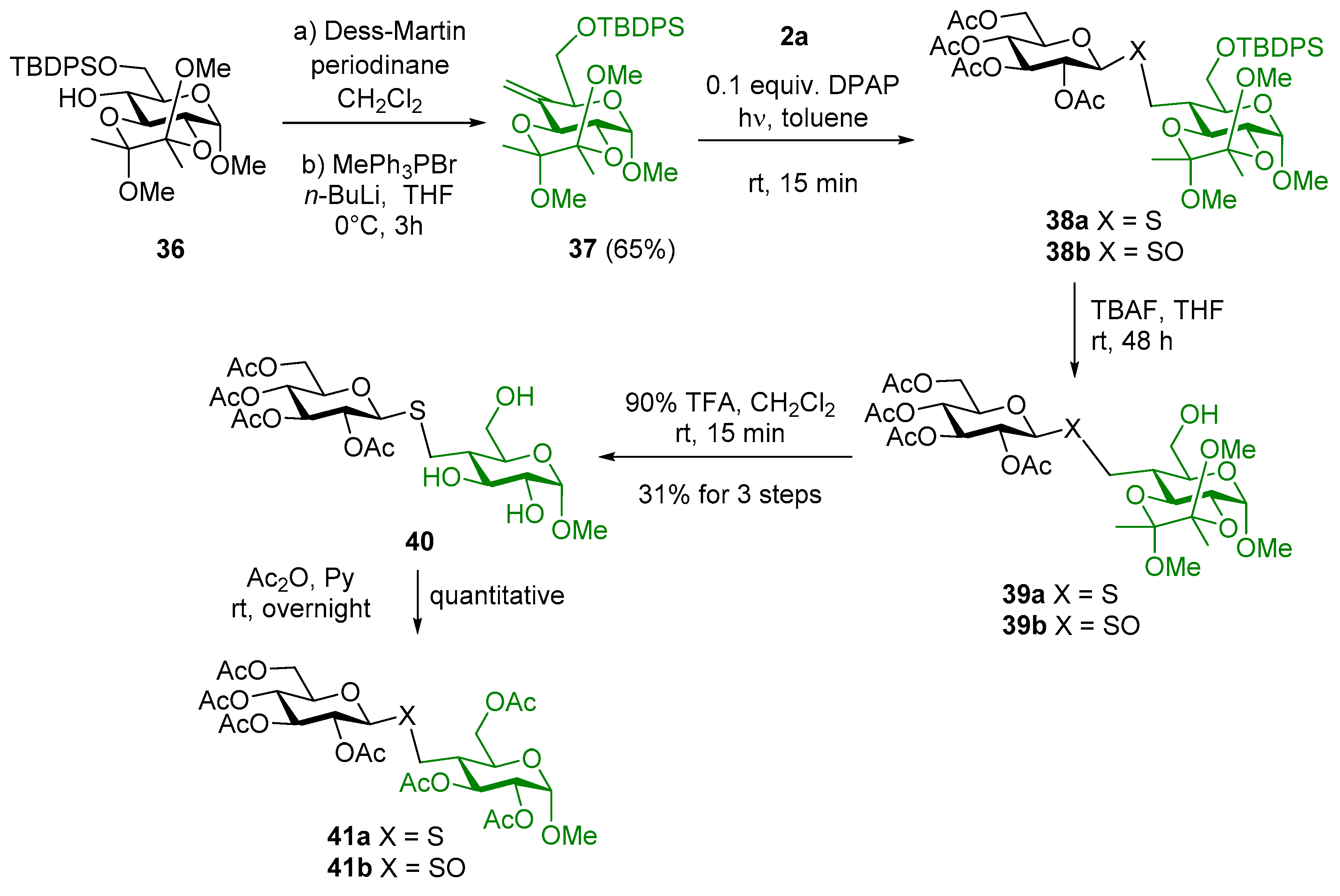
4.2.20. Methyl-6-O-tert-butyldiphenylsilyl-2,3-(2′,3′-dimethoxybutane-2′3′-diyl)-4-deoxy-4-C-methylene-α-d-xylo-hexopyranoside (37)
Compound 36 (2.862 g, 5.235 mmol) was dissolved in abs. CH2Cl2 (29 mL). Dess-Martin periodinane (2.669 g, 6.282 mmol, 1.2 equiv.) was added and the reaction was stirred for one hour. When TLC showed complete disappearance of the starting material, the reaction mixture was diluted with CH2Cl2, aq NaOH solution (28 mL, 1.3 M) was added and the mixture was vigorously stirred for 10 min. In the next step, the organic layer was separated and washed with water, dried over MgSO4, and concentrated in vacuo. The crude product 4-ulose (2.822 mg, 99%) was used in the next step without further purification. Dry tetrahydrofurane (30 mL) was stirred under argon and methyltriphenylphosphonium bromide (2.961 g, 8.289 mmol, 1.6 equiv.) was added. The suspension was cooled to 0 °C and n-butyllithium in hexane (3.316 mL, c = 2.5 M, 8.289 mmol) was added dropwise. After stirring the mixture for 30 min., 4-ulose dissolved in dry tetrahydrofurane (15 mL) was added dropwise. The reaction was monitored by TLC. After thtre hours, ethyl acetate (200 mL) was added, and the organic layer was washed three times with satd aq NH4Cl solution and water, dried over MgSO4, and evaporated in vacuo. The crude product was purified by column chromatography to give 37 (1.831 g).
Yield: 65% (over two steps), colorless syrup. [α]D20 = +21.8 (c 0.1, CHCl3). Rf 0.73 (hexane/acetone 8/2). 1H NMR (400 MHz, CDCl3): δ = 7.72–7.67 (m, 4H, arom), 7.46-7.33 (m, 6H arom), 5.27 (s, 1H, methylene CH2a), 4.95 (s, 1H, methylene CH2b), 4.80 (d, J = 3.6 Hz, 1H, H-1), 4.52 (d, J = 10.3 Hz, 1H, H-3), 4.23 (t, J = 5.5 Hz, 1H, H-5), 4.03 (dd, J = 10.7, 4.9 Hz, 1H, H-6a), 3.88 (dd, J = 10.7, 6.4 Hz, 1H, H-6b), 3.69 (dd, J = 10.3, 3.6 Hz, 1H, H-2), 3.41 (s, 3H, OCH3), 3.26 (s, 3H, OCH3), 3.23 (s, 3H, OCH3), 1.36 (s, 3H, CH3 butanedione), 1.34 (s, 3H CH3 butanedione), 1.06 (d, J = 7.0 Hz, 9H, 3×CH3, t-Bu); 13C NMR (101 MHz, CDCl3): δ = 141.4(C-4), 135.8, 135.8, 134.9, 133.6, 133.6, 129.8, 127.9, 127.8 and 127.8 (arom), 106.2 (CH2), 100.0 and 99.9 (2xCq butanedione), 98.1 (C-1), 71.9, 69.7, 66.4 and 63.8 (C-5, C-3, C-2), 55.0 (C-1-OCH3), 48.0 (2×OCH3 butanedione), 26.9 (3×CH3, t-Bu), 19.4 (Cq, t-Bu), 18.0 and 17.9 (2×CH3 butanedione). Anal. Calcd for C30H42O7Si: C, 66.39; H, 7.80. Found: C, 67.88; H, 7.63.
4.2.21. Methyl-6-O-tert-butyldiphenylsilyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-2,3-(2″,3″-dimethoxybutane-2″3″-diyl)-α-d-glucopyranoside (38a) and Methyl-6-O-tert-butyldiphenylsilyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-sulfinylmethyl-β-d-glucopyranosyl)-2,3-(2″,3″-dimethoxybutane-2″3″-diyl)-α-d-glucopyranoside (38b)
4-Exomethylene
37 (1.380 g, 2.543 mmol), thiol
2a (1.112 g, 3.051 mmol, 1.2 equiv.) and DPAP (65 mg, 0.254 mmol, 0.1 equiv.) were reacted according to the general method with one irradiation cycle to provide compounds
38a and
38b (2.056 g,
38a:
38b ~ 3:2 on the basis of
1H NMR spectrum, see
supplementary file). The two products cannot be separated and used as a mixture in the next step.
Rf 0.27 (hexane/acetone 8/2). 1H NMR (360 MHz, CDCl3) Characteristic peaks: δ 4.76 (d, J = 3.5 Hz, 1H, H-1major), 4.74 (d, J = 3.9 Hz, 0.6H, H-1minor), 4.49 (d, J = 10.1 Hz, 0.6H, H-1’minor), 4.26 (d, J = 9.8 Hz, 1H, H-1′major).
MS (ESI-TOF) m/z: [M + Na]+ 38a: Calcd for C44H62O16SSiNa 929.3420; Found 929.3430; [M + Na]+ 38b: Calcd for C44H62O17SSiNa 945.3369; Found 945.3375.
4.2.22. Methyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-2,3-(2″,3″-dimethoxybutane-2″3″-diyl)-α-d-glucopyranoside (39a) and Methyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-sulfinylmethyl-β-d-glucopyranosyl)-2,3-(2″,3″-dimethoxybutane-2″3″-diyl)-α-d-glucopyranoside (39b)
The mixture of 38a and 38b (2.056 g, 2.3 mmol) were dissolved in tetrahydrofurane (20 mL), 1M tetrabutyl-ammonium-fluoride in tetrahydrofurane (3.400 mL, 3.400 mmol, 1.5 equiv.) was added and the solution was stirred for 12 h. When TLC showed complete disappearance of the starting material the mixture was evaporated in vacuo, the crude product was purified by flash chromatography (hexane/acetone 75/25 → 65:35) to give 39a,b as an inseparable mixture (877 mg).
39a,b: Rf 0.12 (hexane/acetone 7/3). 1H NMR (360 MHz, CDCl3) Characteristic peaks: δ 4.78 (d, J = 3.5 Hz, 1H, H-1major), 4.72 (d, J = 3.9 Hz, 0.5H, H-1minor), 4.55 (d, J = 10.1 Hz, 0.5H, H-1’minor), 4.48 (d, J = 9.8 Hz, 1H, , H-1’major).
MS (ESI-TOF) m/z: [M + Na]+ 39a,b: Calcd for C28H44O16SNa 691.225; Found 691.240; [M + Na]+ 39b: Calcd for C28H44O17SNa 707.220; Found 707.224.
4.2.23. Methyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-α-d-glucopyranoside (40)
The mixture of 39a,b (850 mg) was dissolved in CH2Cl2 and 3 mL 90 v/v% trifluoroacetic acid (2.7 mL trifluoroacetic acid + 0.3 mL water) was added dropwise. After 15 min. toluene (5 mL) was added and the mixture was evaporated in vacuo. The crude product was purified by flash chromatography to provide 40 (443 mg).
Yield: 31% for three steps, colorless syrup. Rf 0.13 (CH2Cl2/acetone 7/3). 1H NMR (360 MHz, CDCl3): δ = 5.23 (t, J = 9.3 Hz, 1H), 5.07 (t, J = 9.8 Hz, 1H) and 5.07 (t, J = 9.7 Hz, 1H) (H-2′, H-3′, H-4′), 4.81 (d, J = 3.8 Hz, 1H, H-1), 4.51 (d, J = 10.0 Hz, 1H, H-1’), 4.27–4.17 (m, 2H, H-6′a, H-6′b), 3.84–3.65 (m, 6H) and 3.51 (dd, J = 9.2, 3.7 Hz, 1H) (H-2, H-3, H-4, H-5, H-6a, H-6b, H-5′), 3.41 (s, 3H, OCH3), 3.02 (dd, J = 13.6, 3.1 Hz, 1H, SCH2a), 2.90 (dd, J = 13.6, 4.2 Hz, 1H, SCH2b), 2.12 (s, 3H, COCH3), 2.08 (s, 3H, COCH3), 2.04 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 13C NMR (91 MHz, CDCl3): δ = 99.8 (C-1), 83.7 (C-1′), 76.2, 73.8, 73.7, 70.8, 69.9, 69.8 and 68.5 (7C, skeleton carbons), 62.5 and 62.2 (C-6 and C-6′), 55.5 (C-4), 42.4 (SCH2), 26.9, 20.9 and 20.7 (4C, 4×COCH3), MS (MALDI-TOF) m/z: [M + Na]+ Calcd for C22H34O14SNa 577.157; Found: 577.169.
4.2.24. Methyl-2,3,6-tri-O-acetyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-α-d-glucopyranoside (41a) and Methyl-2,3,6-tri-O-acetyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-sulfinylmethyl-β-d-glucopyranosyl)-α-d-glucopyranoside (41b)
Compound 40 (430 mg) was dissolved in CH2Cl2, acetic anhydride (0.141 mL, 1.487 mmol, 3.6 equiv.) and pyridine (0.100 mL, 1.237 mmol, 3.0 equiv.) was added. The mixture was stirred overnight. When TLC showed the complete disappearance of the starting material, the solvent was evaporated in vacuo, and the crude product was purified by column chromatography to give 41a and 41b (223 mg). 41a and 41b cannot be separated from each other.
Yield: 69%, colorless syrup. Rf 0.82 (CH2Cl2/acetone 7/3).
1H NMR (400 MHz, CDCl3): δ 5.41 (dd, J = 10.9, 9.7 Hz, 1H, H-3), 5.28 (dd, J = 5.3, 1.7 Hz, 0.3H), 5.21 (t, J = 9.4 Hz, 1H), 5.08 (t, J = 9.7 Hz, 1H), 5.01 (td, J = 9.8, 3.6 Hz, 2H), 4.96–4.91 (m, 1H), 4.89 (d, J = 3.6 Hz, 1H), 4.82 (dd, J = 9.7, 3.6 Hz, 1H), 4.40 (d, J = 9.8 Hz, 2H), 4.36–4.15 (m, 8H), 4.12 (dt, J = 10.7, 3.5 Hz, 1H), 3.81 (ddd, J = 10.1, 4.3, 2.2 Hz, 1H), 3.70 (ddd, J = 10.1, 4.9, 2.3 Hz, 1H), 3.38 (s, 2H), 3.37 (s, 3H), 2.99 (dd, J = 13.1, 3.3 Hz, 1H), 2.84 (dd, J = 13.1, 7.7 Hz, 1H), 2.78 (d, J = 3.9 Hz, 2H), 2.56 (q, J = 4.1 Hz, 1H), 2.22 (ddt, J = 14.9, 11.8, 3.8 Hz, 1H), 2.11 (s, 3H), 2.10 (s, 1H), 2.10 (s, 6H), 2.09 (s, 4H), 2.08 (d, J = 0.8 Hz, 1H), 2.06 (s, 3H), 2.06 (s, 3H), 2.03 (s, 1H), 2.02 (s, 3H), 2.01 (s, 1H), 2.00 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.78, 170.75, 170.69, 170.21, 170.15, 169.68, 169.49, 169.47, 169.45, 97.24, 97.02, 83.84, 82.52, 76.05, 73.80, 72.67, 69.83, 69.58, 69.12, 68.41, 68.28, 68.12, 67.82, 67.15, 64.13, 63.25, 62.04, 61.90, 55.35, 55.15, 41.26, 40.48, 25.54, 22.44, 21.04, 20.92, 20.77, 20.68. MS (MALDI-TOF) m/z: [M + Na]+ 41a: Calcd for C28H40O17SNa 703.188; Found 703.199; 41b: m/z: [M + Na+] Calcd for C28H40O18SNa 719.183; Found 719.195.
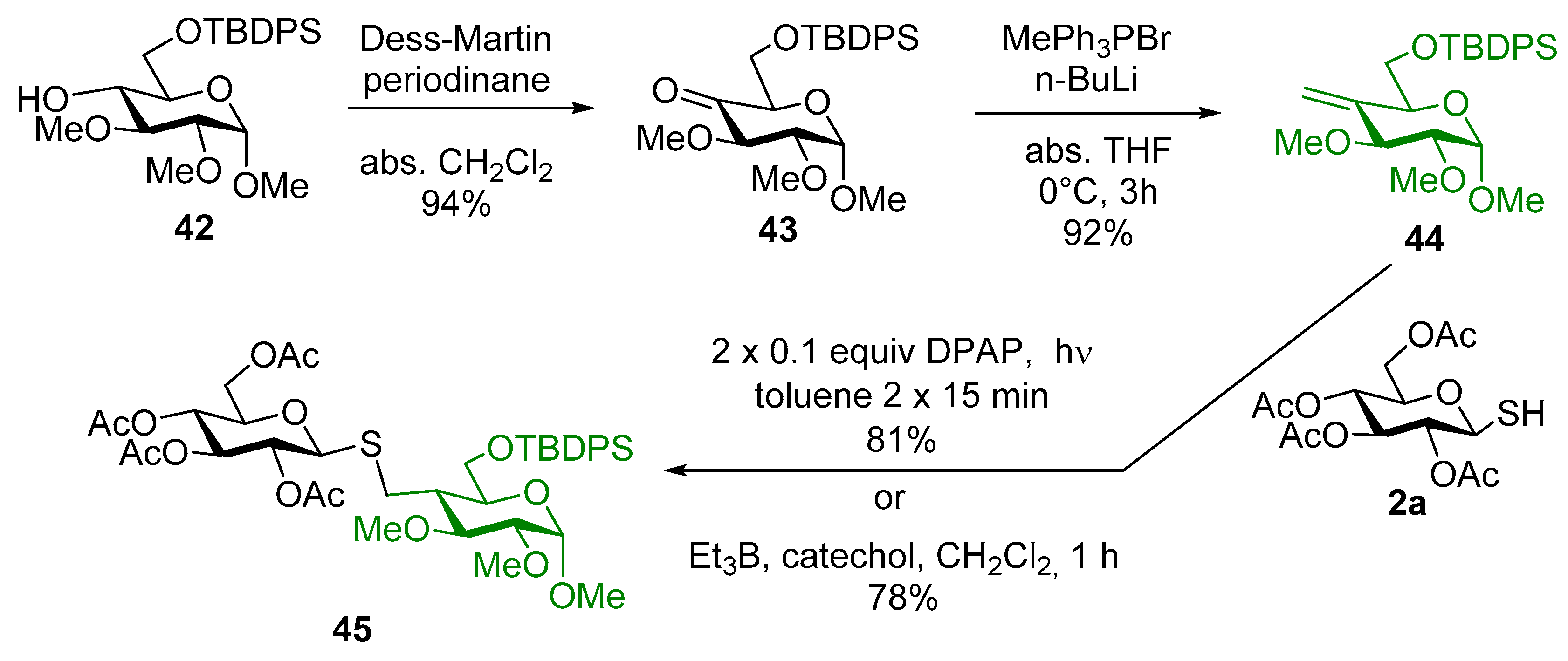
4.2.25. Methyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-glucopyranoside (42)
Methyl-2,3-di-
O-methyl-α-
d-glucopyranoside [
69] was dissolved in abs. CH
2Cl
2 (5 mL) and pyridine (0.653 mL, 6.75 mmol 2 equiv.).
tert-Butyl-diphenylsilyl chloride (1.038 mL, 4.050 mmol, 1.2 equiv.) was added and the mixture was stirred overnight. When the reaction was complete, CH
2Cl
2 was added and the mixture was extracted with 1M HCl, satd aq NaHCO
3 solution and water. The organic phase was dried over MgSO
4 and then concentrated; the crude product was purified by flash chromatography to give
42 (1.180 g).
Yield: 76%, colorless syrup. [α]D20 = +48.7 (c 0.2, CHCl3). Rf 0.77 (CH2Cl2/acetone 8/2). 1H NMR (400 MHz, CDCl3): δ = 7.73–7.35 (m, 10H, arom), 4.82 (d, J = 3.5 Hz, 1H, H-1), 3.93–3.83 (m, 2H, H-6a and H-6b), 3.69–3.63 (m, 1H, H-5), 3.64 (s, 3H, OCH3), 3.56 (dt, J = 9.4, 1.6 Hz, 1H, H-4), 3.50 (s, 3H, OCH3), 3.50–3.45 (m, J = 8.5, 4.5 Hz, 1H, H-3), 3.39 (s, 3H, OCH3), (dd, J = 9.4, 3.6 Hz, 1H, H-2), 2.77 (d, J = 2.0 Hz, 1H, OH), 1.06 (s, 9H, 3×CH3, t-Bu), 13C NMR (101 MHz, CDCl3): δ = 135.7, 133.3, 129.9 and 127.8 (12C, arom), 97.4 (C-1), 83.0, 81.9, 71.7 and 70.8 (4C, skeleton carbons), 64.5 (C-6), 61.3 (OCH3), 58.6 (OCH3), 55.1 (OCH3), 26.9 (3C, 3×CH3, t-Bu), 19.3 (Cq, t-Bu). Anal. Calcd for C25H36O6Si: C, 65.19; H, 7.88. Found: C, 67.02; H, 7.65.
4.2.26. Methyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-xylo-hexopyranoside-4-ulose (43)
Dess–Martin periodinane (1.239 g, 2.915 mmol, 1.2 equiv.) was added and the mixture was stirred for one hour to a solution of
42 [
46] (1.119 g, 2.429 mmol) dissolved in abs. CH
2Cl
2 (10 mL). The mixture was diluted with CH
2Cl
2, aq NaOH solution (19 mL, 1.3M) was added and vigorously stirred for 10 min. The organic phase was separated and washed with water, over MgSO
4 concentrated to give
43 (1.056 g). The crude
43 was used in the next step without further purification.
Yield: 94%, colorless syrup. Rf = 0.56 (hexane/ethyl acetate 6/4). 1H NMR (400 MHz, CDCl3): δ = 7.74–7.34 (m, 10H, arom), 5.03 (d, J = 3.4 Hz, 1H, H-1), 4.19 (dd, J = 6.4, 3.1 Hz, 1H), 4.11–4.06 (m, 2H, H-6a and H-6b), 3.89 (dd, J = 11.3, 6.4 Hz, 1H), 3.58 (s, 3H, OCH3), 3.56 (s, 3H, OCH3), 3.54 (s, 3H, OCH3), 3.53–3.50 (m, J = 6.2, 2.1 Hz, 1H), 1.04 (s, 9H, 3×CH3, t-Bu), 13C NMR (101 MHz, CDCl3): δ = 135.8, 135.7, 133.5, 133.3, 129.8, 127.8, 127.8 (12C, arom), 97.6 (C-1), 84.6, 82.7 and 74.3 (C-2, C-3 and C-5), 62.1 (C-6), 60.3 (OCH3), 59.8 (OCH3), 56.0 (OCH3), 26.8 (3C, 3×CH3, t-Bu), 19.3 (Cq, t-Bu).
4.2.27. Methyl-6-O-tert-butyldiphenylsilyl-4-deoxy-4-methylene-2,3-di-O-methyl-α-d-xylo-hexopyranoside (44)
Dry tetrahydrofurane (10 mL) was stirred under argon and methyltriphenylphosphonium bromide (1.255 g, 3.513 mmol) was added. The suspension was cooled to 0 °C and n-butyllithium in hexane (1.405 mL, c = 2.5 M, 3.513 mmol) was added dropwise. After stirring the mixture for 30 min., 43 (1.007 g, 2.196 mmol) dissolved in dry tetrahydrofurane (5 mL) was added dropwise. The reaction was monitored by TLC. After three hours, ethyl acetate (100 mL) was added, and the organic layer was washed three times with satd aq NH4Cl solution and water, over MgSO4 evaporated in vacuo. The crude product was purified by column chromatography to give 44 (920 mg).
Yield: 92%, colorless syrup. [α]D20 = +95.0 (c 0.2, CHCl3). Rf 0.46 (hexane/ethyl acetate 7/3). 1H NMR (400 MHz, CDCl3): δ = 7.73–7.35 (m, 10H, arom), 5.20 (s, 1H, =CH2a), 4.96 (s, 1H, =CH2b), 4.88 (d, J = 3.6 Hz, 1H, H-1), 4.17 (t, J = 5.7 Hz, 1H, H-5), 4.02–3.94 (m, 2H, H-6a and H-3), 3.88 (dd, J = 10.6, 6.6 Hz, 1H, H-6b), 3.53 (s, 3H, OCH3), 3.53 (s, 3H, OCH3), 3.42 (s, 3H, OCH3), 3.18 (dd, J = 9.5, 3.6 Hz, 1H, H-2), 1.05 (s, 9H, 3×CH3, t-Bu); 13C NMR (101 MHz, CDCl3): δ = 142.5 (C-4), 135.8, 135.8, 133.6, 132.6, 129.9, 127.8 and 127.8 (12C, arom), 107.4 (=CH2), 97.9 (C-1), 83.8, 81.2 and 69.6 (C-2, C-3 and C-5), 63.9 (C-6), 59.5 (OCH3), 59.4 (OCH3), 55.2 (OCH3), 26.9 (3C, 3×CH3, t-Bu), 19.4 (Cq, t-Bu). Anal. Calcd for C26H36O5Si: C, 68.39; H, 7.95. Found: C, 66.74; H, 8.18.
4.2.28. Methyl-6-O-tert-butyldiphenylsilyl-4-deoxy-4-C-(2′,3′,4′,6′-tetra-O-acetyl-1′-thiomethyl-β-d-glucopyranosyl)-2,3-di-O-methyl-α-d-glucopyranoside (45)
Preparation by photoinduced free radical hydrothiolation: 44 (106 mg, 0.232 mmol), and 2a (100 mg, 0.278 mmol, 1.2 equiv.) were reacted according to the general method with two irradiation cycle, to give compound 45 (153 mg, 81%).
The preparation by Et3B-catechol initiated free radical hydrothiolation: 44 (100 mg, 0.262 mmol) and 2a (95 mg, 0.182 mmol, 1.2 equiv.) were dissolved in CH2Cl2 (2 mL) and catechol (29 mg, 0.263 mmol, 1.2 equiv.) and Et3B in hexane (307 μL, c = 1 M, 0.307 mmol, 1.4 equiv.) were added. After 1 h, the solvents was evaporated and the crude product was purified by column chromatography to give 45 (116 mg, 78%).
Yield: 81%, [α]D20 = +1.30 (c = 0.23, CHCl3). Rf 0.33 (CH2Cl2/acetone 95/5). 1H NMR (400 MHz, CDCl3): δ = 7.74–7.35 (m, 10H, arom), 5.19 (t, J = 9.4 Hz, 1H, H-3′), 5.03 (t, J = 9.7 Hz, 1H, H-4’), 4.95 (t, J = 9.7 Hz, 1H, H-2′), 4.86 (d, J = 3.5 Hz, 1H, H-1), 4.38 (d, J = 10.1 Hz, 1H, H-1′), 4.17 (dd, J = 12.4, 4.9 Hz, 1H, H-6′a), 4.01 (dd, J = 12.4, 2.2 Hz, 1H, H-6′b), 3.89 (dd, J = 11.2, 2.2 Hz, 1H, H-6b), 3.74 (dd, J = 11.3, 4.7 Hz, 1H, H-6a), 3.68 (ddd, J = 10.5, 4.4, 2.2 Hz, 1H, H-5), 3.62–3.59 (m, 1H, H-5′), 3.58 (s, 3H, OCH3), 3.52 (s, 3H, OCH3), 3.46–3.39 (m, 1H, H-3), 3.39 (s, 3H, OCH3), 3.23 (dd, J = 9.2, 3.5 Hz, 1H, H-2), 2.82 (dd, J = 13.1, 4.6 Hz, 1H, SCH2a), 2.71 (dd, J = 13.1, 2.8 Hz, 1H, SCH2b), 2.02 (s, 3H, COCH3), 2.01 (s, 3H, COCH3), 2.00 (s, 3H, COCH3), 1.98–1.94 (m, 1H, H-4), 1.93 (s, 3H, COCH3), 1.06 (s, 9H, 3×CH3, t-Bu), 13C NMR (101 MHz, CDCl3): δ = 170.5, 170.2, 169.3 and 169.1 (4×COCH3), 135.7, 135.7, 135.7, 133.5, 133.4, 129.8, 129.7, 127.7 and 127.7 (10C, arom), 97.6 (C-1), 83.6 (C-2), 83.4 (C-1′), 78.7 (C-3), 75.8 (C-5′), 73.9 (C-3′), 71.1 (C-5), 69.8 (C-2′), 68.3 (C-4′), 64.1 (C-6), 62.0 (C-6′), 61.1, 58.5 and 55.0 (3×OCH3), 43.3 (C-4), 26.8 (4C, 3×CH3, t-Bu and SCH2), 20.6, 20.6, 20.6 and 20.5 (4C, 4×COCH3), 19.3 (Cq, t-Bu). Anal. Calcd for C40H56O14SSi: C, 58.53; H, 6.88; S, 3.89. Found: C, 59.90; H, 7.05; S, 3.60.
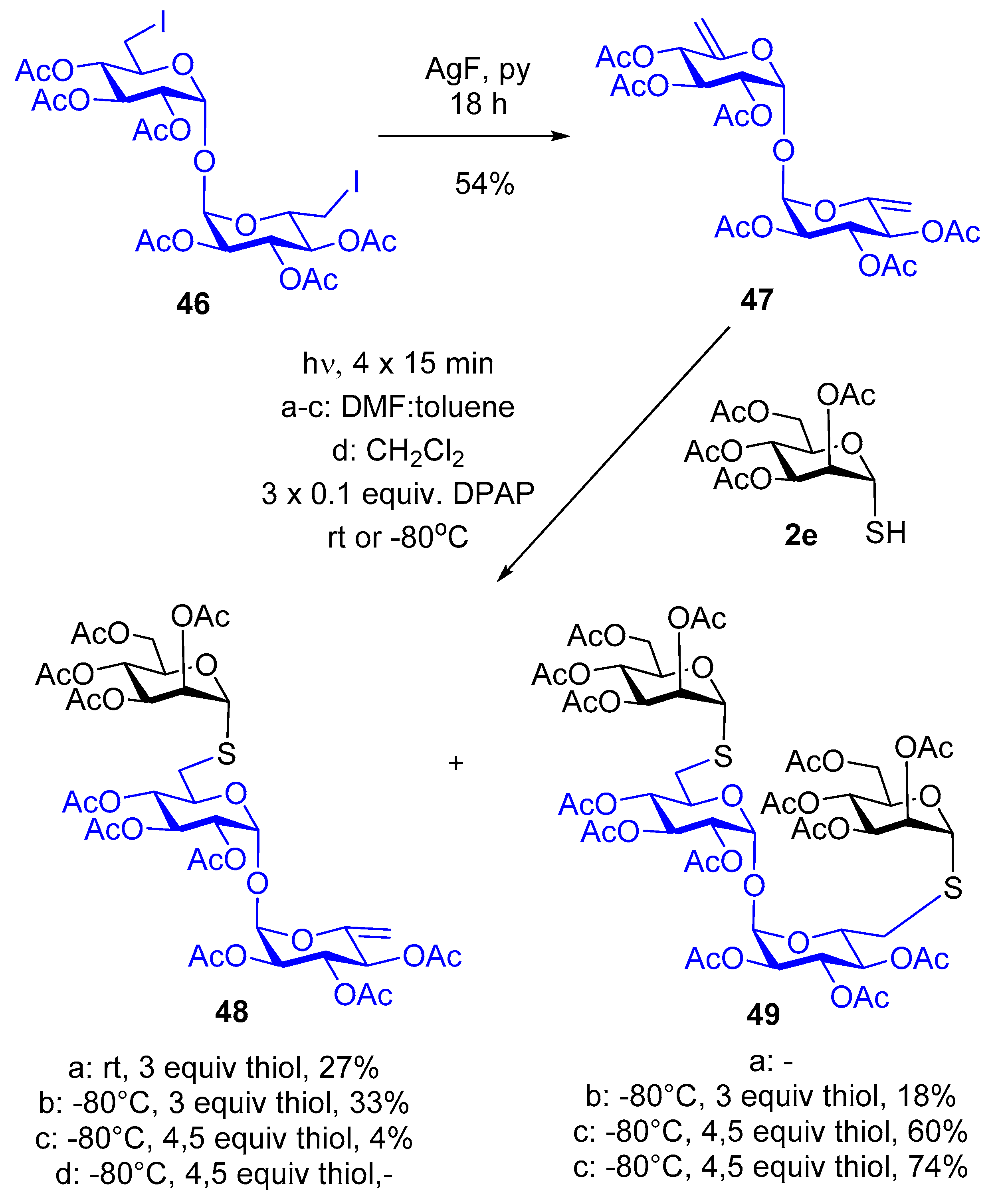
4.2.29. 2,3,4-Tri-O-acetyl-α-d-xylo-hex-5-enopyranosyl-(1→1)-2,3,4-tri-O-acetyl-α-d-xylo-hex-5-enopyranoside (47)
To a solution of 46 (1.63 g, 2 mmol) in pyridine (50 mL) AgF (3.16 g, 25 mmol) was added and the mixture was stirred in the dark overnight, and then the completion of the reaction was controlled by TLC (CH2Cl2:acetone 95:5). Et2O (320 mL) was added to the mixture and the organic layer was washed with 10% aq Na2S2O3 solution and water. The organic phase was dried over Na2SO4, evaporated under reduced pressure, and the residue was purified by column chromatography to give 47 (608 mg).
Yield: 54%, white needles. m.p.: 214.3 °C [α]D20 = + 11.2 (c 0.08, CHCl3). Rf 0.35 (hexane/acetone 7/3). 1H NMR (400 MHz, CDCl3): δ = 5.52 (d, J = 3.6 Hz, 1H, H-1), 5.50-5.35 (m, 2H, H-3 and H-4), 5.02 (dd, J = 8.6, 3.6, Hz, 1H, H-2), 4.66 (t, J = 2.0 Hz, 1H, =CH2a), 4.55 (t, J = 1.8 Hz, 1H, =CH2b), 2.11 (s, 3H, CH3CO), 2.06 (s, 3H, CH3CO), 2.04 (s, 3H, CH3CO).13C NMR (101 MHz, CDCl3): δ = 170.1, 169.5, 169.3, (3C, 3 x CO) 149.7 (C-5), 97.7 (C-6), 90.3 (C-1), 69.7, 69.3, 69.2 (3C, C-2, C-3 and C-4), 20.7, 20.7, 20.6 (3C, 3 x CH3CO). MS (MALDI-TOF) m/z: [M + Na]+ Calcd for C24H30 NaO15 581.1482; Found: 581.323.
4.2.30. 2,3,4-Tri-O-acetyl-6-thio-6-S-(2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl)-α-d-glucopyranosyl-(1→1)-2,3,4-tri-O-acetyl-α-d-xylo-hex-5-enopyranoside (48) and 2,3,4-Tri-O-acetyl-6-thio-6-S-(2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl)-α-d-glucopyranosyl-(1→1)-2,3,4-tri-O-acetyl-6-thio-6-S-(2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl)-α-d-glucopyranoside (49)
Diene 47 (100 mg, 0.18 mmol), thiol 2e (195 mg, 0.54 mmol, 3.0 equiv.) and DPAP (5 mg, 0.1 equiv.) were reacted according to the general method in DMF:toluene 3:1 (3 mL) with four irradiation cycles. The crude product was purified by column chromatography (hexane/acetone 6/4) to give compound 48 (44 mg, 27%) as a colorless foam. The reaction was also carried out in the same scale in DMF:toluene 3:1 at −80 °C to give compounds 48 (55 mg, 33%) and 49 (41 mg, 18%). The reaction was repeated while using 4.5 equiv. of thiol 2e in DMF:toluene 3:1 at −80 °C to give compounds 48 (7 mg, 4 %) and 49 (135 mg, 60%). The reaction was repeated using 4.5 equiv. of thiol 2e in dichloromethane to give compound 49 (164 mg, 74%).
48: Yield: 33%, colorless foam. [α]D20 = +196.6 (c = 0.18, CHCl3). Rf 0.40 (hexane/acetone 6/4). 1H NMR (400 MHz, CDCl3) δ (ppm) 5.51 (d, 1H, J = 3.6 Hz, 1H, H-1enoside), 5.47–5.43 (m, 2H), 5.41 (d, 1H, J = 3.6 Hz, 1H, H-1Glcp), 5.34 (dd, J = 3.2, 1.4 Hz, 1H), 5.33–5.25 (m, 5H), 5.16 (ddd, J = 9.2, 3.6, 0.8 Hz, 1H), 5.06–4.99 (m, 1H), 4.91 (dd, J = 10.2, 3.9 Hz, 1H), 4.67 (t, J = 1.9 Hz, 1H, =CH2a), 4.55 (t, J = 1.8 Hz, 1H, =CH2b), 4.33 (dd, J = 12.2, 5.0 Hz, 1H, H-6aManp), 4.26 (ddd, J = 9.5, 4.9, 2.1 Hz, 1H, H-5Manp), 4.07 (dd, J = 12.2, 2.2 Hz, 1H, H-6bManp), 3.95 (ddd, J = 10.0, 6.9, 3.0 Hz, 1H, H-5Glcp), 2.81 (dd, J = 14.0, 3.1 Hz, 1H, SCH2a), 2.65 (dd, J = 14.0, 7.0 Hz, 1H, SCH2b), 2.18 (s, 3H, CH3CO), 2.13 (s, 3H, CH3CO), 2.12 (s, 3H, CH3CO), 2.11 (s, 3H, CH3CO), 2.05 (s, 3H, CH3CO), 2.05–2.04 (m, 9H, 3×CH3CO), 2.03 (s, 3H, CH3CO), 2.00 (s, 3H, CH3CO). 13C NMR (101 MHz, CDCl3) δ 170.7, 170.247, 169.9, 169.9, 169.8, 169.7, 169.7, 169.4 (10C, 10×CO), 149.8 (C-5), 98.0 (C-6), 91.4, 90.9 (2C, 2×C-1trehalose), 82.6 (C-1Manp), 71.2, 70.9, 70.0, 69.6, 69.8, 69.7, 69.5, 69.4, 69.2 (2×), 66.2 (11C, skeleton), 62.3 (C-6Manp), 31.3 (SCH2), 21.0, 20.9 (2×), 20.8 (5×), 20.7 (2×) (10C, 10×CH3CO). MS (MALDI-TOF) m/z: [M + Na]+ Calcd for C38H50NaO24S 945.231; Found: 945.230.
49: Yield: 74%, colorless foam. [α]D20 = +49.3 (c = 0.15, CHCl3). Rf 0.24 (hexane/acetone 6/4). 1H NMR (400 MHz, CDCl3) δ (ppm) 5.46 (t, J = 9.8 Hz, 1H), 5.36–5.21 (m, 5H), 5.05 (dt, J = 9.6, 6.9 Hz, 2H), 4.39–4.23 (m, 2H), 4.14–3.96 (m, 2H), 2.82 (dd, J = 13.9, 3.0 Hz, 1H, SCH2a), 2.67 (dd, J = 14.0, 6.6 Hz, 1H, SCH2b), 2.17 (s, 3H, CH3CO), 2.11 (s, 6H, 2 x CH3CO), 2.06 (s, 6H, 2×CH3CO), 2.03 (s, 3H, CH3CO), 1.99 (s, 3H, CH3CO).13C NMR (101 MHz, CDCl3) δ 170.6, 170.0, 169.9, 169.8, 169.7, 169.7 (2×) (7×CO), 92.1 (1C, C-1trehalose), 82.4 (C-1Manp), 71.1, 70.8, 70.0, 69.9, 69.5, 69.4, 69.3, 66.2 (8C, skeleton), 62.4 (C-6Manp), 31.3 (SCH2), 20.9, 20.8, 20.7 (3×), 20.6 (7C, 7 x CH3CO). MS (MALDI-TOF) m/z: [M + Na]+ Calcd for C52H70NaO33S2 1309.314; Found: 1309.319.
4.2.31. Methyl (5-acetamido-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-2-thio-d-glycero-α-d-galacto-non-2-ulopyranosyl)-onate-(2→7)-6’-deoxy-1′,2′:3′,4′-di-O-isopropylidene-7′-S-α-d-galacto-heptopyranose (51)
Exomethylene 50 (22 mg, 0.11 mmol) and thiol 2f (66 mg, 0.13 mmol) were reacted according to the general method at −80 °C with three irradiation cycles to give compound 51 (65 mg).
Yield: 69%, colorless syrup. [α]D20 = −4.0 (c = 0.5, CHCl3). Rf 0.29 (CH2Cl2/aceton 8/2). 1H NMR (500 MHz, CDCl3): δ = 5.49 (d, J = 5.0 Hz, 1H, H-1′), 5.38 (ddd, J = 8.0, 5.1, 2.7 Hz, 1H, H-8), 5.31 (dd, J = 8.5, 2.2 Hz, 1H, H-7), 5.16 (d, J = 10.1 Hz, 1H, NH), 4.86 (ddd, J = 11.7, 10.5, 4.6 Hz, 1H, H-4), 4.59 (dd, J = 7.9, 2.3 Hz, 1H, H-3′), 4.32–4.27 (m, 2H, H-9a, H-2′), 4.17 (dd, J = 7.9, 1.8 Hz, 1H, H-4′), 4.11 (dd, J = 12.5, 5.2 Hz, 1H, H-9b), 4.04 (q, J = 10.4 Hz, 1H, H-5), 3.86 (ddd, J = 8.9, 4.0, 1.4 Hz, 1H, H-5’), 3.82 (dd, J = 10.8, 2.2 Hz, 1H, H-6), 3.80 (s, 3H, OCH3), 2.84–2.68 (m, 3H, H-7′a, H-7′b, H-3a), 2.14 (s, 6H, 2×COCH3), 2.03 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 2.01–1.96 (m, 1H, H-3b), 1.93–1.88 (m, 1H, H-6’a), 1.87 (s, 3H, COCH3), 1.81–1.74 (m, 1H, H-6’b), 1.59 (s, 3H, CH3, i-Pr), 1.44 (s, 3H, CH3, i-Pr), 1.34 (s, 3H, CH3, i-Pr), 1.32 (s, 3H, CH3, i-Pr); 13C NMR (91 MHz, CDCl3) δ (ppm) 171.1, 170.8, 170.3 and 170.0 (6C, 6×CO), 109.2 and 108.6 (2C, 2Cq, i-Pr), 96.6 (C-1′), 83.4 (C-2), 74.3 (C-6), 72.7 (C-4′), 71.1 (C-3’), 70.7 (C-2’), 69.8 (C-4), 68.6 (C-8), 67.4 (C-7), 66.3 (C-5’), 62.3 (C-9), 53.1 (OCH3), 49.5 (C-5), 38.2 (C-3), 30.8 (C-6’), 26.2 (CH3, i-Pr), 26.1 (CH3, i-Pr), 25.4 (C-7’), 25.2 (CH3, i-Pr), 24.5 (CH3, i-Pr), 23.4, 21.2, 21.0 and 20.9 (5C, 5×COCH3); MS (ESI-QTOF) m/z: [M+Na]+ Calcd for C33H49NNaO17S 786.2619; Found 786.2616.



 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
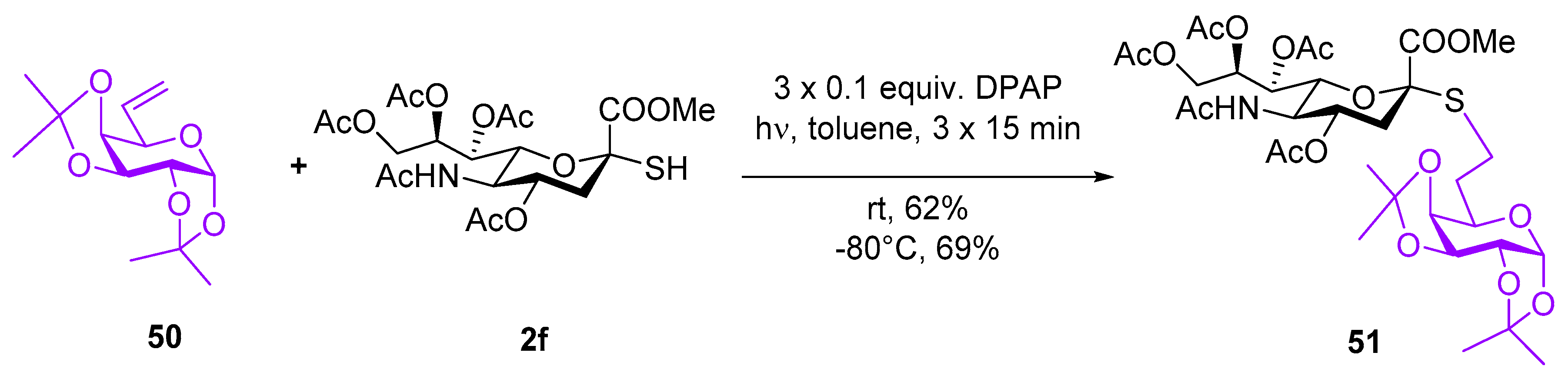{kind=link}
{kind=link}
{kind=link}











