Development of a Protein Scaffold for Arginine Sensing Generated through the Dissection of the Arginine-Binding Protein from Thermotoga maritima





Abstract
:1. Introduction
2. Results
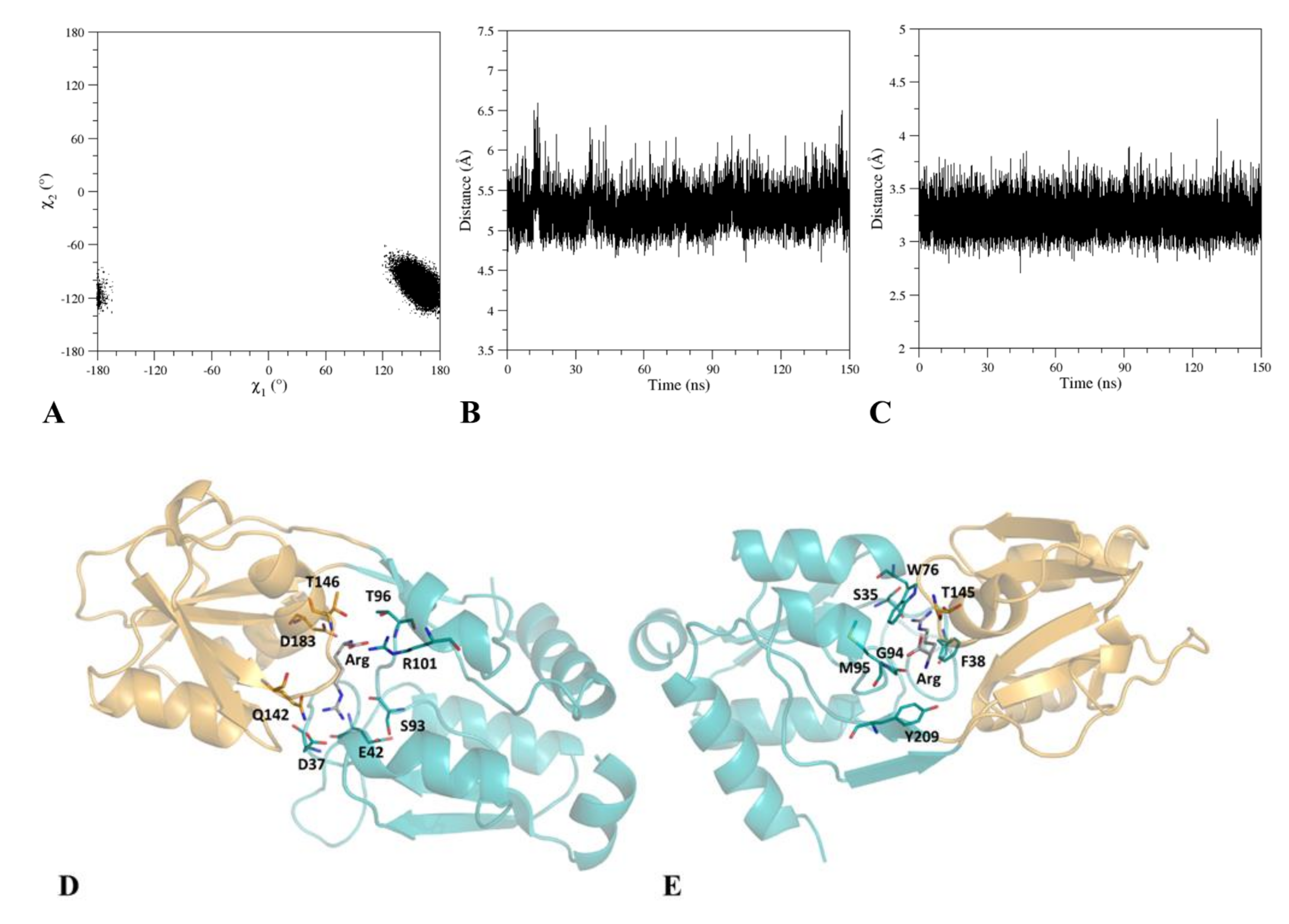
2.1. Design of a Potentially Fluorescent Mutant and its Characterization by Computational Techniques
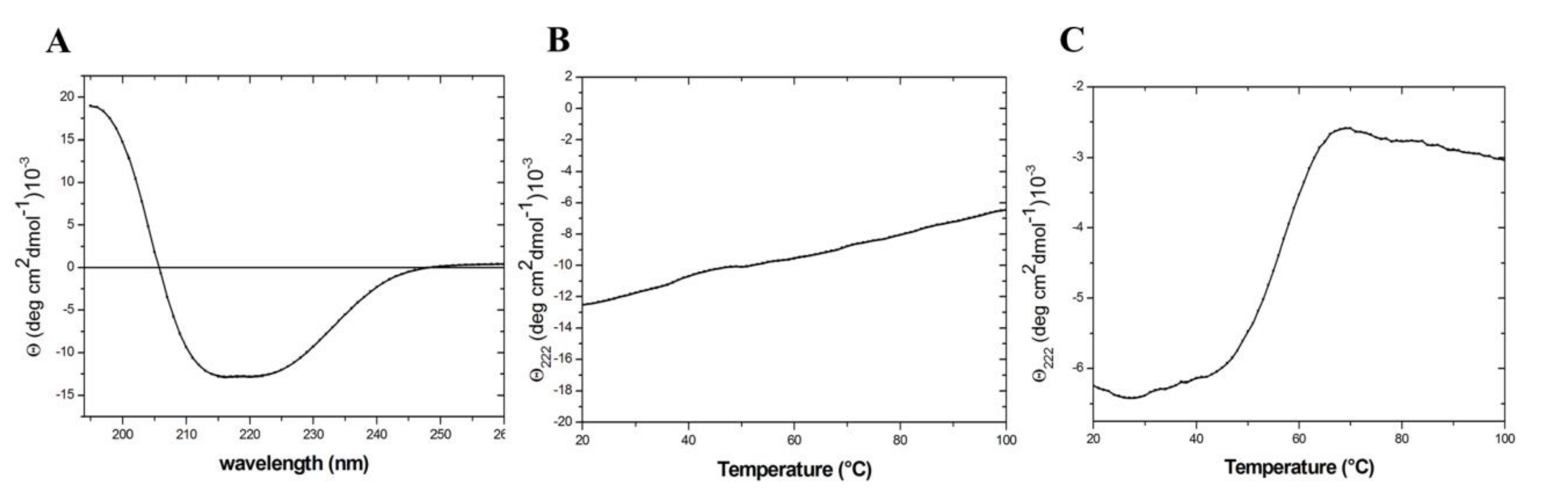
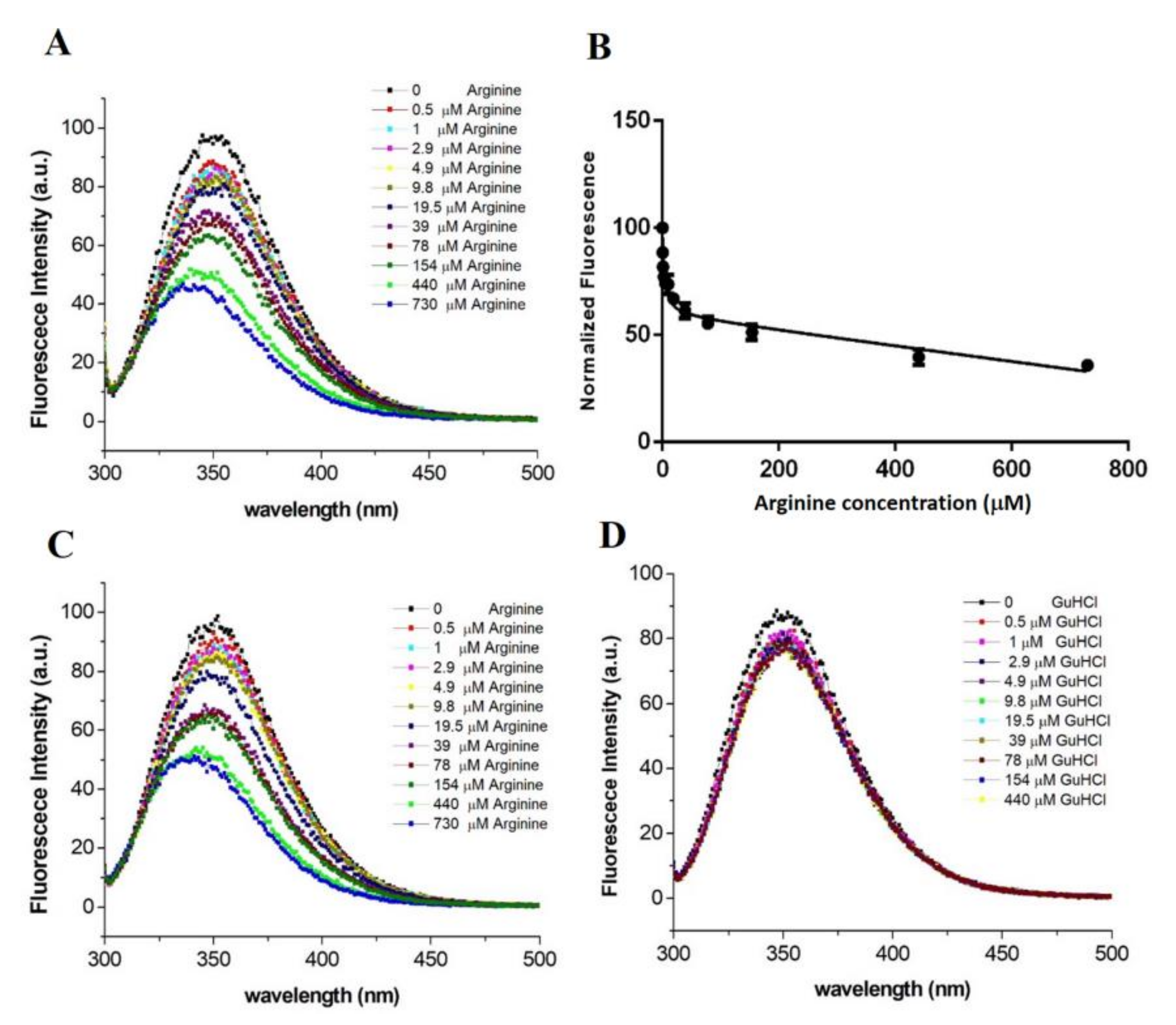
2.2. Expression and Characterization of the Mutant TmArgBP20−233_F76W
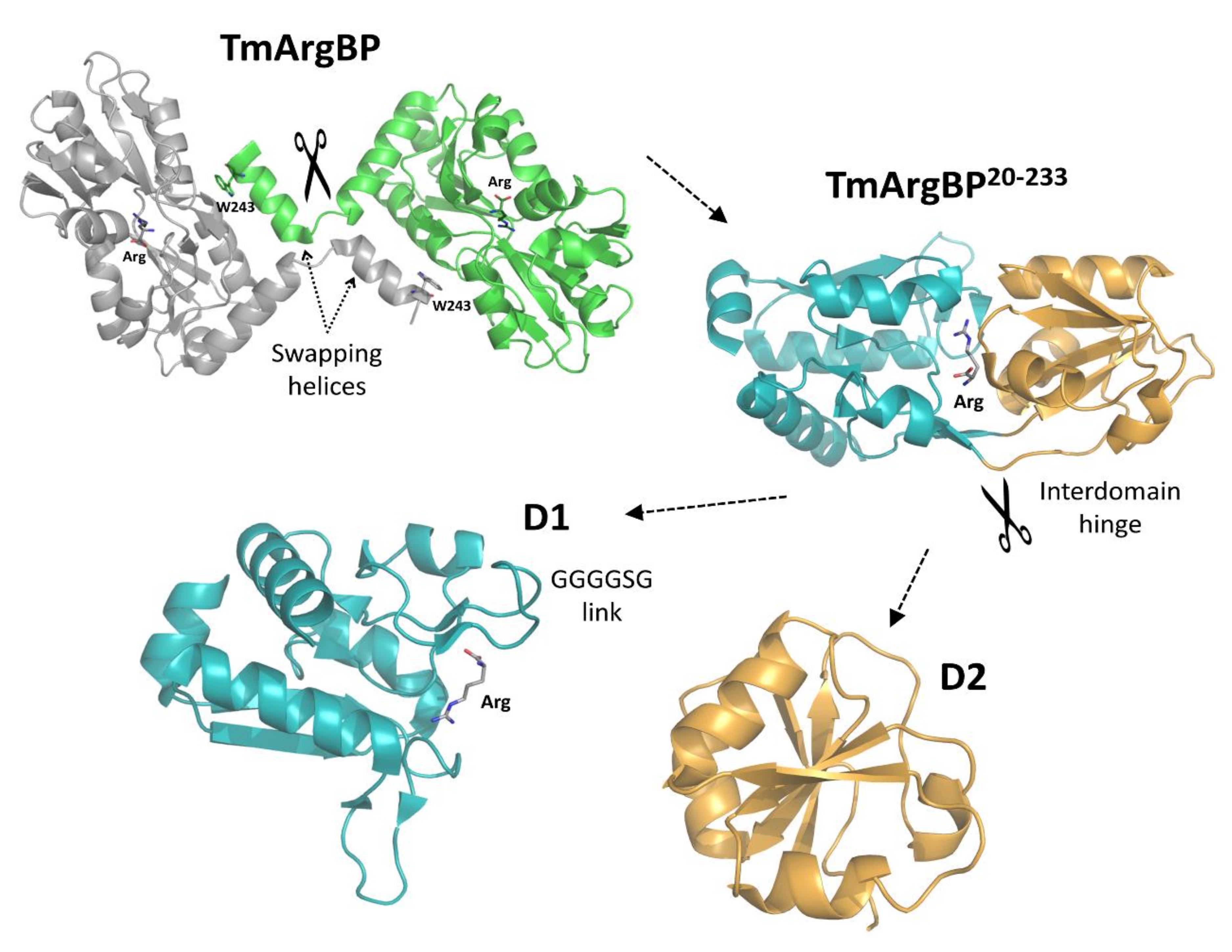
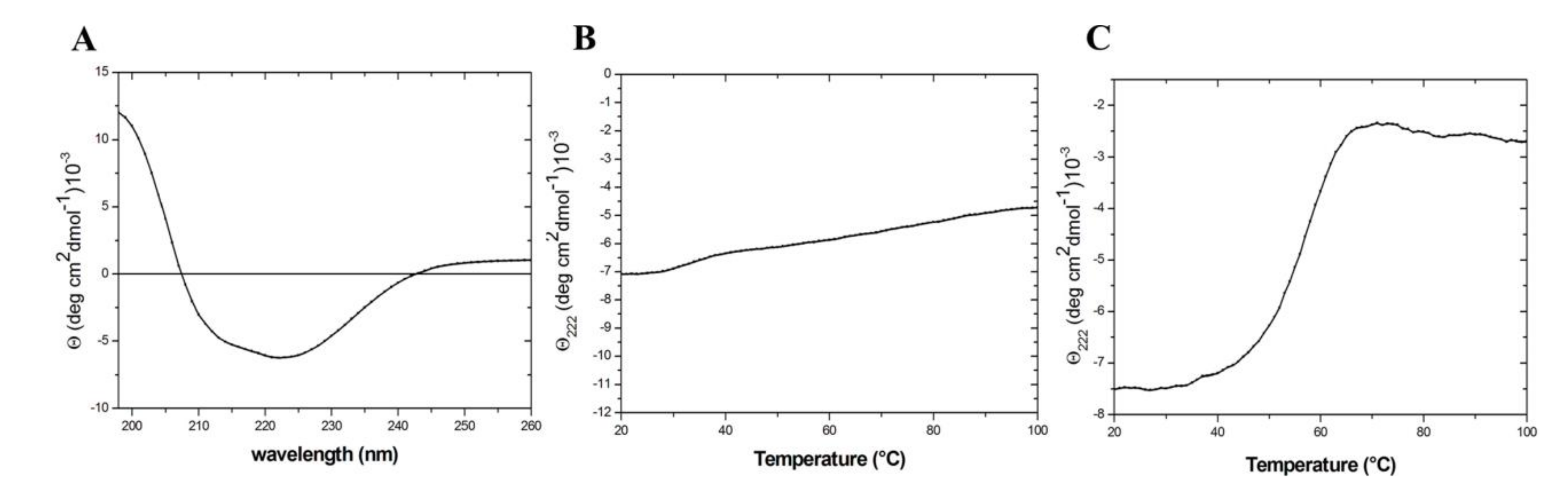
2.3. From the Monomeric to a Single Domain Scaffold
2.4. Crystal Structure of D1F57W
3. Discussion
4. Materials and Methods
4.1. Notations
4.2. Molecular Dynamics: Models and Protocol
4.3. Cloning, Expression and Purification of the Mutants
4.4. Circular Dichroism (CD)
4.5. Tryptofan Fluorescence Spectroscopy (TFS)
4.6. Protein Crystallization, Data Collection and Structure Refinement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TmArgBP TmArgBP20−233 D1 | Arginine-binding protein from Thermotoga maritima Region encompassing residues 20–233 of TmArgBP Domain 1 of TmArgBP (residues 20–114 and 207–233 linked by a GGGGSG segment) |
| SBP | Substrate-binding proteins |
| MD | Molecular dynamics |
| GuHCl | Guanidinium chloride |
| PDB | Protein Data Bank |
| RMSD | Root-Mean-Square Deviation |
References
- Tapiero, H.; Mathé, G.; Couvreur, P.; Tew, K.D.I. Arginine. Biomed. Pharmacother. 2002, 56, 439–445. [Google Scholar] [CrossRef]
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531. [Google Scholar] [CrossRef]
- Wheatley, D.N.; Scott, L.; Lamb, J.; Smith, S. Single amino acid (arginine) restriction: Growth and death of cultured HeLa and human diploid fibroblasts. Cell. Physiol. Biochem. 2000, 10, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Bednarz-Misa, I.; Fortuna, P.; Fleszar, M.G.; Lewandowski, Ł.; Diakowska, D.; Rosińczuk, J.; Krzystek-Korpacka, M. Esophageal Squamous Cell Carcinoma Is Accompanied by Local and Systemic Changes in L-arginine/NO Pathway. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Xiong, L.; Teng, J.L.; Botelho, M.G.; Lo, R.C.; Lau, S.K.; Woo, P.C. Arginine Metabolism in Bacterial Pathogenesis and Cancer Therapy. Int. J. Mol. Sci. 2016, 17, 363. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.M. Arginine Metabolism Revisited. J. Nutr. 2016, 146, 2579S–2586S. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F.; Lee, B. Clinical, biochemical, and molecular spectrum of hyperargininemia due to arginase I deficiency. Am. J. Med. Genet. C Semin Med. Genet. 2006, 142C, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Jęśko, H.; Wilkaniec, A.; Cieślik, M.; Hilgier, W.; Gąssowska, M.; Lukiw, W.J.; Adamczyk, A. Altered Arginine Metabolism in Cells Transfected with Human Wild-Type Beta Amyloid Precursor Protein (βAPP). Curr. Alzheimer Res. 2016, 13, 1030–1039. [Google Scholar] [CrossRef]
- Ruggiero, A.; Dattelbaum, J.D.; Staiano, M.; Berisio, R.; D’Auria, S.; Vitagliano, L. A loose domain swapping organization confers a remarkable stability to the dimeric structure of the arginine binding protein from Thermotoga maritima. PLoS ONE 2014, 9, e96560. [Google Scholar] [CrossRef] [Green Version]
- Smaldone, G.; Berisio, R.; Balasco, N.; D’Auria, S.; Vitagliano, L.; Ruggiero, A. Domain swapping dissection in Thermotoga maritima arginine binding protein: How structural flexibility may compensate destabilization. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 952–962. [Google Scholar] [CrossRef]
- Smaldone, G.; Balasco, N.; Vigorita, M.; Ruggiero, A.; Cozzolino, S.; Berisio, R.; Del Vecchio, P.; Graziano, G.; Vitagliano, L. Domain communication in Thermotoga maritima Arginine Binding Protein unraveled through protein dissection. Int. J. Biol. Macromol. 2018, 119, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Jaworek, M.W.; Ruggiero, A.; Graziano, G.; Winter, R.; Vitagliano, L. On the extraordinary pressure stability of the Thermotoga maritima arginine binding protein and its folded fragments—A high-pressure FTIR spectroscopy study. Phys. Chem. Chem. Phys. 2020, 22, 11244–11248. [Google Scholar] [CrossRef] [PubMed]
- Balasco, N.; Smaldone, G.; Vigorita, M.; Del Vecchio, P.; Graziano, G.; Ruggiero, A.; Vitagliano, L. The characterization of Thermotoga maritima Arginine Binding Protein variants demonstrates that minimal local strains have an important impact on protein stability. Sci. Rep. 2019, 9, 6617. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, G.; Vigorita, M.; Ruggiero, A.; Balasco, N.; Dattelbaum, J.D.; D’Auria, S.; Del Vecchio, P.; Graziano, G.; Vitagliano, L. Proline 235 plays a key role in the regulation of the oligomeric states of Thermotoga maritima Arginine Binding Protein. Biochim. Biophys. Acta 2016, 1864, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Smaldone, G.; Ruggiero, A.; Balasco, N.; Abuhammad, A.; Autiero, I.; Caruso, D.; Esposito, D.; Ferraro, G.; Gelardi, E.L.M.; Moreira, M.; et al. The non-swapped monomeric structure of the arginine-binding protein from Thermotoga maritima. Acta Crystallogr. F Struct. Biol. Commun. 2019, 75, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Deacon, L.J.; Billones, H.; Galyean, A.A.; Donaldson, T.; Pennacchio, A.; Iozzino, L.; D’Auria, S.; Dattelbaum, J.D. Tryptophan-scanning mutagenesis of the ligand binding pocket in Thermotoga maritima arginine-binding protein. Biochimie 2014, 99, 208–214. [Google Scholar] [CrossRef]
- Cozzolino, S.; Balasco, N.; Vigorita, M.; Ruggiero, A.; Smaldone, G.; Del Vecchio, P.; Vitagliano, L.; Graziano, G. Guanidinium binding to proteins: The intriguing effects on the D1 and D2 domains of Thermotoga maritima Arginine Binding Protein and a comprehensive analysis of the Protein Data Bank. Int. J. Biol. Macromol. 2020, 163, 375–385. [Google Scholar] [CrossRef]
- Scirè, A.; Marabotti, A.; Staiano, M.; Iozzino, L.; Luchansky, M.S.; Der, B.S.; Dattelbaum, J.D.; Tanfani, F.; D’Auria, S. Amino acid transport in thermophiles: Characterization of an arginine-binding protein in Thermotoga maritima. 2. Molecular organization and structural stability. Mol. Biosyst. 2010, 6, 687–698. [Google Scholar] [CrossRef]
- Luchansky, M.S.; Der, B.S.; D’Auria, S.; Pocsfalvi, G.; Iozzino, L.; Marasco, D.; Dattelbaum, J.D. Amino acid transport in thermophiles: Characterization of an arginine-binding protein in Thermotoga maritima. Mol. Biosyst. 2010, 6, 142–151. [Google Scholar] [CrossRef]
- Donaldson, T.; Iozzino, L.; Deacon, L.J.; Billones, H.; Ausili, A.; D’Auria, S.; Dattelbaum, J.D. Engineering a switch-based biosensor for arginine using a Thermotoga maritima periplasmic binding protein. Anal. Biochem. 2017, 525, 60–66. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The penultimate rotamer library. Proteins 2000, 40, 389–408. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Fink, A.L. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta 2007, 1768, 1862–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, N.; Singh, A.K.; Singh, M. L-arginine biosensors: A comprehensive review. Biochem. Biophys. Rep. 2017, 12, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res. 2002, 62, 5443–5450. [Google Scholar]
- Wheatley, D.N.; Philip, R.; Campbell, E. Arginine deprivation and tumour cell death: Arginase and its inhibition. Mol. Cell Biochem. 2003, 244, 177–185. [Google Scholar] [CrossRef]
- Wheatley, D.N.; Campbell, E. Arginine deprivation, growth inhibition and tumour cell death: 3. Deficient utilisation of citrulline by malignant cells. Br. J. Cancer 2003, 89, 573–576. [Google Scholar] [CrossRef] [Green Version]
- Bruch-Gerharz, D.; Schnorr, O.; Suschek, C.; Beck, K.F.; Pfeilschifter, J.; Ruzicka, T.; Kolb-Bachofen, V. Arginase 1 overexpression in psoriasis: Limitation of inducible nitric oxide synthase activity as a molecular mechanism for keratinocyte hyperproliferation. Am. J. Pathol. 2003, 162, 203–211. [Google Scholar] [CrossRef]
- Meurs, H.; Maarsingh, H.; Zaagsma, J. Arginase and asthma: Novel insights into nitric oxide homeostasis and airway hyperresponsiveness. Trends Pharmacol. Sci. 2003, 24, 450–455. [Google Scholar] [CrossRef]
- Ginésy, M.; Enman, J.; Rusanova-Naydenova, D.; Rova, U. Simultaneous Quantification of L-arginine and Monosaccharides during Fermentation: An Advanced Chromatography Approach. Molecules 2019, 24. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.Y.; Chung, S.F.; Chen, Y.W.; So, Y.H.; So, P.K.; Cheong, W.L.; Wong, K.Y.; Leung, Y.C. Design of a structure-based fluorescent biosensor from bioengineered arginine deiminase for rapid determination of L-arginine. Int. J. Biol. Macromol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ausili, A.; Staiano, M.; Dattelbaum, J.; Varriale, A.; Capo, A.; D’Auria, S. Periplasmic Binding Proteins in Thermophiles: Characterization and Potential Application of an Arginine-Binding Protein from Thermotoga maritima: A Brief Thermo-Story. Life (Basel) 2013, 3, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, J.H.; Zhang, W.H.; Herde, M.K.; Clifton, B.E.; Radziejewski, J.; Janovjak, H.; Henneberger, C.; Jackson, C.J. Construction of a robust and sensitive arginine biosensor through ancestral protein reconstruction. Protein Sci. 2015, 24, 1412–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput Chem 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Amadei, A.; Ceruso, M.A.; Di Nola, A. On the convergence of the conformational coordinates basis set obtained by the essential dynamics analysis of proteins’ molecular dynamics simulations. Proteins 1999, 36, 419–424. [Google Scholar] [CrossRef]
- Merlino, A.; Vitagliano, L.; Ceruso, M.A.; Mazzarella, L. Subtle functional collective motions in pancreatic-like ribonucleases: From ribonuclease A to angiogenin. Proteins 2003, 53, 101–110. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.J.; Perrakis, A.; Lamzin, V.S. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 2003, 374, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Improta, R.; Vitagliano, L.; Esposito, L. Peptide bond distortions from planarity: New insights from quantum mechanical calculations and peptide/protein crystal structures. PLoS ONE 2011, 6, e24533. [Google Scholar] [CrossRef] [Green Version]
- Balasco, N.; Esposito, L.; Vitagliano, L. Factors affecting the amplitude of the τ angle in proteins: A revisitation. Acta Crystallogr. D Struct. Biol. 2017, 73, 618–625. [Google Scholar] [CrossRef]
- Balasco, N.; Esposito, L.; Thind, A.S.; Guarracino, M.R.; Vitagliano, L. Dissection of Factors Affecting the Variability of the Peptide Bond Geometry and Planarity. Biomed. Res. Int. 2017, 2017, 2617629. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | TmArgBP D1F57W |
|---|---|
| X-ray device | Rigaku FR007HF with CCD detector |
| Space group | P212121 |
| a, b, c (Å) | 37.48, 67.22, 102.41 |
| Resolution range (Å) | 50.00–1.79 |
| Wavelength (Å) | 1.54 |
| Average redundancy | 4.0 (2.6) |
| Unique reflections | 25021 (3013) |
| Completeness (%) | 99.1 (97.7) |
| R merge (%) | 6.1 (34.2) |
| Average I/σ(I) | 27.4 (4.0) |
| Asymmetric unit | Two molecules |
| R/R-free | 0.178/0.233 |
| No. of atoms | 2353 |
| No. of residues | 259 |
| No. of water molecules | 297 |
| Mean B value (Å2) | 24.745 |
| R.m.s. bonds (Å) | 0.018 |
| R.m.s. angles (°) | 1.841 |
| MolProbity statistics | |
| MolProbity score | 1.47 |
| Clashscore, all atoms: | 3.86 |
| Ramachandran Allowed | 257/257 |
| Ramachandran Favored | 246/257 |
| Ramachandran Outliers | 0/257 |
| Favored rotamers | 212/221 |
| Poor rotamers | 2/221 |
| Cβ deviation outliers | 2/237 |
| Bad bonds | 2/2074 |
| Bad angles | 1/2795 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smaldone, G.; Ruggiero, A.; Balasco, N.; Vitagliano, L. Development of a Protein Scaffold for Arginine Sensing Generated through the Dissection of the Arginine-Binding Protein from Thermotoga maritima. Int. J. Mol. Sci. 2020, 21, 7503. https://doi.org/10.3390/ijms21207503
Smaldone G, Ruggiero A, Balasco N, Vitagliano L. Development of a Protein Scaffold for Arginine Sensing Generated through the Dissection of the Arginine-Binding Protein from Thermotoga maritima. International Journal of Molecular Sciences. 2020; 21(20):7503. https://doi.org/10.3390/ijms21207503
Chicago/Turabian StyleSmaldone, Giovanni, Alessia Ruggiero, Nicole Balasco, and Luigi Vitagliano. 2020. "Development of a Protein Scaffold for Arginine Sensing Generated through the Dissection of the Arginine-Binding Protein from Thermotoga maritima" International Journal of Molecular Sciences 21, no. 20: 7503. https://doi.org/10.3390/ijms21207503
APA StyleSmaldone, G., Ruggiero, A., Balasco, N., & Vitagliano, L. (2020). Development of a Protein Scaffold for Arginine Sensing Generated through the Dissection of the Arginine-Binding Protein from Thermotoga maritima. International Journal of Molecular Sciences, 21(20), 7503. https://doi.org/10.3390/ijms21207503