Enaminone Substituted Resorcin[4]arene—Sealing of an Upper-Rim with a Directional System of Hydrogen-Bonds

 , and
, and
Abstract
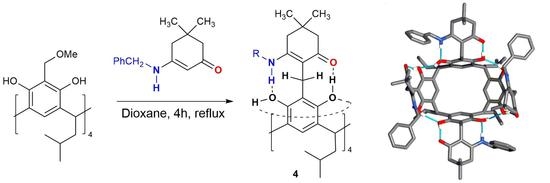
1. Introduction
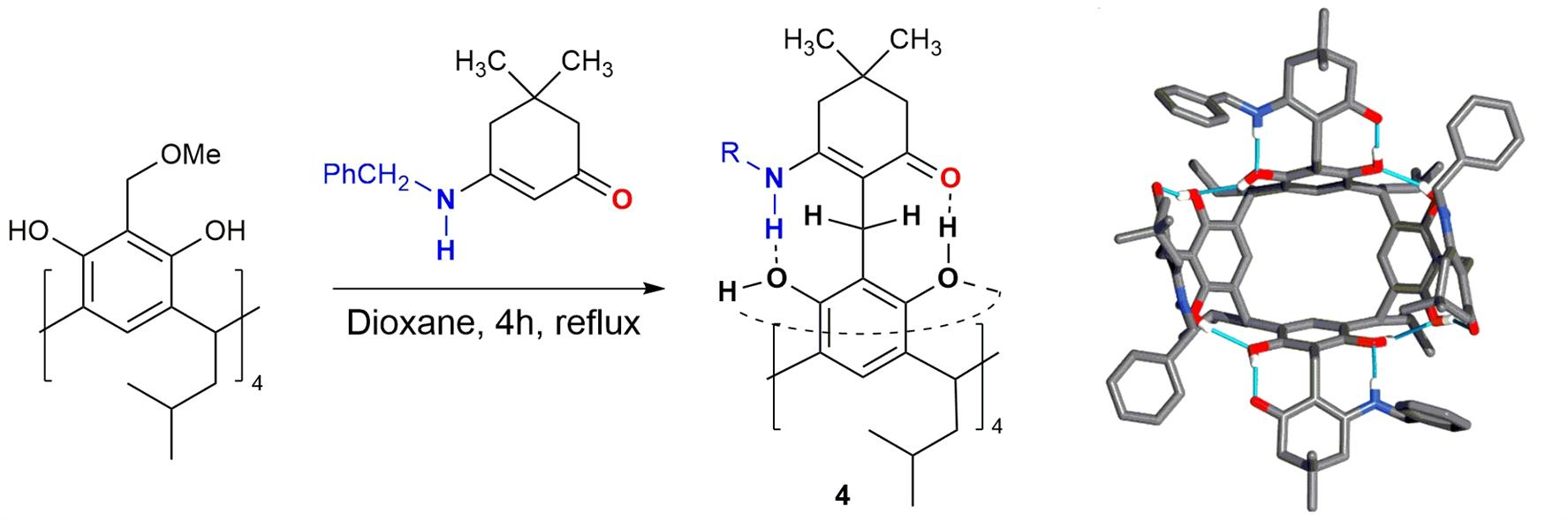
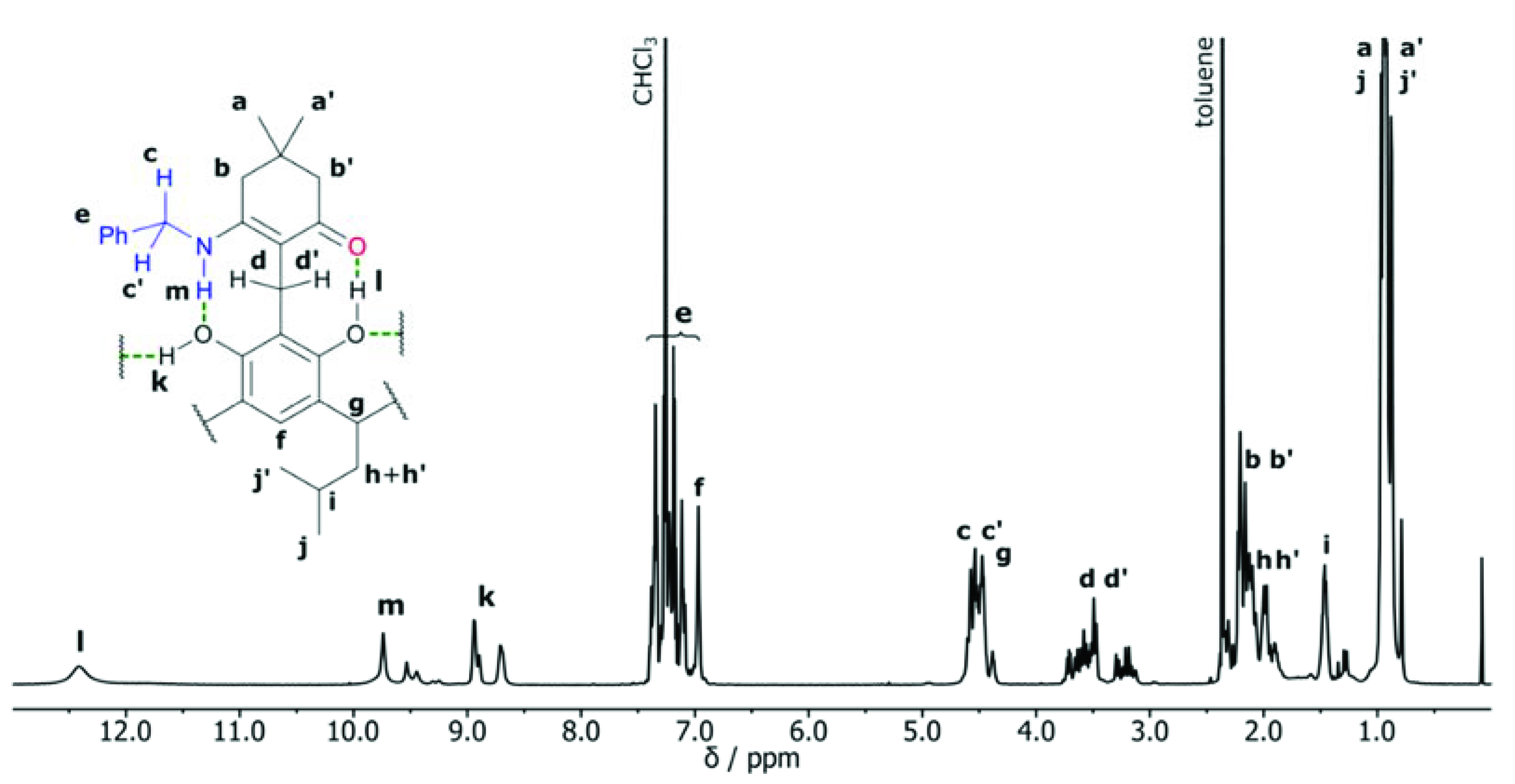
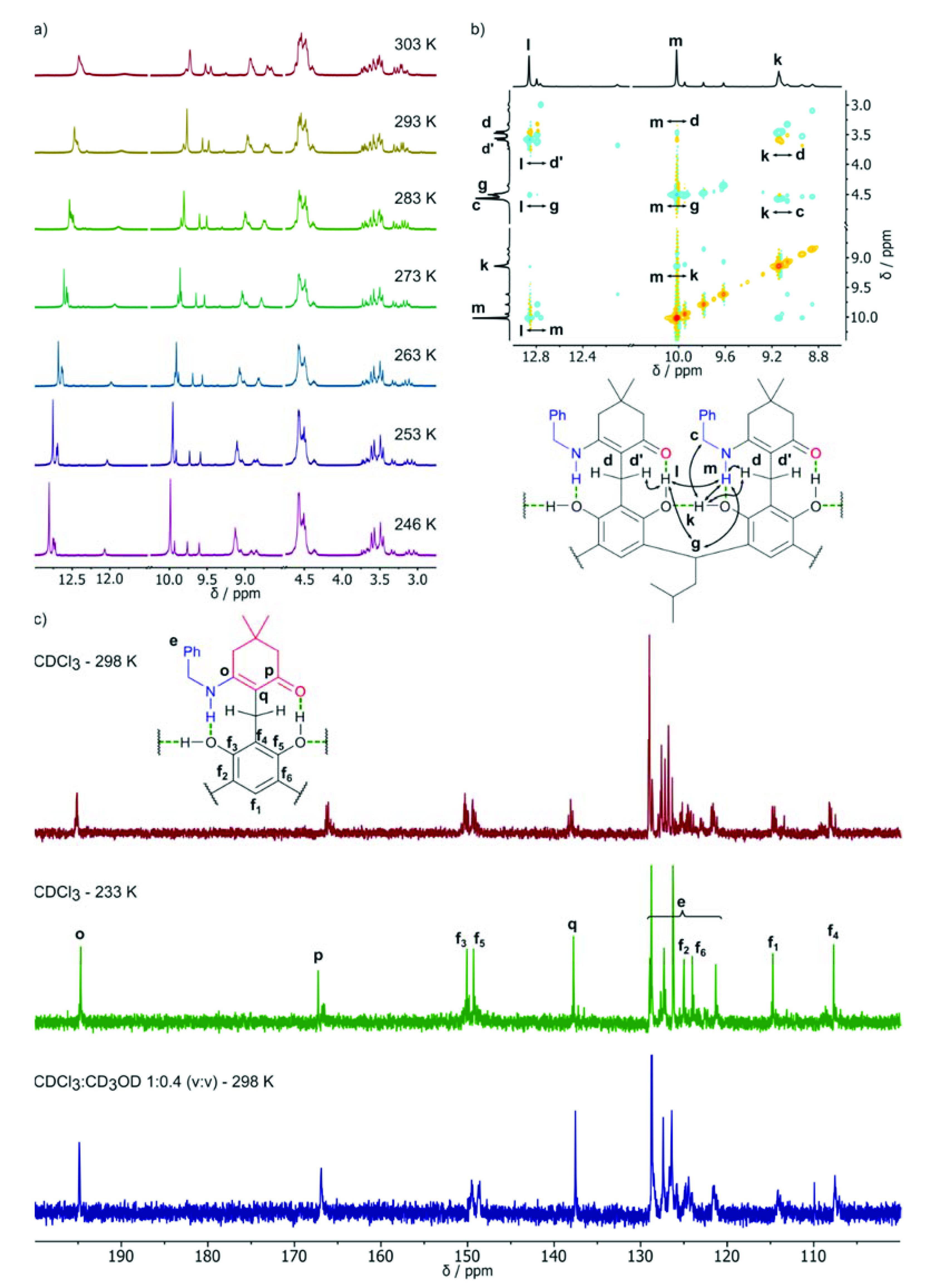
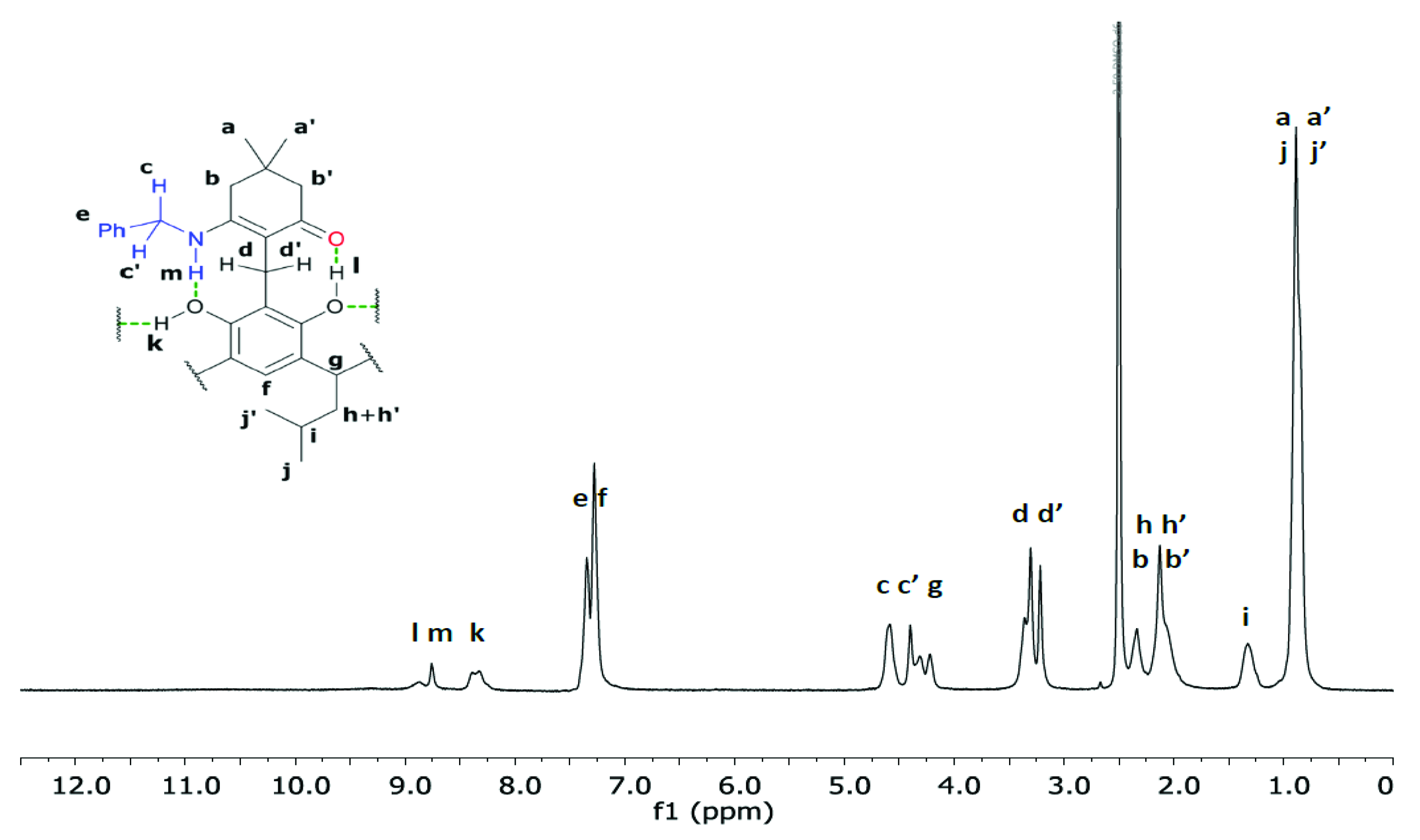
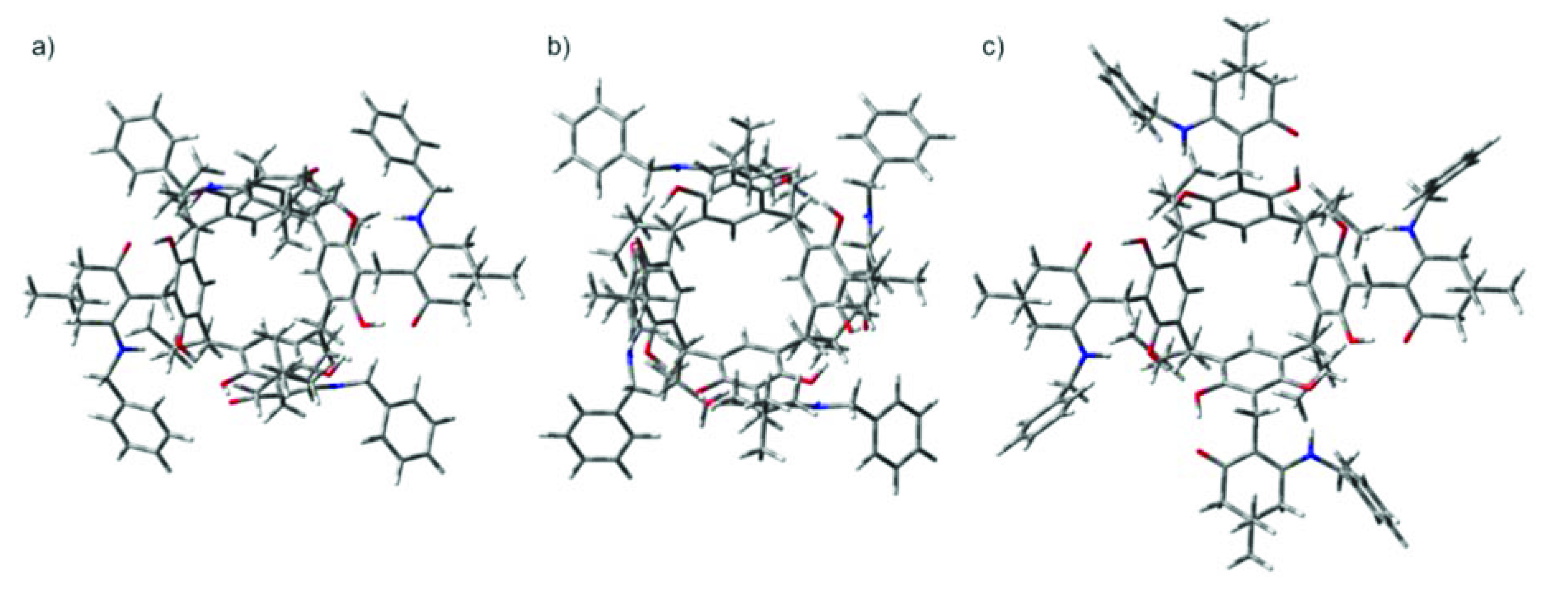
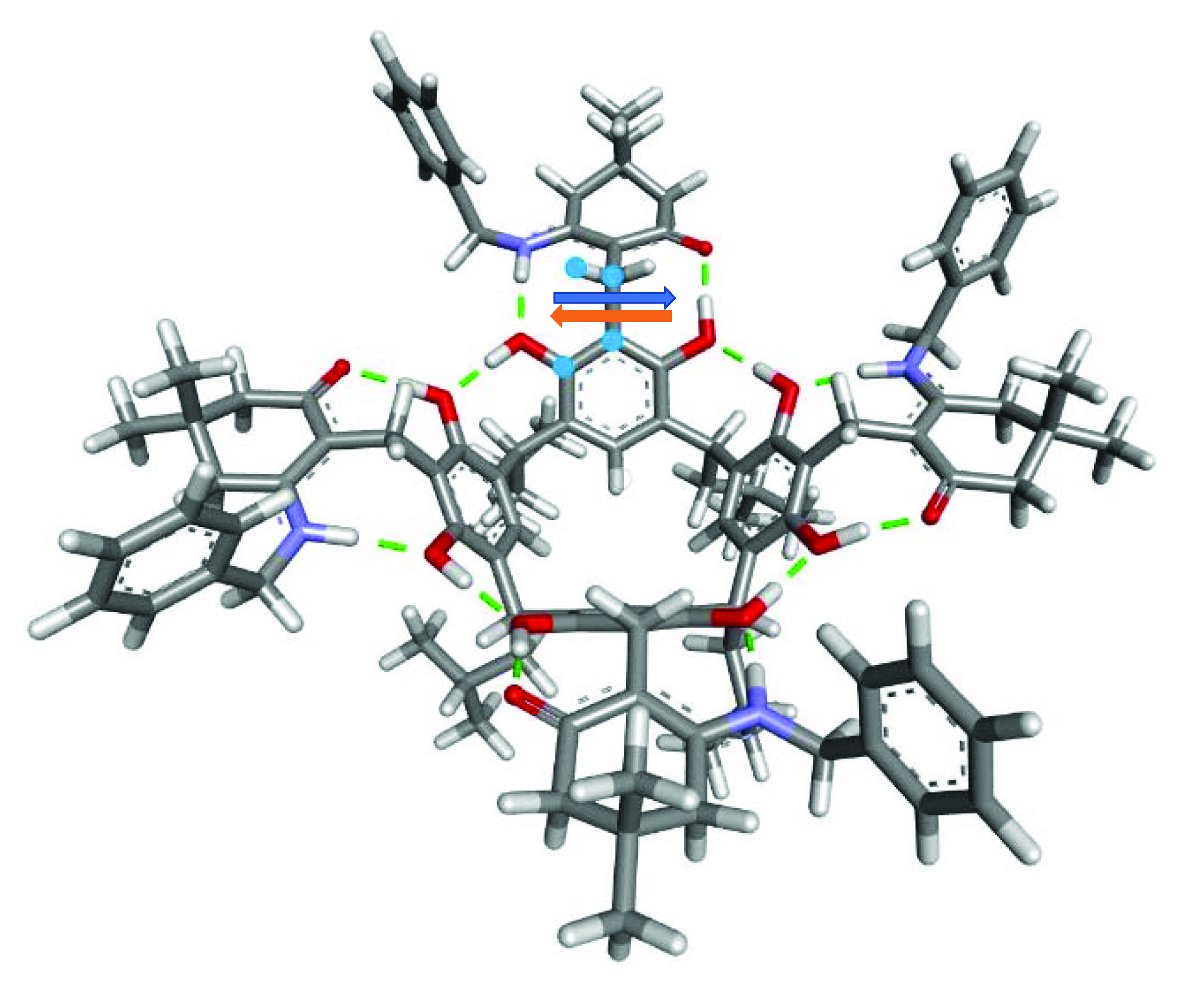
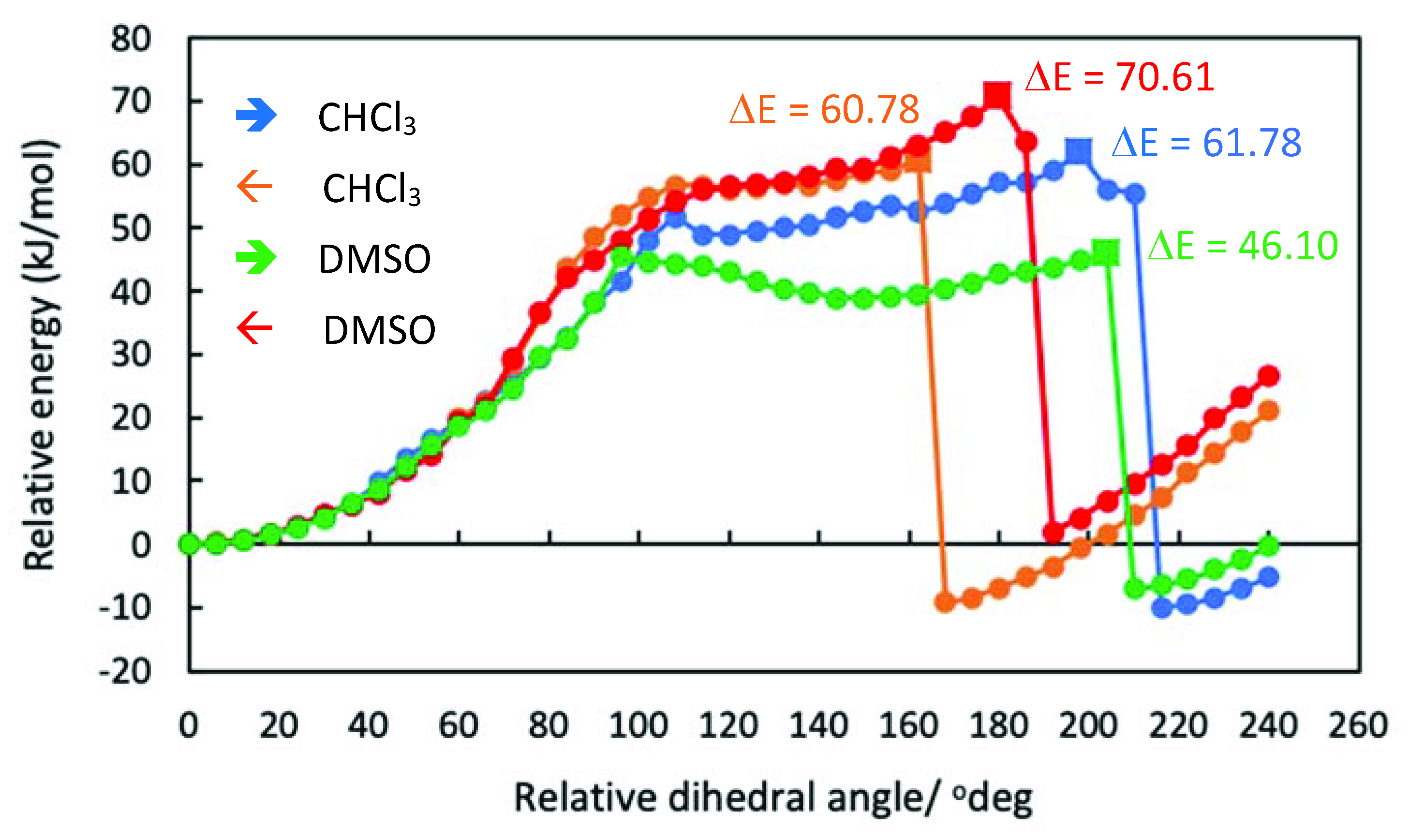
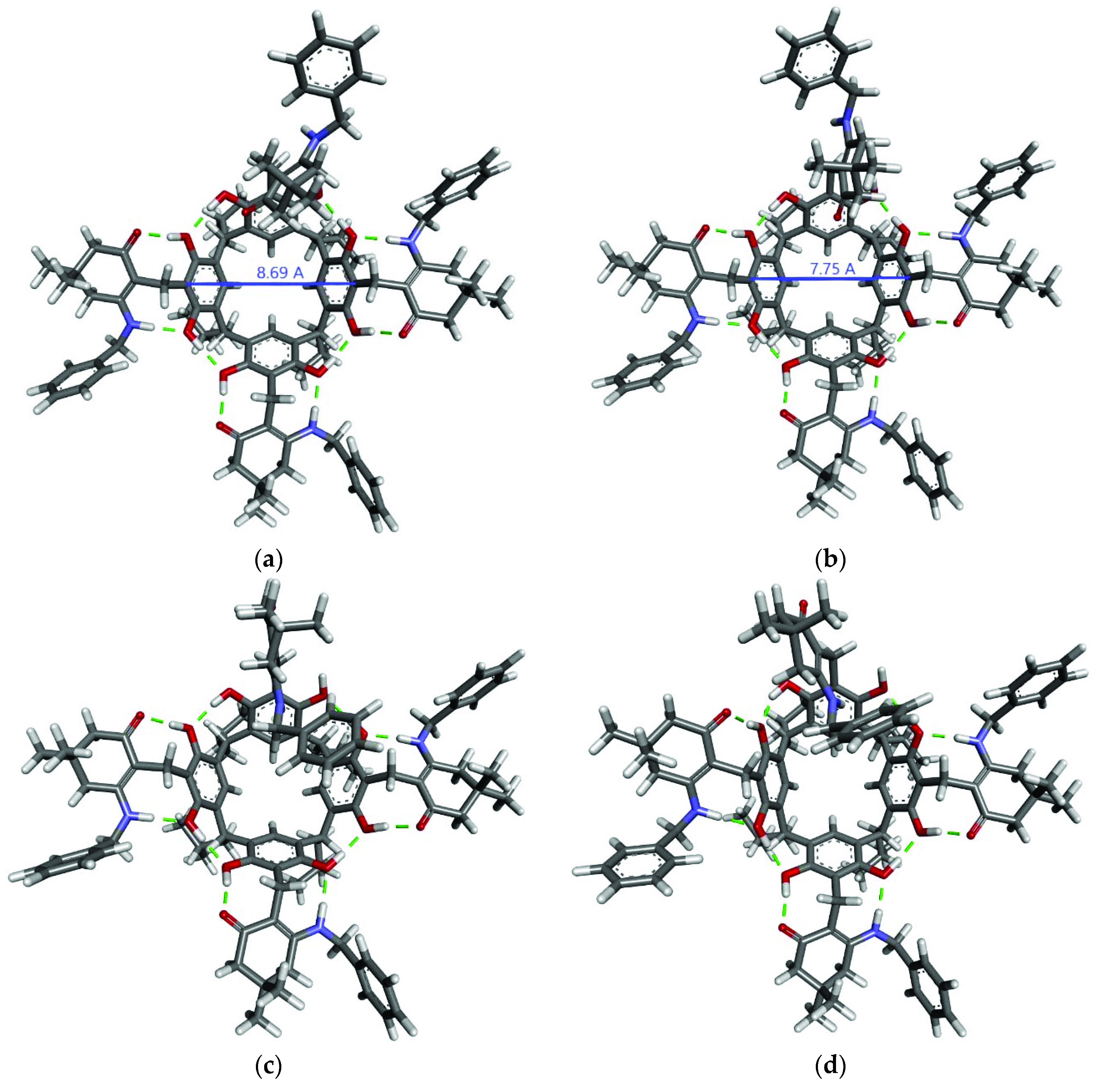
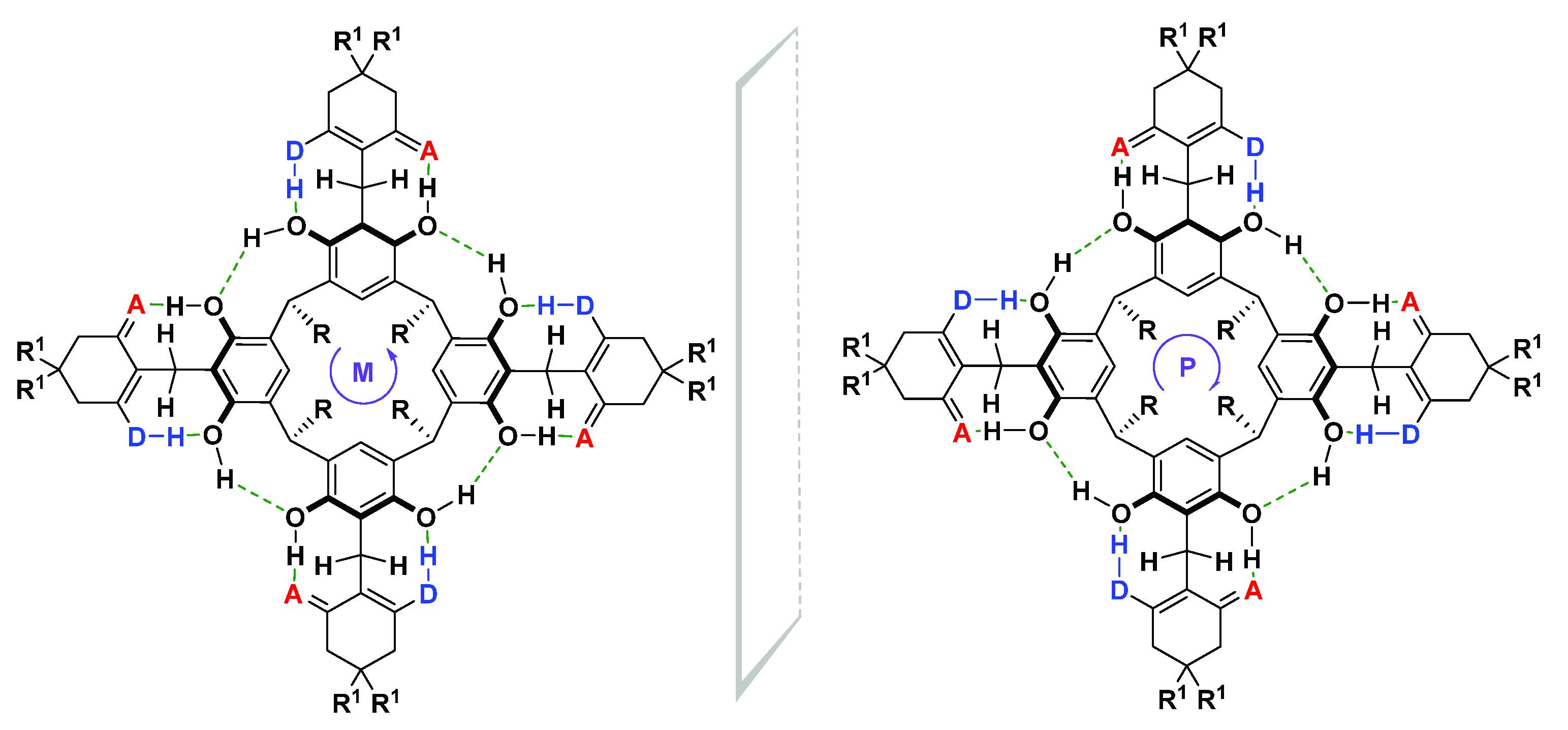
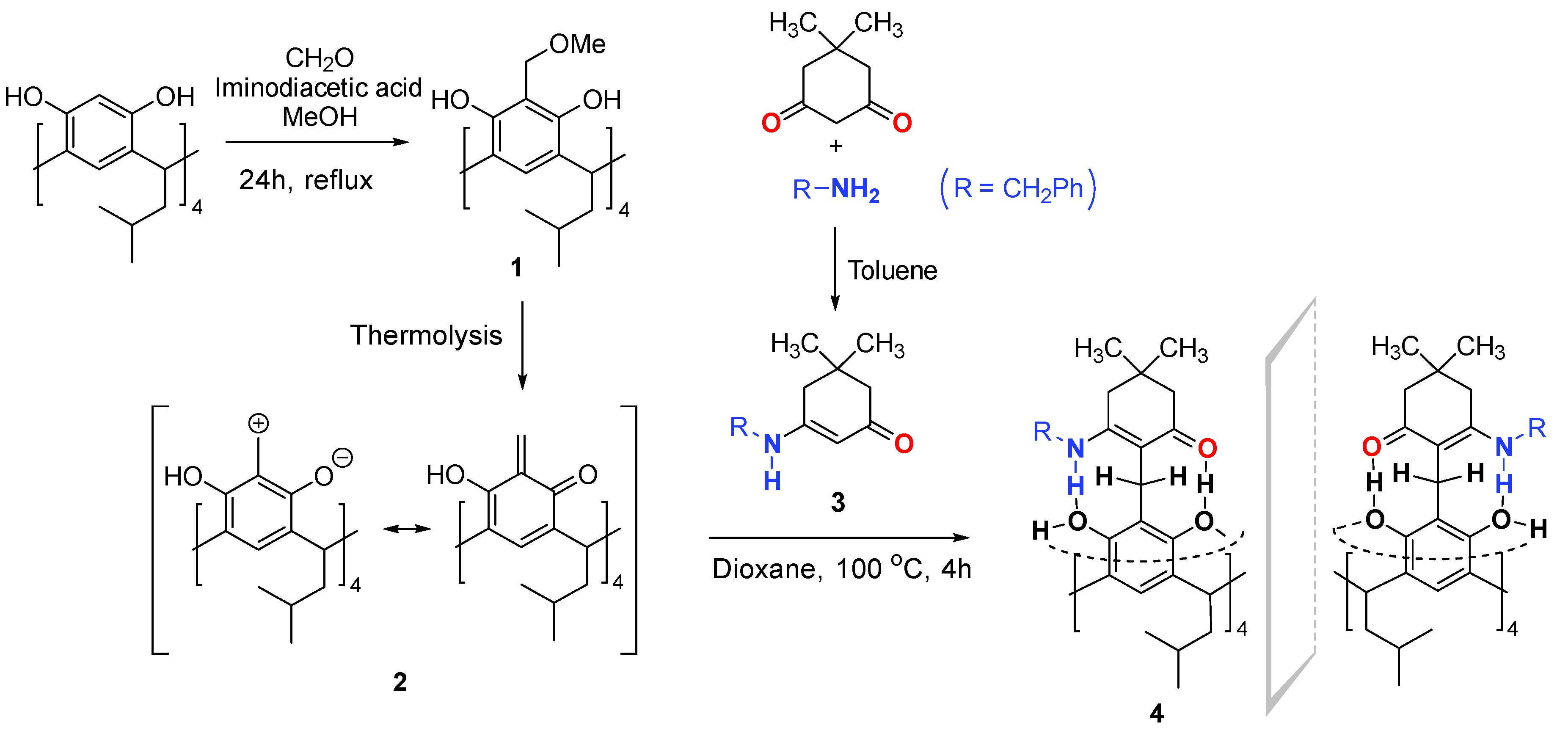
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cram, D.J.; Cram, J.M. Container Molecules and Their Guests, Monographs in Supramolecular Chemistry; Stoddart, J.F., Ed.; The Royal Society of Chemistry: Cambridge, UK, 1994. [Google Scholar]
- Timmerman, P.; Verboom, W.; Reinhoudt, D.N. Resorcinarenes. Tetrahedron 1996, 52, 2663–2704. [Google Scholar] [CrossRef]
- Neri, P.; Sessler, J.; Wang, M.-X. (Eds.) Calixarenes and Beyond; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Iwanek, W.; Wzorek, A. Introduction to the chirality of Resorcinarenes. Min. Rev. Org. Chem. 2009, 6, 398–411. [Google Scholar] [CrossRef]
- Moran, J.R.; Ericson, J.L.; Dalcanale, E.; Bryant, J.A.; Knobler, C.B.; Cram, D.J. Vases and Kites as Cavitands. J. Am. Chem. Soc. 1991, 113, 5707–5714. [Google Scholar] [CrossRef]
- Gropp, C.; Quigley, B.L.; Diederich, F. Molecular Recognition With Resorcin[4]arene Cavitands: Switching, Halogen-Bonded Capsules, and Enantioselective Complexation. J. Am. Chem. Soc. 2018, 140, 2705–2717. [Google Scholar] [CrossRef] [PubMed]
- Iwanek, W.; Fröhlich, R.; Schwab, P.; Schurig, V. The synthesis and crystallographic structures of novel bora-oxazino-oxazolidine derivatives of resorcarene. Chem. Comm. 2002, 2516–2517. [Google Scholar] [CrossRef]
- Skinner, P.J.; Cheetham, A.G.; Beeby, A.; Gramlich, V.; Diederich, F. Conformational switching of resorcin [4]arene cavitands by protonation, preliminary communication. Helv. Chim. Acta 2001, 84, 2146–2153. [Google Scholar] [CrossRef]
- Roncucci, P.; Pirondini, L.; Paderni, G.; Massera, C.; Dalcanale, E.; Azov, V.A.; Diederich, F. Conformational Behavior of Pyrazine-Bridged and Mixed-Bridged Cavitands: A General Model for Solvent Effects on Thermal “Vase–Kite” Switching. Chem. Eur. J. 2006, 12, 4775–4784. [Google Scholar] [CrossRef]
- Azov, V.A.; Schlegel, A.; Diederich, F. Geometrically Precisely Defined Multinanometer Expansion/Contraction Motions in a Resorcin[4]arene Cavitand Based Molecular Switch. Angew. Chem. Int. Ed. 2005, 44, 4635–4638. [Google Scholar] [CrossRef]
- Szumna, A. Inherently chiral concave molecules-from synthesis to applications. Chem. Soc. Rev. 2010, 39, 4274–4285. [Google Scholar] [CrossRef]
- Wiegmann, S.; Mattay, J. Inherently chiral resorcin[4]arenes: A new concept for improving the functionality. Org. Lett. 2011, 13, 3226–3228. [Google Scholar] [CrossRef]
- Pamuła, M.; Nissinen, M.; Helttunen, K. Correlating Solution- and Solid-State Structures of Conformationally Flexible Resorcinarenes: Significance of a Sulfonyl Group in Intramolecular Self-Inclusion. Chem. Eur. J. 2020, 26, 7374–7383. [Google Scholar] [CrossRef]
- Iwanek, W. Chiral calixarene derived from resorcinol. Part 4: Diastereoselective closure of the oxazine ring. Tetrahedron Asymmetry 1998, 9, 4289–4290. [Google Scholar] [CrossRef]
- Iwanek, W.; Mattay, J. Chiral calixarenes derived from resorcinol. Eur. J. Org. Chem. 1995, 1995, 1463–1466. [Google Scholar] [CrossRef]
- Stefańska, K.; Jędrzejewska, H.; Wierzbicki, M.; Szumna, A.; Iwanek, W. The inverse demand oxa-Diels-Alder reaction of resorcinarenes: An experimental and theoretical analysis of regioselectivity and diastereoselectivity. J. Org. Chem. 2016, 81, 6018–6025. [Google Scholar] [CrossRef] [PubMed]
- Stefańska, K.; Szafraniec, A.; Szymański, M.; Wierzbicki, M.; Szumna, A.; Iwanek, W. Chiral chromane[4]arenes synthesised by cycloaddition reactions of o-quinomethine resorcin[4]arenes. New J. Chem. 2019, 43, 2687–2693. [Google Scholar] [CrossRef]
- Wzorek, A.; Mattay, J.; Iwanek, W. Synthesis and structural investigation of the cyclochiral boron resorcinarenes obtained from l-amino acids and phenylboronic acid. Tetrahedron Asymmetry 2012, 23, 271–277. [Google Scholar] [CrossRef]
- Kumar, P.; Venkatakrishnan, P. Coumarin[4]arene: A Fluorescent Macrocycle. Org. Lett. 2018, 20, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Szumna, A. Cyclochiral conformers of resorcin[4]arenes stabilized by hydrogen bonds. Org. Biomol. Chem. 2007, 5, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Grajda, M.; Wierzbicki, M.; Cmoch, P.; Szumna, A. Inherently Chiral Iminoresorcinarenes through Regioselective Unidirectional Tautomerization. J. Org. Chem. 2013, 78, 11597–11601. [Google Scholar] [CrossRef]
- Jędrzejewska, H.; Kwit, M.; Szumna, A. Switching of inherent chirality driven by self-assembly. Chem. Commun. 2015, 51, 13799–13801. [Google Scholar] [CrossRef]
- Chithanna, S.; Vyasamudri, S.; Yang, D.-H. Application of Dimedone Enamines as Protecting Groups for Amines and Peptides. Org. Lett. 2020, 22, 2391–2395. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, Y.; Li, A.; Liu, K.; Li, L.; Yang, T.; Zhou, C. One-Pot Synthesis of 5-Acyl-1,2,3-Thiadiazoles from Enaminones, Tosylhydrazine, and Elemental Sulfur under Transition-Metal-Free Conditions. J. Org. Chem. 2019, 84, 16262–16267. [Google Scholar] [CrossRef]
- Wang, Y.F.; Izawa, T.; Kobayashi, S.; Ohno, M. Stereocontrolled synthesis of (ţ)-negamycin from an acyclic homoallylamine by 1,3-asymmetric induction. J. Am. Chem. Soc. 1982, 104, 6465–6466. [Google Scholar] [CrossRef]
- Michael, J.P.; de Koning, C.B.; Hosken, G.D.; Stanbury, T.V. Reformatskii reactions with N-arylpyrrolidine-2-thiones: Synthesis of tricyclic analogues of quinolone antibacterial agents. Tetrahedron 2001, 57, 9635–9648. [Google Scholar] [CrossRef]
- Boger, D.L.; Ishizaki, T.; Wysocki, J.R., Jr.; Munk, S.A.; Kitos, P.A.; Suntornwat, O. Total synthesis and evaluation of (+-)-N-(tert-butoxycarbonyl)-CBI, (+-)-CBI-CDPI1, and (+-)-CBI-CDPI2: CC-1065 functional agents incorporating the equivalent 1,2,9,9a-tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) left-hand subunit. J. Am. Chem. Soc. 1989, 111, 6461–6463. [Google Scholar] [CrossRef]
- Osipov, D.V.; Osyanin, V.A.; Klimochkin, Y.N. Ortho-Quinone methides as key intermediates in cascade heterocyclizations. Russ. Chem. Rev. 2017, 86, 625–687. [Google Scholar] [CrossRef]
- Lukashenko, A.V.; Osyanin, V.A.; Osipov, D.V.; Klimochkin, Y.N. Reaction of Push-Pull Enaminoketones and in Situ Generated ortho-Quinone Methides: Synthesis of 3-Acyl-4H-chromenes and 2-Acyl-1H-benzo[f]chromenes as Precursors for Hydroxybenzylated Heterocycles. J. Org. Chem. 2017, 82, 1517–1528. [Google Scholar] [CrossRef]
- Tero, T.-R.; Suhonen, A.; Suhonen, A.; Salorinne, K.; Campos-Barbosa, H.; Nissinen, M. The Missing Member of the Partially O-Alkylated Resorcinarene Family: Synthesis and Conformation of Methyl Tetramethoxy Resorcinarene. Org. Lett. 2013, 15, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- McIldowie, M.J.; Mocerino, M.; Skelton, B.W.; White, A.H. Facile Lewis Acid Catalyzed Synthesis of C4 Symmetric Resorcinarenes. Org. Lett. 2000, 2, 3869–3871. [Google Scholar] [CrossRef] [PubMed]
- Mcildowie, M.J.; Mocerino, M.; Skelton, B.W.; White, A.H. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, UK, 2009. [Google Scholar]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB An Accurate and Broadly Parametrized Self Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, M.; Iwanek, W. Synthesis of alkoxymethyl derivatives of resorcinarene via the Mannich reaction catalysed with iminodiacetic acid. Tetrahedron 2006, 62, 1508–1511. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical Extended Tight-Binding Program Package xTB. Version 6.32. 2020. Available online: https://github.com/grimme-lab/xtb (accessed on 16 March 2020).
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chloroform | DMSO | |||
|---|---|---|---|---|
| conformation (symmetry) | DFT/B3LYP ∆E/kJ·mol−1 (population %) | DFTB/GFN2-xTB ∆E/kJ·mol−1 (population%) | DFT/B3LYP ∆E/kJ·mol−1 (population %) | DFTB/GFN2-xTB ∆E/kJ·mol−1 (population%) |
| all-in (C4) | 0 (85%) | 18.86 (<1%) | 0 (83%) | 16.08 (<1%) |
| in-out (C2) | 5.55 (9%) | 0.68 (43%) | 4.71 (13%) | 18.47 (<1%) |
| all-out (C4) | 6.51 (6%) | 0 (57%) | 7.17 (4%) | 0 (100%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szafraniec, A.; Grajda, M.; Jędrzejewska, H.; Szumna, A.; Iwanek, W. Enaminone Substituted Resorcin[4]arene—Sealing of an Upper-Rim with a Directional System of Hydrogen-Bonds. Int. J. Mol. Sci. 2020, 21, 7494. https://doi.org/10.3390/ijms21207494
Szafraniec A, Grajda M, Jędrzejewska H, Szumna A, Iwanek W. Enaminone Substituted Resorcin[4]arene—Sealing of an Upper-Rim with a Directional System of Hydrogen-Bonds. International Journal of Molecular Sciences. 2020; 21(20):7494. https://doi.org/10.3390/ijms21207494
Chicago/Turabian StyleSzafraniec, Anna, Marcin Grajda, Hanna Jędrzejewska, Agnieszka Szumna, and Waldemar Iwanek. 2020. "Enaminone Substituted Resorcin[4]arene—Sealing of an Upper-Rim with a Directional System of Hydrogen-Bonds" International Journal of Molecular Sciences 21, no. 20: 7494. https://doi.org/10.3390/ijms21207494
APA StyleSzafraniec, A., Grajda, M., Jędrzejewska, H., Szumna, A., & Iwanek, W. (2020). Enaminone Substituted Resorcin[4]arene—Sealing of an Upper-Rim with a Directional System of Hydrogen-Bonds. International Journal of Molecular Sciences, 21(20), 7494. https://doi.org/10.3390/ijms21207494