The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases

Abstract
1. Introduction
2. The Human Cardiac Action Potential
2.1. General Properties of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs)
2.1.1. hiPSC Differentiation into Cardiomyocytes
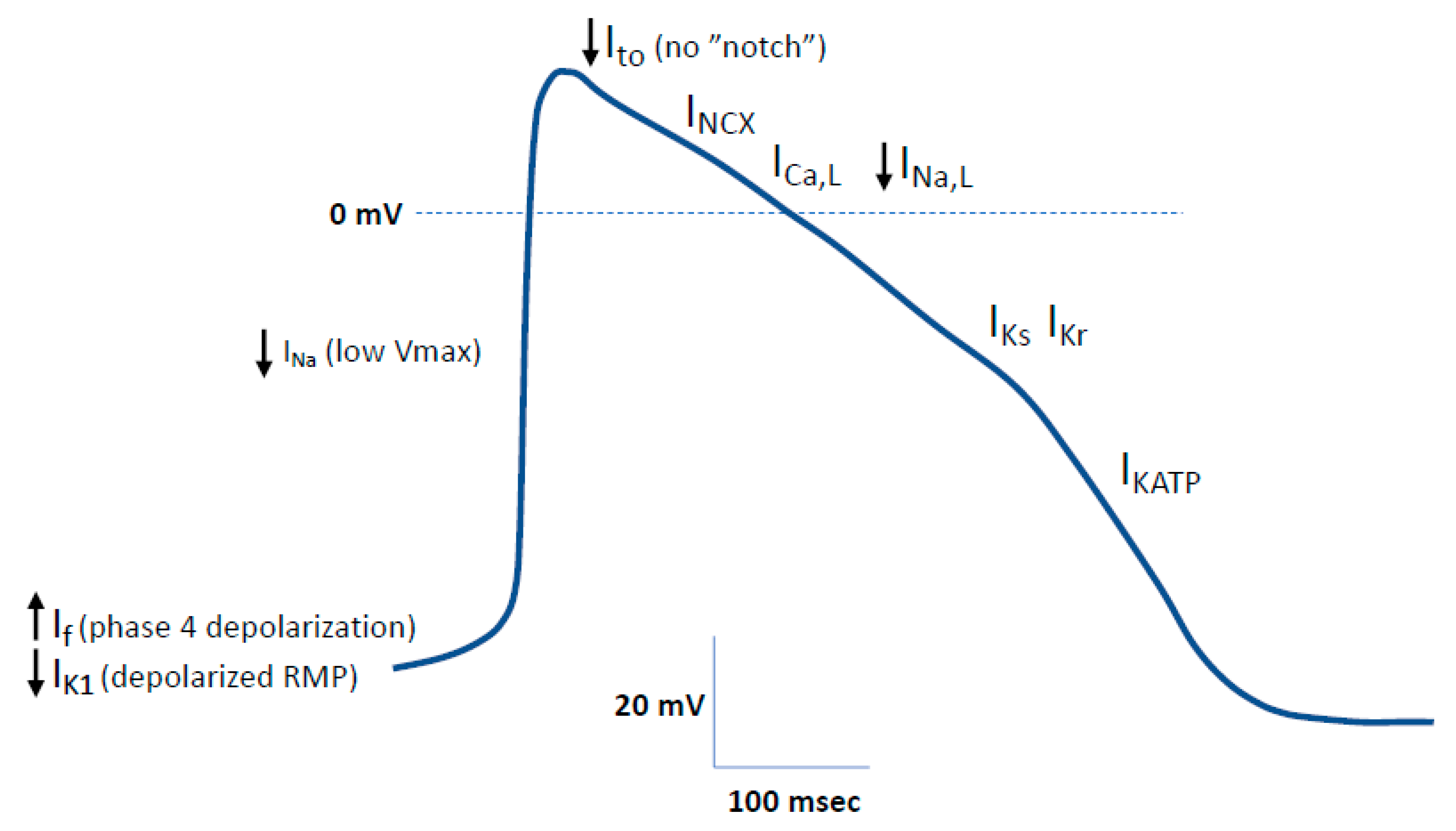
2.1.2. Electrophysiological Phenotype of hiPSC-CMs
2.2. Technical Considerations for the Electrophysiological Characterization of hiPSC-CMs
3. Inherited Arrhythmogenic Diseases
3.1. Congenital Long QT Syndrome (LQTS)
3.2. The Brugada Syndrome (BrS)
3.3. Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
3.4. Short QT Syndrome (SQTS)
3.5. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
3.6. Hypertrophy Cardiomyopathy (HCM)
3.7. Dilated Cardiomyopathy (DCM)
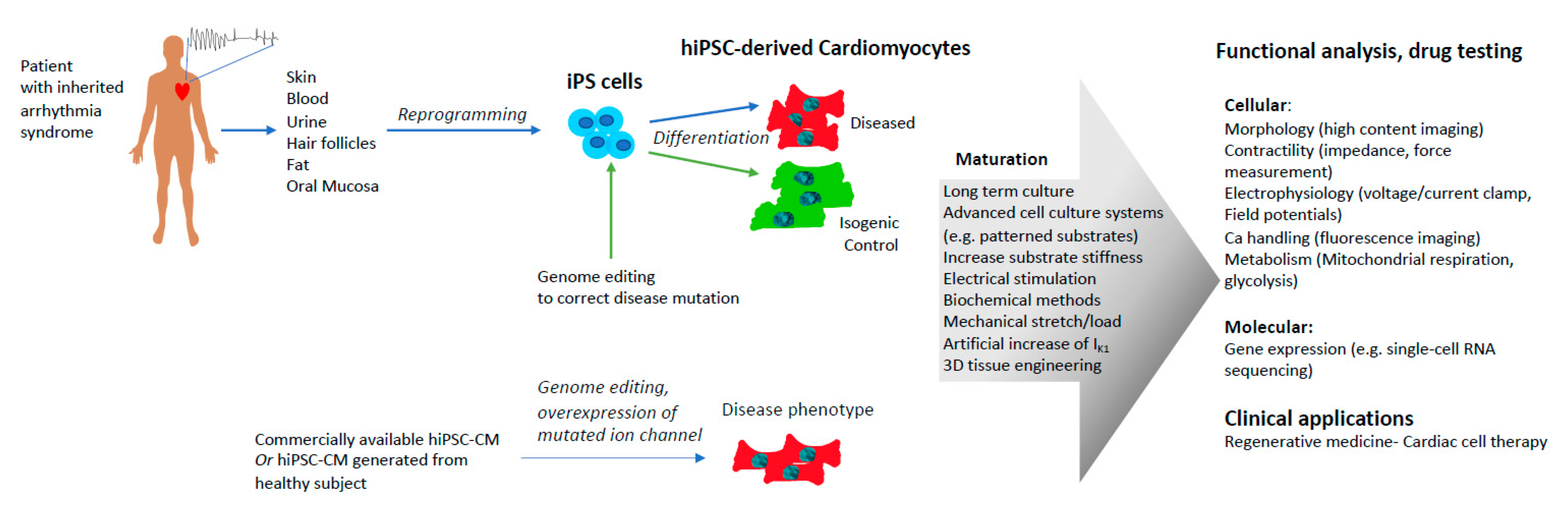
4. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Model to Study Inherited Arrhythmogenic Cardiac Diseases
4.1. Patient-Specific hiPSC-CMs
4.2. Genome Editing
4.3. Overexpression of Mutated Genes in hiPSC-CMs
5. Limitations of Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) to Model Inherited Arrhythmogenic Cardiac Diseases
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C. Induced pluripotent stem cell-derived cardiomyocytes: Boutique science or valuable arrhythmia model? Circ. Res. 2013, 112, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Gnecchi, M.; Schwartz, P.J. Long QT Syndrome Modelling with Cardiomyocytes Derived from Human-induced Pluripotent Stem Cells. Arrhythmia Electrophysiol. Rev. 2019, 8, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Strauss, D.G.; Blinova, K. Clinical Trials in a Dish. Trends Pharmacol. Sci. 2017, 38, 4–7. [Google Scholar] [CrossRef]
- Fermini, B.; Coyne, S.T.; Coyne, K.P. Clinical Trials in a Dish: A Perspective on the Coming Revolution in Drug Development. SLAS Discov. 2018, 23, 765–776. [Google Scholar] [CrossRef]
- Blinova, K.; Schocken, D.; Patel, D.; Daluwatte, C.; Vicente, J.; Wu, J.C.; Strauss, D.G. Clinical Trial in a Dish: Personalized Stem Cell–Derived Cardiomyocyte Assay Compared With Clinical Trial Results for Two QT-Prolonging Drugs. Clin. Transl. Sci. 2019, 12, 687–697. [Google Scholar] [CrossRef]
- Clauss, S.; Bleyer, C.; Schüttler, D.; Tomsits, P.; Renner, S.; Klymiuk, N.; Wakili, R.; Massberg, S.; Wolf, E.; Kääb, S. Animal models of arrhythmia: Classic electrophysiology to genetically modified large animals. Nat. Rev. Cardiol. 2019, 16, 457–475. [Google Scholar] [CrossRef]
- Casini, S.; Verkerk, A.O.; Remme, C.A. Human iPSC-Derived Cardiomyocytes for Investigation of Disease Mechanisms and Therapeutic Strategies in Inherited Arrhythmia Syndromes: Strengths and Limitations. Cardiovasc. Drugs Ther. 2017, 31, 325–344. [Google Scholar] [CrossRef]
- Garg, P.; Garg, V.; Shrestha, R.; Sanguinetti, M.C.; Kamp, T.J.; Wu, J.C. Human induced pluripotent stem cell-derived cardiomyocytes as models for cardiac channelopathies: A primer for non-electrophysiologists. Circ. Res. 2018, 123, 224–243. [Google Scholar] [CrossRef]
- Nerbonne, J.M.; Kass, R.S. Molecular Physiology of Cardiac Repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef]
- Endoh, M. Force-frequency relationship in intact mammalian ventricular myocardium: Physiological and pathophysiological relevance. Eur. J. Pharmacol. 2004, 500, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Bers, D.M.; Despa, S. Cardiac Myocytes Ca2+ and Na+ Regulation in Normal and Failing Hearts. J. Pharmacol. Sci. 2006, 100, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Schram, G.; Pourrier, M.; Melnyk, P.; Nattel, S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ. Res. 2002, 90, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Matsa, E.; Wu, J.C. Induced pluripotent stem cells: At the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 2016, 13, 333–349. [Google Scholar] [CrossRef]
- Burridge, P.W.; Diecke, S.; Matsa, E.; Sharma, A.; Wu, H.; Wu, J.C. Modeling Cardiovascular Diseases with Patient-Specific Human Pluripotent Stem Cell-Derived Cardiomyocytes. Methods Mol. Biol. 2016, 1353, 119–130. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yoshida, Y.; Yamanaka, S. iPS cells: A source of cardiac regeneration. J. Mol. Cell. Cardiol. 2011, 50, 327–332. [Google Scholar] [CrossRef]
- Shiba, Y.; Hauch, K.D.; Laflamme, M.A. Cardiac applications for human pluripotent stem cells. Curr. Pharm. Des. 2009, 15, 2791–2806. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, A.S.T.; Macadangdang, J.; Leung, W.; Laflamme, M.A.; Kim, D.H. Human iPSC-derived cardiomyocytes and tissue engineering strategies for disease modeling and drug screening. Biotechnol. Adv. 2017, 35, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically Defined and Small Molecule-Based Generation of Human Cardiomyocytes. Nat. Methods 2014, 11, 855. [Google Scholar] [CrossRef]
- Weng, Z.; Kong, C.W.; Ren, L.; Karakikes, I.; Geng, L.; He, J.; Chow, M.Z.Y.; Mok, C.F.; Harvey, C.; Sarah, W.; et al. A simple, cost-effective but highly efficient system for deriving ventricular cardiomyocytes from human pluripotent stem cells. Stem Cells Dev. 2014, 23, 1704–1716. [Google Scholar] [CrossRef] [PubMed]
- Cyganek, L.; Tiburcy, M.; Sekeres, K.; Gerstenberg, K.; Bohnenberger, H.; Lenz, C.; Henze, S.; Stauske, M.; Salinas, G.; Zimmermann, W.H.; et al. Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 2018, 3, e99941. [Google Scholar] [CrossRef]
- Protze, S.I.; Liu, J.; Nussinovitch, U.; Ohana, L.; Backx, P.H.; Gepstein, L.; Keller, G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017, 35, 56–68. [Google Scholar] [CrossRef]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva, S.M.; Lopes, S.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–230. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef]
- Gibson, J.K.; Yue, Y.; Bronson, J.; Palmer, C.; Numann, R. Human stem cell-derived cardiomyocytes detect drug-mediated changes in action potentials and ion currents. J. Pharmacol. Toxicol. Methods 2014, 70, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Mummery, C.L.; Wilde, A.A.M.; Bezzina, C.R.; Verkerk, A.O. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front. Physiol. 2012, 3, 346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lan, H.; El-Battrawy, I.; Li, X.; Buljubasic, F.; Sattler, K.; Yücel, G.; Lang, S.; Tiburcy, M.; Zimmermann, W.-H.; et al. Ion Channel Expression and Characterization in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Int. 2018, 2018, 6067096. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kryshtal, D.O.; Feaster, T.K.; Sánchez-Freire, V.; Zhang, J.; Kamp, T.J.; Hong, C.C.; Wu, J.C.; Knollmann, B.C. Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J. Mol. Cell. Cardiol. 2015, 85, 79–88. [Google Scholar] [CrossRef]
- Pieske, B.; Maier, L.S.; Bers, D.M.; Hasenfuss, G. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ. Res. 1999, 85, 38–46. [Google Scholar] [CrossRef]
- Germanguz, I.; Sedan, O.; Zeevi-Levin, N.; Shtrichman, R.; Barak, E.; Ziskind, A.; Eliyahu, S.; Meiry, G.; Amit, M.; Itskovitz-Eldor, J.; et al. Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J. Cell. Mol. Med. 2011, 15, 38–51. [Google Scholar] [CrossRef]
- Ruan, J.-L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 2016, 134, 1557–1567. [Google Scholar] [CrossRef]
- Maier, L.S.; Bers, D.M.; Pieske, B. Differences in Ca(2+)-handling and sarcoplasmic reticulum Ca(2+)-content in isolated rat and rabbit myocardium. J. Mol. Cell. Cardiol. 2000, 32, 2249–2258. [Google Scholar] [CrossRef]
- Crumb, W.J.; Vicente, J.; Johannesen, L.; Strauss, D.G. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods 2016, 81, 251–262. [Google Scholar] [CrossRef]
- Gilchrist, K.H.; Lewis, G.F.; Gay, E.A.; Sellgren, K.L.; Grego, S. High-throughput cardiac safety evaluation and multi-parameter arrhythmia profiling of cardiomyocytes using microelectrode arrays. Toxicol. Appl. Pharmacol. 2015, 288, 249–257. [Google Scholar] [CrossRef]
- Pradhapan, P.; Kuusela, J.; Viik, J.; Aalto-Setälä, K.; Hyttinen, J. Cardiomyocyte MEA Data Analysis (CardioMDA)—A Novel Field Potential Data Analysis Software for Pluripotent Stem Cell Derived Cardiomyocytes. PLoS ONE 2013, 8, e73637. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Stohlman, J.; Dang, Q.; Strauss, D.G.; Blinova, K. Assessment of Proarrhythmic Potential of Drugs in Optogenetically Paced Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 2019, 170, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Ferenczi, E.A.; Tan, X.; Huang, C.L.-H. Principles of Optogenetic Methods and Their Application to Cardiac Experimental Systems. Front. Physiol. 2019, 10, 1096. [Google Scholar] [CrossRef] [PubMed]
- Laurila, E.; Ahola, A.; Hyttinen, J.; Aalto-Setälä, K. Methods for in vitro functional analysis of iPSC derived cardiomyocytes—Special focus on analyzing the mechanical beating behavior. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Long QT syndrome: Reduced repolarization reserve and the genetic link. J. Intern. Med. 2006, 259, 59–69. [Google Scholar] [CrossRef]
- Roden, D.M. Long-QT Syndrome. N. Engl. J. Med. 2008, 358, 169–176. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef]
- Skinner, J.R.; Winbo, A.; Abrams, D.; Vohra, J.; Wilde, A.A. Channelopathies That Lead to Sudden Cardiac Death: Clinical and Genetic Aspects. Heart Lung Circ. 2019, 28, 22–30. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.M.; Ackerman, M.J.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009, 6, 1297–1303. [Google Scholar] [CrossRef]
- Liang, W.; Gasparyan, L.; AlQarawi, W.; Davis, D.R. Disease modeling of cardiac arrhythmias using human induced pluripotent stem cells. Expert Opin. Biol. Ther. 2019, 19, 313–333. [Google Scholar] [CrossRef]
- Marban, E. Cardiac channelopathies 1. Nature 2002, 415, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Cranefield, P.F. Action potentials, afterpotentials, and arrhythmias. Circ. Res. 1977, 41, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.R.; Park, K.; Alweis, R. Long QT Syndrome: A Comprehensive Review of the Literature and Current Evidence. Curr. Probl. Cardiol. 2019, 44, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Modell, S.M.; Lehmann, M.H. The long QT syndrome family of cardiac ion channelopathies: A HuGE review. Genet. Med. 2006, 8, 143–155. [Google Scholar] [CrossRef]
- Claridge, S.; Yue, A. Cardiac channelopathies. Medicine (United Kingdom) 2018, 46, 618–621. [Google Scholar] [CrossRef]
- Camm, A.J.; Janse, M.J.; Roden, D.M.; Rosen, M.R.; Cinca, J.; Cobbe, S.M. Congenital and acquired long QT syndrome. Eur. Heart J. 2000, 21, 1232–1237. [Google Scholar] [CrossRef]
- Kass, R.S.; Moss, A.J. Long QT syndrome: Novel insights into the mechanisms of cardiac arrhythmias. J. Clin. Investig. 2003, 112, 810–815. [Google Scholar] [CrossRef]
- Shah, M.; Akar, F.G.; Tomaselli, G.F. Molecular basis of arrhythmias. Circulation 2005, 112, 2517–2529. [Google Scholar] [CrossRef]
- Fernández-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology (Basel) 2017, 6, 7. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Crotti, L.; Insolia, R. Long-QT Syndrome. Circ. Arrhythmia Electrophysiol. 2012, 5, 868–877. [Google Scholar] [CrossRef]
- Lu, J.T.; Kass, R.S. Recent progress in congenital long QT syndrome. Curr. Opin. Cardiol. 2010, 25, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Kotta, M.-C.; Sala, L.; Ghidoni, A.; Badone, B.; Ronchi, C.; Parati, G.; Zaza, A.; Crotti, L. Calmodulinopathy: A Novel, Life-Threatening Clinical Entity Affecting the Young. Front. Cardiovasc. Med. 2018, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Yan, G.X.; Antzelevitch, C. Cellular basis for the electrocardiographic J wave. Circulation 1996, 93, 372–379. [Google Scholar] [CrossRef]
- Savio-Galimberti, E.; Argenziano, M.; Antzelevitch, C. Cardiac arrhythmias related to sodium channel dysfunction. In Handbook of Experimental Pharmacology; Springer New York LLC: New York, NY, USA, 2018; Volume 246, pp. 331–354. [Google Scholar]
- Tukkie, R.; Sogaard, P.; Vleugels, J.; de Groot, I.K.L.M.; Wilde, A.A.M.; Tan, H.L. Delay in right ventricular activation contributes to Brugada syndrome. Circulation 2004, 109, 1272–1277. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient outward current (I to) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011, 8, 1024–1032. [Google Scholar] [CrossRef]
- Medeiros-domingo, A.; Tan, B.; Crotti, L.; Tester, D.J.; Eckhardt, L.; Cuoretti, A.; Kroboth, S.L.; Song, C.; Zhou, Q.; Kopp, D.; et al. Gain-of-Function Mutation, S422L, in the KCNJ8-Encoded Cardiac KATP Channel Kir6.1 as a Pathogenic Substrate for J Wave Syndromes. Heart Rhythm 2010, 7, 1466–1471. [Google Scholar] [CrossRef]
- Tse, G.; Liu, T.; Li, K.H.C.; Laxton, V.; Chan, Y.W.F.; Keung, W.; Li, R.A.; Yan, B.P. Electrophysiological Mechanisms of Brugada Syndrome: Insights from Pre-clinical and Clinical Studies. Front. Physiol. 2016, 7, 467. [Google Scholar] [CrossRef]
- Zhao, Y.-T.; Valdivia, C.R.; Gurrola, G.B.; Powers, P.P.; Willis, B.C.; Moss, R.L.; Jalife, J.; Valdivia, H.H. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc. Natl. Acad. Sci. USA 2015, 112, E1669–E1677. [Google Scholar] [CrossRef]
- Roston, T.M.; Yuchi, Z.; Kannankeril, P.J.; Hathaway, J.; Vinocur, J.M.; Etheridge, S.P.; Potts, J.E.; Maginot, K.R.; Salerno, J.C.; Cohen, M.I.; et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: Findings from an international multicentre registry. Europace 2018, 20, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; Ngoc, D.D.; Coumel, P. Catecholaminergic Polymorphic Ventricular Tachycardia in Children. Circulation 1995, 91, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Chen, S.R.W. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Venetucci, L.; Denegri, M.; Napolitano, C.; Priori, S.G. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 2012, 9, 561–575. [Google Scholar] [CrossRef]
- Wit, A.L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin. Electrophysiol. 2018, 41, 883–896. [Google Scholar] [CrossRef]
- Gaita, F.; Giustetto, C.; Bianchi, F.; Wolpert, C.; Schimpf, R.; Riccardi, R.; Grossi, S.; Richiardi, E.; Borggrefe, M. Short QT Syndrome. Circulation 2003, 108, 965–970. [Google Scholar] [CrossRef]
- Anttonen, O.; Junttila, M.J.; Rissanen, H.; Reunanen, A.; Viitasalo, M.; Huikuri, H.V. Prevalence and Prognostic Significance of Short QT Interval in a Middle-Aged Finnish Population. Circulation 2007, 116, 714–720. [Google Scholar] [CrossRef]
- Funada, A.; Hayashi, K.; Ino, H.; Fujino, N.; Uchiyama, K.; Sakata, K.; Masuta, E.; Sakamoto, Y.; Tsubokawa, T.; Yamagishi, M. Assessment of QT Intervals and Prevalence of Short QT Syndrome in Japan. Clin. Cardiol. 2008, 31, 270–274. [Google Scholar] [CrossRef]
- Kobza, R.; Roos, M.; Niggli, B.; Abächerli, R.; Lupi, G.A.; Frey, F.; Schmid, J.J.; Erne, P. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009, 6, 652–657. [Google Scholar] [CrossRef]
- Mazzanti, A.; O’Rourke, S.; Ng, K.; Miceli, C.; Borio, G.; Curcio, A.; Esposito, F.; Napolitano, C.; Priori, S.G. The usual suspects in sudden cardiac death of the young: A focus on inherited arrhythmogenic diseases. Expert Rev. Cardiovasc. Ther. 2014, 12, 499–519. [Google Scholar] [CrossRef]
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Recent Advances in Short QT Syndrome. Front. Cardiovasc. Med. 2018, 5, 149. [Google Scholar] [CrossRef] [PubMed]
- Hancox, J.C.; Whittaker, D.G.; Du, C.; Stuart, A.G.; Zhang, H. Emerging therapeutic targets in the short QT syndrome. Expert Opin. Ther. Targets 2018, 22, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Calkins, H. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy—Three Decades of Progress—. Circ. J. 2015, 79, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Morgan, R.D.; Chambers, J.C.; McKenna, W.J. Arrhythmogenic Cardiomyopathy: Etiology, Diagnosis, and Treatment. Annu. Rev. Med. 2010, 61, 233–253. [Google Scholar] [CrossRef]
- Bauce, B.; Rampazzo, A.; Basso, C.; Mazzotti, E.; Rigato, I.; Steriotis, A.; Beffagna, G.; Lorenzon, A.; De Bortoli, M.; Pilichou, K.; et al. Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm 2011, 8, 1686–1695. [Google Scholar] [CrossRef]
- Coppini, R.; Ferrantini, C.; Yao, L.; Fan, P.; Del Lungo, M.; Stillitano, F.; Sartiani, L.; Tosi, B.; Suffredini, S.; Tesi, C.; et al. Late Sodium Current Inhibition Reverses Electromechanical Dysfunction in Human Hypertrophic Cardiomyopathy. Circulation 2013, 127, 575–584. [Google Scholar] [CrossRef]
- Moore, B.; Semsarian, C.; Chan, K.H.; Sy, R.W. Sudden Cardiac Death and Ventricular Arrhythmias in Hypertrophic Cardiomyopathy. Heart Lung Circ. 2019, 28, 146–154. [Google Scholar] [CrossRef]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, epidemiology, and global burden of cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genom. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef]
- Lam, C.K.; Wu, J.C. Disease modelling and drug discovery for hypertrophic cardiomyopathy using pluripotent stem cells: How far have we come? Eur. Heart J. 2018, 39, 3893–3895. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Maron, B.A.; Loscalzo, J. Moving Beyond the Sarcomere to Explain Heterogeneity in Hypertrophic Cardiomyopathy: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Luedde, M.; Katus, H.A. Mechanisms of disease: Hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Goff, Z.D.; Calkins, H. Sudden death related cardiomyopathies—Hypertrophic cardiomyopathy. Prog. Cardiovasc. Dis. 2019, 62, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996, 93, 841–842. [CrossRef]
- Wu, T.J.; Ong, J.J.; Hwang, C.; Lee, J.J.; Fishbein, M.C.; Czer, L.; Trento, A.; Blanche, C.; Kass, R.M.; Mandel, W.J.; et al. Characteristics of wave fronts during ventricular fibrillation in human hearts with dilated cardiomyopathy: Role of increased fibrosis in the generation of reentry. J. Am. Coll. Cardiol. 1998, 32, 187–196. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-Specific Induced Pluripotent Stem -Cell Models for Long-QT Syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef]
- Egashira, T.; Yuasa, S.; Suzuki, T.; Aizawa, Y.; Yamakawa, H.; Matsuhashi, T.; Ohno, Y.; Tohyama, S.; Okata, S.; Seki, T.; et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc. Res. 2012, 95, 419–429. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef]
- Mehta, A.; Ramachandra, C.J.A.; Singh, P.; Chitre, A.; Lua, C.H.; Mura, M.; Crotti, L.; Wong, P.; Schwartz, P.J.; Gnecchi, M.; et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. Heart J. 2018, 39, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Malan, D.; Friedrichs, S.; Fleischmann, B.K.; Sasse, P. Cardiomyocytes obtained from induced pluripotent stem cells with Long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ. Res. 2011, 109, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; Casini, S.; Van Den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Van Oostwaard, D.W.; Wilde, A.A.M.; Bezzina, C.R.; et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wei, H.; Lu, J.; Ho, S.; Zhang, G.; Sun, X.; Oh, Y.; Tan, S.H.; Ng, M.L.; Shim, W.; et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Terrenoire, C.; Wang, K.; Chan Tung, K.W.; Chung, W.K.; Pass, R.H.; Lu, J.T.; Jean, J.-C.; Omari, A.; Sampson, K.J.; Kotton, D.N.; et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J. Gen. Physiol. 2012, 141, 61–72. [Google Scholar] [CrossRef]
- Fatima, A.; Kaifeng, S.; Dittmann, S.; Xu, G.; Gupta, M.K.; Linke, M.; Zechner, U.; Nguemo, F.; Milting, H.; Farr, M.; et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long qt syndrome type 3 patients. PLoS ONE 2013, 8, e83005. [Google Scholar] [CrossRef]
- Ma, D.; Wei, H.; Zhao, Y.; Lu, J.; Li, G.; Sahib, N.B.E.; Tan, T.H.; Wong, K.Y.; Shim, W.; Wong, P.; et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int. J. Cardiol. 2013, 168, 5277–5286. [Google Scholar] [CrossRef]
- Bezzina, C.; Veldkamp, M.W.; van Den Berg, M.P.; Postma, A.V.; Rook, M.B.; Viersma, J.W.; van Langen, I.M.; Tan-Sindhunata, G.; Bink-Boelkens, M.T.; van Der Hout, A.H.; et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 1999, 85, 1206–1213. [Google Scholar] [CrossRef]
- Okata, S.; Yuasa, S.; Suzuki, T.; Ito, S.; Makita, N.; Yoshida, T.; Li, M.; Kurokawa, J.; Seki, T.; Egashira, T.; et al. Embryonic type Na + channel β-subunit, SCN3B masks the disease phenotype of Brugada syndrome. Sci. Rep. 2016, 6, 34198. [Google Scholar] [CrossRef]
- Kuroda, Y.; Yuasa, S.; Watanabe, Y.; Ito, S.; Egashira, T.; Seki, T.; Hattori, T.; Ohno, S.; Kodaira, M.; Suzuki, T.; et al. Flecainide ameliorates arrhythmogenicity through NCX flux in Andersen-Tawil syndrome-iPS cell-derived cardiomyocytes. Biochem. Biophys. Rep. 2017, 9, 245–256. [Google Scholar] [CrossRef]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using iPS cells to investigate cardiac phenotypes in patients with Timothy Syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Makiyama, T.; Harita, T.; Sasaki, K.; Wuriyanghai, Y.; Hayano, M.; Nishiuchi, S.; Kohjitani, H.; Hirose, S.; Chen, J.; et al. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum. Mol. Genet. 2017, 26, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.; Sala, L.; Dreizehnter, L.; Crotti, L.; Sinnecker, D.; Mura, M.; Pane, L.S.; Altomare, C.; Torre, E.; Mostacciuolo, G.; et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 531–541. [Google Scholar] [CrossRef]
- Limpitikul, W.B.; Dick, I.E.; Tester, D.J.; Boczek, N.J.; Limphong, P.; Yang, W.; Choi, M.H.; Babich, J.; Disilvestre, D.; Kanter, R.J.; et al. A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome. Circ. Res. 2017, 120, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Johnson, C.N.; Graf, E.; De Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin Mutations Associated With Recurrent Cardiac Arrest in Infants. Circulation 2013, 127, 1009–1017. [Google Scholar] [CrossRef]
- Fatima, A.; Xu, G.; Shao, K.; Papadopoulos, S.; Lehmann, M.; Arnáiz-cot, J.J.; Rosa, A.O.; Matzkies, M.; Dittmann, S.; Stone, S.L.; et al. Cellular Physiology Biochemistry and Biochemistr y In vitro Modeling of Ryanodine Receptor 2 Dysfunction Using Human Induced Pluripotent Stem Cells. Cell. Physiol. Biochem. 2011, 28, 579–592. [Google Scholar] [CrossRef]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef]
- Jung, C.B.; Moretti, A.; Mederos y Schnitzler, M.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef]
- Novak, A.; Barad, L.; Zeevi-Levin, N.; Shick, R.; Shtrichman, R.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Cardiomyocytes generated from CPVT D307H patients are arrhythmogenic in response to β-adrenergic stimulation. J. Cell. Mol. Med. 2012, 16, 468–482. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Lodola, F.; Miragoli, M.; Denegri, M.; Avelino-Cruz, J.E.; Buonocore, M.; Nakahama, H.; Portararo, P.; Bloise, R.; Napolitano, C.; et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013, 4, e843-11. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Haviland, S.; Wei, H.; Šarić, T.; Fatima, A.; Hescheler, J.; Cleemann, L.; Morad, M. Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell Calcium 2013, 54, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Paavola, J.; Väänänen, H.; Larsson, K.; Penttinen, K.; Toivonen, L.; Kontula, K.; Laine, M.; Aalto-Setälä, K.; Swan, H.; Viitasalo, M. Slowed depolarization and irregular repolarization in catecholaminergic polymorphic ventricular tachycardia: A study from cellular Ca2+ transients and action potentials to clinical monophasic action potentials and electrocardiography. Europace 2016, 18, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.L.; Horváth, A.; Di Mauro, V.; Koivumäki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Krämer, E.; et al. Disease modeling of a mutation in a -actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, e11115, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Schick, R.; Mekies, L.N.; Shemer, Y.; Eisen, B.; Hallas, T.; Ben Jehuda, R.; Ben-Ari, M.; Szantai, A.; Willi, L.; Shulman, R.; et al. Functional abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS ONE 2018, 13, e0205719. [Google Scholar] [CrossRef]
- Ma, D.; Wei, H.; Lu, J.; Huang, D.; Liu, Z.; Loh, L.J.; Islam, O.; Liew, R.; Shim, W.; Cook, S.A. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2015, 6, 39. [Google Scholar] [CrossRef]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef]
- Takaki, T.; Inagaki, A.; Chonabayashi, K.; Inoue, K.; Miki, K.; Ohno, S.; Makiyama, T.; Horie, M.; Yoshida, Y. Optical Recording of Action Potentials in Human Induced Pluripotent Stem Cell-Derived Cardiac Single Cells and Monolayers Generated from Long QT Syndrome Type 1 Patients. Stem Cells Int. 2019, 2019, 7532657. [Google Scholar] [CrossRef]
- Zhang, M.; D’Aniello, C.; Verkerk, A.O.; Wrobel, E.; Frank, S.; Ward-Van Oostwaard, D.; Piccini, I.; Freund, C.; Rao, J.; Seebohm, G.; et al. Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: Disease mechanisms and pharmacological rescue. Proc. Natl. Acad. Sci. USA 2014, 111, E5383–E5392. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, P.; Lan, F.; Wu, H.; Lisowski, L.; Gu, M.; Hu, S.; Kay, M.A.; Urnov, F.D.; Shinnawi, R.; et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J. Am. Coll. Cardiol. 2014, 64, 451–459. [Google Scholar] [CrossRef]
- Matsa, E.; Rajamohan, D.; Dick, E.; Young, L.; Mellor, I.; Staniforth, A.; Denning, C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur. Heart J. 2011, 32, 952–962. [Google Scholar] [CrossRef]
- Jouni, M.; Si-Tayeb, K.; Es-Salah-Lamoureux, Z.; Latypova, X.; Champon, B.; Caillaud, A.; Rungoat, A.; Charpentier, F.; Loussouarn, G.; Baró, I.; et al. Toward personalized medicine: Using cardiomyocytes differentiated from urine-derived pluripotent stem cells to recapitulate electrophysiological characteristics of type 2 long QT syndrome. J. Am. Heart Assoc. 2015, 4, e002159. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.; Casini, S.; Davis, R.P.; D’Aniello, C.; Haas, J.; Ward-van Oostwaard, D.; Tertoolen, L.G.J.; Jung, C.B.; Elliott, D.A.; Welling, A.; et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013, 32, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Pourrier, M.; Dou, Y.; Luerman, G.; Fedida, D. Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) to model arrhythmogenic diseases. J. Pharmacol. Toxicol. Methods 2018, 93, 118. [Google Scholar] [CrossRef]
- Gélinas, R.; El Khoury, N.; Chaix, M.A.; Beauchamp, C.; Alikashani, A.; Ethier, N.; Boucher, G.; Villeneuve, L.; Robb, L.; Latour, F.; et al. Characterization of a Human Induced Pluripotent Stem Cell-Derived Cardiomyocyte Model for the Study of Variant Pathogenicity: Validation of a KCNJ2 Mutation. Circ. Cardiovasc. Genet. 2017, 10, e001755. [Google Scholar] [CrossRef] [PubMed]
- Chavali, N.V.; Kryshtal, D.O.; Parikh, S.S.; Wang, L.; Glazer, A.M.; Blackwell, D.J.; Kroncke, B.M.; Shoemaker, M.B.; Knollmann, B.C. The patient-independent human iPSC model—A new tool for rapid determination of genetic variant pathogenicity in long QT syndrome. Heart Rhythm 2019, 16, 1686–1695. [Google Scholar] [CrossRef]
- Guo, F.; Sun, Y.; Wang, X.; Wang, H.; Wang, J.; Gong, T.; Chen, X.; Zhang, P.; Su, L.; Fu, G.; et al. Patient-Specific and Gene-Corrected Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Short QT Syndrome. Circ. Res. 2019, 124, 66–78. [Google Scholar] [CrossRef]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Vermglinchan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell–Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef]
- Caspi, O.; Huber, I.; Gepstein, A.; Arbel, G.; Maizels, L.; Boulos, M.; Gepstein, L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ. Cardiovasc. Genet. 2013, 6, 557–568. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal Calcium Handling Properties Underlie Familial Hypertrophic Cardiomyopathy Pathology in Patient-Specific Induced Pluripotent Stem Cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef]
- Hallas, T.; Eisen, B.; Shemer, Y.; Ben Jehuda, R.; Mekies, L.N.; Naor, S.; Schick, R.; Eliyahu, S.; Reiter, I.; Vlodavsky, E.; et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell–derived cardiomyocytes. J. Cell. Mol. Med. 2018, 22, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm 2018, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra47. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.F.; Ho, J.C.Y.; Choi, S.W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.W.; Simpson, M.A.; Lai, W.H.; Chan, Y.C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef] [PubMed]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Shemer, Y.; Baskin, P.; Reiter, I.; Willi, L.; Freimark, D.; Gherghiceanu, M.; et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. J. Cell. Mol. Med. 2019, 23, 2125–2135. [Google Scholar] [CrossRef]
- Benzoni, P.; Campostrini, G.; Landi, S.; Bertini, V.; Marchina, E.; Iascone, M.; Ahlberg, G.; Olesen, M.S.; Crescini, E.; Mora, C.; et al. Human iPSC modeling of a familial form of atrial fibrillation reveals a gain of function of If and ICaL in patient-derived cardiomyocytes. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Crombie, D.E.; Curl, C.L.; Raaijmakers, A.J.; Sivakumaran, P.; Kulkarni, T.; Wong, R.C.; Minami, I.; Evans-Galea, M.V.; Lim, S.Y.; Delbridge, L.; et al. Friedreich’s ataxia induced pluripotent stem cell-derived cardiomyocytes display electrophysiological abnormalities and calcium handling deficiency. Aging (Albany NY) 2017, 9, 1440–1449. [Google Scholar] [CrossRef]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Corrigan-Curay, J.; O’Reilly, M.; Kohn, D.B.; Cannon, P.M.; Bao, G.; Bushman, F.D.; Carroll, D.; Cathomen, T.; Joung, J.K.; Roth, D.; et al. Genome editing technologies: Defining a path to clinic. Mol. Ther. 2015, 23, 796–806. [Google Scholar] [CrossRef]
- Matsa, E.; Ahrens, J.H.; Wu, J.C. Human Induced Pluripotent Stem Cells as a Platform for Personalized and Precision Cardiovascular Medicine. Physiol. Rev. 2016, 96, 1093–1126. [Google Scholar] [CrossRef] [PubMed]
- Moehle, E.A.; Rock, J.M.; Lee, Y.L.; Jouvenot, Y.; DeKelver, R.C.; Gregory, P.D.; Urnov, F.D.; Holmes, M.C. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc. Natl. Acad. Sci. USA 2007, 104, 3055–3060. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lütge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.; Porteus, M.; Wu, J.C. Genome Editing in Cardiovascular Biology. Circ. Res. 2017, 120, 778–780. [Google Scholar] [CrossRef]
- Strong, A.; Musunuru, K. Genome editing in cardiovascular diseases. Nat. Rev. Cardiol. 2017, 14, 11–20. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Zhang, D.; Pu, W.T. Modeling Inherited Arrhythmia Disorders Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. J. 2016, 81, 12–21. [Google Scholar] [CrossRef]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human iPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- Jiang, Y.; Park, P.; Hong, S.-M.; Ban, K. Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells: Current Strategies and Limitations. Mol. Cells 2018, 41, 613–621. [Google Scholar]
- Feric, N.T.; Radisic, M. Towards adult-like human engineered cardiac tissue: Maturing human pluripotent stem cell-derived cardiomyocytes in human engineered cardiac tissues. Adv. Drug Deliv. Rev. 2016, 96, 110–134. [Google Scholar] [CrossRef]
- Scuderi, G.J.; Butcher, J. Naturally engineered maturation of cardiomyocytes. Front. Cell Dev. Biol. 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Insights into Molecular, Cellular, and Functional Phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Lundy, S.D.; Zhu, W.-Z.; Regnier, M.; Laflamme, M.A. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Lieu, D.K.; Liu, J.; Siu, C.W.; McNerney, G.P.; Tse, H.F.; Abu-Khalil, A.; Huser, T.; Li, R.A. Absence of transverse tubules contributes to non-uniform Ca2+ wavefronts in mouse and human embryonic stem cell-derived cardiomyocytes. Stem Cells Dev. 2009, 18, 1493–1500. [Google Scholar] [CrossRef]
- Li, M.; Kanda, Y.; Ashihara, T.; Sasano, T.; Nakai, Y.; Kodama, M.; Hayashi, E.; Sekino, Y.; Furukawa, T.; Kurokawa, J. Overexpression of KCNJ2 in induced pluripotent stem cell-derived cardiomyocytes for the assessment of QT-prolonging drugs. J. Pharmacol. Sci. 2017, 134, 75–85. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Markandeya, Y.S.; Kamp, T.J.; Makielski, J.C.; January, C.T.; Eckhardt, L.L. IK1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: An improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am. J. Physiol. Circ. Physiol. 2016, 310, H1611–H1621. [Google Scholar] [CrossRef]
- Lewandowski, J.; Rozwadowska, N.; Kolanowski, T.J.; Malcher, A.; Zimna, A.; Rugowska, A.; Fiedorowicz, K.; Łabędź, W.; Kubaszewski, Ł.; Chojnacka, K.; et al. The impact of in vitro cell culture duration on the maturation of human cardiomyocytes derived from induced pluripotent stem cells of myogenic origin. Cell Transplant. 2018, 27, 1047–1067. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Masse, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A new platform for maturation of human pluripotent stem cell derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering adolescence: Maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef]
- Keung, W.; Boheler, K.R.; Li, R.A. Developmental cues for the maturation of metabolic, electrophysiological and calciu handling properties of human pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2014, 5, 17. [Google Scholar] [CrossRef]
- Ribeiro, A.J.S.; Guth, B.D.; Engwall, M.; Eldridge, S.; Foley, C.M.; Guo, L.; Gintant, G.; Koerner, J.; Parish, S.T.; Pierson, J.B.; et al. Considerations for an In Vitro, Cell-Based Testing Platform for Detection of Drug-Induced Inotropic Effects in Early Drug Development. Part 2: Designing and Fabricating Microsystems for Assaying Cardiac Contractility With Physiological Relevance Using Hum. Front. Pharmacol. 2019, 10, 934. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587. [Google Scholar] [CrossRef]
- Karakikes, I.; Senyei, G.D.; Hansen, J.; Kong, C.-W.; Azeloglu, E.U.; Stillitano, F.; Lieu, D.K.; Wang, J.; Ren, L.; Hulot, J.-S.; et al. Small Molecule-Mediated Directed Differentiation of Human Embryonic Stem Cells Toward Ventricular Cardiomyocytes. Stem Cells Transl. Med. 2014, 3, 18–31. [Google Scholar] [CrossRef]
- Xu, C. Differentiation and enrichment of cardiomyocytes from human pluripotent stem cells. J. Mol. Cell. Cardiol. 2012, 52, 1203–1212. [Google Scholar] [CrossRef]
- Okano, S.; Shiba, Y. Therapeutic Potential of Pluripotent Stem Cells for Cardiac Repair after Myocardial Infarction. Biol. Pharm. Bull. 2019, 42, 524–530. [Google Scholar] [CrossRef]
- Hulot, J.S. Modeling Cardiac Arrhythmias With Organoids. J. Am. Coll. Cardiol. 2019, 73, 2325–2327. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Yeager, K.; Teles, D.; Chen, T.; Ma, S.; Song, L.J.; Morikawa, K.; Wobma, H.M.; Vasciaveo, A.; Ruiz, E.C.; et al. Engineering of human cardiac muscle electromechanically matured to an adult-like phenotype. Nat. Protoc. 2019, 14, 2781–2817. [Google Scholar] [CrossRef]
- Zuppinger, C. 3D Cardiac Cell Culture: A Critical Review of Current Technologies and Applications. Front. Cardiovasc. Med. 2019, 6, 87. [Google Scholar] [CrossRef]
- Li, R.A.; Keung, W.; Cashman, T.J.; Backeris, P.C.; Johnson, B.V.; Bardot, E.S.; Wong, A.O.T.; Chan, P.K.W.; Chan, C.W.Y.; Costa, K.D. Bioengineering an electro-mechanically functional miniature ventricular heart chamber from human pluripotent stem cells. Biomaterials 2018, 163, 116–127. [Google Scholar] [CrossRef]
- Eder, A.; Vollert, I.; Hansen, A.; Eschenhagen, T. Human engineered heart tissue as a model system for drug testing. Adv. Drug Deliv. Rev. 2016, 96, 214–224. [Google Scholar] [CrossRef]
- Breckwoldt, K.; Letuffe-Brenière, D.; Mannhardt, I.; Schulze, T.; Ulmer, B.; Werner, T.; Benzin, A.; Klampe, B.; Reinsch, M.C.; Laufer, S.; et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat. Protoc. 2017, 12, 1177–1197. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.J.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef]
- Maiullari, F.; Costantini, M.; Milan, M.; Pace, V.; Chirivì, M.; Maiullari, S.; Rainer, A.; Baci, D.; Marei, H.E.S.; Seliktar, D.; et al. A multi-cellular 3D bioprinting approach for vascularized heart tissue engineering based on HUVECs and iPSC-derived cardiomyocytes. Sci. Rep. 2018, 8, 13532. [Google Scholar] [CrossRef]
- Lee, J.M.; Sing, S.L.; Tan, E.Y.S.; Yeong, W.Y. Bioprinting in cardiovascular tissue engineering: A review. Int. J. Bioprinting 2017, 2, 27–36. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease Modeled | Gene Mutation | Main Findings | Approach | Ref |
|---|---|---|---|---|
| LQTS1 | KCNQ1 R190Q | Decreased IKs due to trafficking defect, altered channel activation and deactivation, APD prolongation, increased susceptibility to catecholamine-induced tachyarrhythmias, attenuated by treatment with beta blockade. | Patient-specific hiPSC-CMs | [99] |
| LQTS1 | KCNQ1 1893delC | Decreased IKs due to trafficking defect, increased cFPD, E4031-induced EADs and polymorphic ventricular tachycardia-like arrhythmias, insensitivity to chromanol 293B. | Patient-specific hiPSC-CMs | [100] |
| LQTS1 | KCNQ1 308~344del | Decreased IKs, APD prolongation reversed by ML277. | Patient-specific hiPSC-CMs | [126] |
| LQTS1 | KCNQ1 G269S | Increased APD50,70,90. High incidence of arrhythmias including EADs. Increased sensitivity to cardiotoxicity caused by hERG blockade. | Patient-specific hiPSC-CMs | [127] |
| LQTS1 | KCNQ1 c.1022C>T | Reduced chromanol 293B-sensitive IKs, Increased APD | Patient-specific hiPSC-CMs | [128] |
| LQTS1 | KCNQ1 c.478-2A>T c.1781G>A | APD and FPD prolongation and decreased or no IKs | Patient-specific hiPSC-CMs and CRISPR/Cas9 | [129] |
| LQTS1 LQTS2 | KCNQ1 G269S KCNQ1 R190Q KCNQ1 G345E KCNH2 A614V | Increased APD, EADs Pharmacological characterization with nifedipine, pinacidil | Zinc finger nuclease (ZFN)-mediated targeted gene addition into AAVS in iPSCs and patient specific iPSC | [130] |
| LQTS2 | KCNH2 A614V | APD prolongation, reduction of IKr, EADs and triggered arrhythmias. Increased cFPD. Evaluation of potency of pharmacological agents on disease phenotype. | Patient-specific hiPSC-CMs | [29] |
| LQTS2 | KCNH2 G1681A | APD and FPD prolongation. E4031 and isoprenaline-induced EADs. AP shortening in the presence of potassium channel openers and in some cases, abolition of EADs. | Patient-specific hiPSC-CMs | [131] |
| LQTS2 | KCNH2 R176W | APD prolongation, reduced IKr. Increased sensitivity to sotalol and E4031, more pronounced inverse correlation between the beating rate and repolarization time compared with control cells. | Patient-specific hiPSC-CMs | [127] |
| LQTS2 | KCNH2 A561P | Decreased IKr due to hERG trafficking defects, APD prolongation, higher incidence of EADs and increased sensitivity to E4031. | Patient-specific hiPSC-CMs | [132] |
| LQTS2 | KCNH2 A614V N996I | Decreased IKr, Prolongation of FPD, increased sensitivity to E4031 (hERG inhibitor). Targeted gene correction rescued the wild type phenotype. | Patient-specific hiPSC-CMs. | [133] |
| LQTS2 | KCNH2 G628S | Increased APD90, increased sensitivity to dofetilide | Overexpression of mutated ion channel in Cor.4U cells | [134] |
| LQTS2 | KCNH2 S428X R366X A561V IVS9-28A/G | Increased FPDc, Ca-handling defects and proarrhythmic events at 2 Hz. Lumacaftor restored trafficking in trafficking deficient mutants. | Patient-specific hiPSC-CMs | [102] |
| LQT3 | SCN5A ΔKPQ | Faster recovery from Na channel inactivation, increased late INa, prolonged APDs and EADs at low pacing rates. | hiPSC-CMs from a mouse model | [103] |
| LQT3, BrS | SCN5A 1795insD | Decreased peak INa, smaller Vmax and longer APD90, increased late INa. | Patient-specific hiPSC-CMs | [104] |
| LQTS3 | SCN5A V1763M | Increased APD, increased TTX-sensitive late INa, positive shift of steady state inactivation and faster recovery from inactivation. Mexiletine reversed the LQT3 phenotype. | Patient-specific hiPSC-CMs | [108] |
| LQTS3 | SCN5A F1473C | Increased late INa. Positive shift of steady state inactivation, faster recovery from inactivation. Pronounced rate dependence of INa: reduced late INa and increased late INa block by mexiletine with increasing pacing rate. | Patient-specific hiPSC-CMs | [106] |
| LQTS3 | SCN5A V240M SCN5A R535Q | Increased APD50 and APD90, longer time to peak and longer time to 90% inactivation in INa recordings. | Patient-specific hiPSC-CMs | [107] |
| LQTS3, BrS | SCN5A E1784K | Increased cFPD and APD90 (ventricular-type cells). Increased in late INa, no change in peak INa. LQTS3/BrS iPSC-CM recapitulate the LQT3 phenotype, not the BrS phenotype. Knockdown of SCN3B in LQTS3/BrS showed decreased peak INa, negative shift of the steady state inactivation, thus unmasking the BrS phenotype in LQTS3/BrS iPSCs. | Patient-specific hiPSC-CMs; siRNA used for knockdown | [110] |
| LQTS7 (ATS) | KCNJ2 R218W | Higher incidence of irregular Ca release, strong arrhythmic events, reversed by flecainide through the modulation of INCX. | Patient-specific hiPSC-CMs | [111] |
| LQTS7 | KCNJ2 G52V | Native IK1 induced by electrical pacing resulted in more negative RMP. G52V abolished native IK1, depolarized RMP, decreased cell excitability. In transfected cells that generated an AP: Increased APD, arrhythmic activity (EADs and spontaneous activity). | Transient transfection of Cor.4U hiPSC with TransIT-LT1 | [135] |
| LQTS8 (Timothy syndrome) | CACNA1C G406R | Irregular contraction, excess calcium influx, increased APD, irregular electrical activity and abnormal calcium transients in ventricular-like cells | Patient-specific hiPSC-CMs | [112] |
| LQTS8 | CACNA1C N639T | Increase in ERP (in optically stimulated cells), increased APD, slower voltage-dependent inactivation of Cav1.2. | CRISPR/Cas9 | [136] |
| LQTS15 | CALM2-N98S | Lower beating rates, increased APD, impaired inactivation of L-type Ca channels. Specific ablation of the mutant allele using CRISPR-Cas9 rescued the electrophysiological abnormalities of LQTS15-hiPSC-CMs. | Patient-specific hiPSC-CMs, CRISPR/Cas9 | [113] |
| LQTS15 | CALM1 F142L | Increased ICa,L due to severe impairment of ICa,L Ca-dependent inactivation, Prolonged FPD and APD, failure of repolarization to adapt to high rates. Verapamil reversed the mutation-induced repolarization abnormalities. | Patient-specific hiPSC-CMs | [114] |
| Short QTS | KCNH2 T618I | Increased IKr (current density and enhanced membrane expression). Shorter APD, increased beat to beat variability. IKr inhibition restored a normal phenotype. | Patient-specific hiPSC-CMs, CRISPR/Cas9 | [137] |
| BrS | SCN5A R620H and R811H SCN5A 4189delT | Decreased INa (lower membrane expression), abnormal AP profiles, closely coupled single trigger beat, sustained triggered activity, reduced Vmax, Ca transient abnormalities. | Patient-specific hiPSC-CMs, CRISPR/Cas9 | [138] |
| CPVT | RyR2 F2483I | Catecholaminergic stimulation-induced DADs, higher amplitudes and longer durations of spontaneous Ca release events at basal state. CICR events continued after repolarization and were abolished by increased cytosolic cAMP levels. | Patient-specific hiPSC-CMs | [117] |
| CPVT | RYR2 M4109R | Higher incidence of DADs, increased frequency and magnitude of after-depolarizations in the presence of isoproterenol and forskolin, triggered activity. Flecainide and Thapsigargin eliminated DADs. Whole cell Ca transient irregularities worsened with adrenergic stimulation and Ca overload, improved with beta blockers. Store-overload-induced Ca release. | Patient-specific hiPSC-CMs | [118] |
| CPVT | RYR2 S406L | Increased diastolic Ca concentrations, reduced sarcoplasmic reticulum Ca content, increased susceptibility to DADs and arrhythmia in the presence of catecholaminergic stress. Increased frequency and duration of elementary Ca sparks. Dantrolene restored normal Ca spark phenotype and reversed the arrhythmogenic phenotype. | Patient-specific hiPSC-CMs | [119] |
| CPVT | CASQ2 D307H | Isoproterenol-induced DADs, oscillatory arrhythmic prepotentials, after contractions and elevation of diastolic Ca concentrations. CPVT iPSC-CMs had a more immature phenotype than control cells (less organized myofibrils, enlarged sarcoplasmic reticulum cisternae and reduced number of caveolae. | Patient-specific hiPSC-CMs | [120] |
| CPVT | RYR2 E2311D | DADs in resting state and in the presence of isoproterenol. Non-homogeneous spreading of calcium transients (aggravated with isoproterenol). KN-93, a CaMKII inhibitor reversed the arrhythmic phenotype. | Patient-specific hiPSC-CMs | [121] |
| CPVT | RyR2 F2483I | Aberrant unitary calcium signaling, smaller calcium stores, higher CICR gains, and sensitized adrenergic regulation. | Patient-specific hiPSC-CMs | [122] |
| CPVT | RyR2 P2328S | Increased non-alternating variability of Ca transients in response to isoproterenol. Epinephrine decreased AP Vmax. | Patient-specific hiPSC-CMs | [123] |
| ARVD/C | PKP2 c.2484C>T | Exaggerated lipogenesis and apoptosis, calcium handling deficits (prolonged Ca transient relaxation), corrected by introducing the wild type PKP2 gene back into mutant iPSC-CMs. | Patient-specific hiPSC-CMs | [139] |
| ARVD/C | PKP2 c.972InsT/N | Prolonged field potential rise time, widened and distorted desmosomes. Clusters of lipid droplets were identified in ARVC iPSCs with the most severe desmosomal pathology. Exposure of the cells to apidogenic stimuli augmented desmosomal distortion and lipid accumulation. | Patient-specific hiPSC-CMs | [140] |
| ARVC | PKP2 c.1841T>C | Reduced cell surface localization of desmosomal proteins, adipogenic phenotype. | Patient-specific hiPSC-CMs | [105] |
| HCM | MYH7 R663H | Dysregulation of Ca cycling, elevation of intracellular Ca concentration, cellular enlargement, arrhythmias (DADs). | Patient-specific hiPSC-CMs | [141] |
| HCM | MYH7 R663H | High incidence of arrhythmias, including DADs. Increased sensitivity to cardiotoxicity caused by hERG blockade. | Patient-specific hiPSC-CMs | [127] |
| HCM | SCO2 E140K SCO2 G193S | Ultrastructural abnormalities, diminished response to isoproterenol and caffeine, DADs, increased beat rate variability | Patient-specific hiPSC-CMs | [142] |
| HCM, WPW | PRKAG2 R302Q | Abnormal firing patterns, DADs, triggered arrhythmias, increased beat to beat variability. CRISPR correction reversed the mutation phenotype. | Patient-specific hiPSC-CMs CRISPR/Cas9 | [143] |
| HCM | T247M | Impaired relaxation, Increased myofilament Ca sensitivity, APD prolongation, Increased ICa,L | Patient-specific hiPSC-CMs | [124] |
| DCM | TNNT2 R173W | Altered Ca handling, decreased contractility, abnormal sarcomeric α-actinin distribution, isoproterenol-induced reduced beating rates, compromised contraction, and more cells with abnormal sarcomeric α-actinin distribution. | Patient-specific hiPSC-CMs | [144] |
| DCM | TNNT2 R173W | Very low frequencies of irregular electrophysiological waveforms. Increased sensitivity to APD90 shortening by nicorandil. | Patient-specific hiPSC-CMs | [127] |
| DCM | DES A285V | Decreased maximum rate of Ca ion reuptake, slower spontaneous beating rate, diminished response to isoproterenol. | Patient-specific hiPSC-CMs | [145] |
| DCM | PLN R14del in | Ca handling abnormalities, electrical instability, abnormal cytoplasmic distribution of PLN protein and increases expression of molecular markers of cardiac hypertrophy in iPSC-CMs. | Patient-specific hiPSC-CMs | [146] |
| DCM | TTN c.8607dupA TTN c.70690dupAT | Decreased response to isoproterenol, elevated external Ca concentration and Angiotensin-II. Altered response to caffeine. | Patient-specific hiPSC-CMs | [125] |
| DMD | DMD c.5899C>T | Slower spontaneous firing rates, decreased If, DADs, prolonged APD, increased ICa,L | Patient-specific hiPSC-CMs | [147] |
| Familial AF | Multiple genetic variants | Higher beating rate, increased ICa,L and If. Prolonged APD, no changes in Ca handling. Stress-induced DADs. | Patient-specific hiPSC-CMs. | [148] |
| Friedreich’s ataxia | FXN Expanded GAA repeat mutation | Increased beating rate variability, calcium handling defect as implied by decrease Ca transients. | Patient-specific hiPSC-CMs | [149] |
| Barth syndrome | TAZ c.517delG TAZ c.328T>C | Sparse and irregular sarcomeres, contraction abnormalities | Patient-specific hiPSC-CMs CRISPR/Cas9 | [150] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pourrier, M.; Fedida, D. The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases. Int. J. Mol. Sci. 2020, 21, 657. https://doi.org/10.3390/ijms21020657
Pourrier M, Fedida D. The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases. International Journal of Molecular Sciences. 2020; 21(2):657. https://doi.org/10.3390/ijms21020657
Chicago/Turabian StylePourrier, Marc, and David Fedida. 2020. "The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases" International Journal of Molecular Sciences 21, no. 2: 657. https://doi.org/10.3390/ijms21020657
APA StylePourrier, M., & Fedida, D. (2020). The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases. International Journal of Molecular Sciences, 21(2), 657. https://doi.org/10.3390/ijms21020657