ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
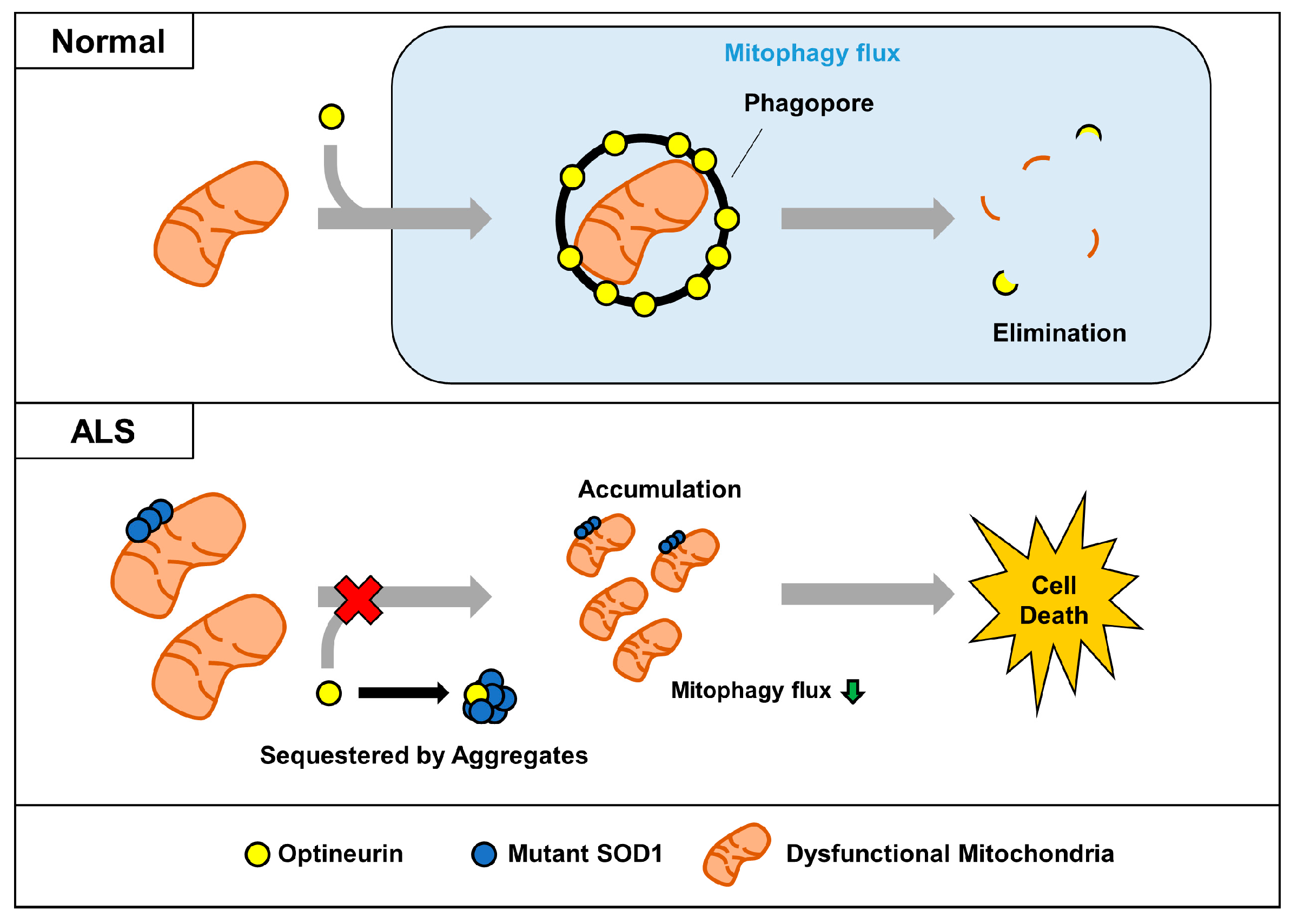
Abstract
:1. Introduction
2. Results
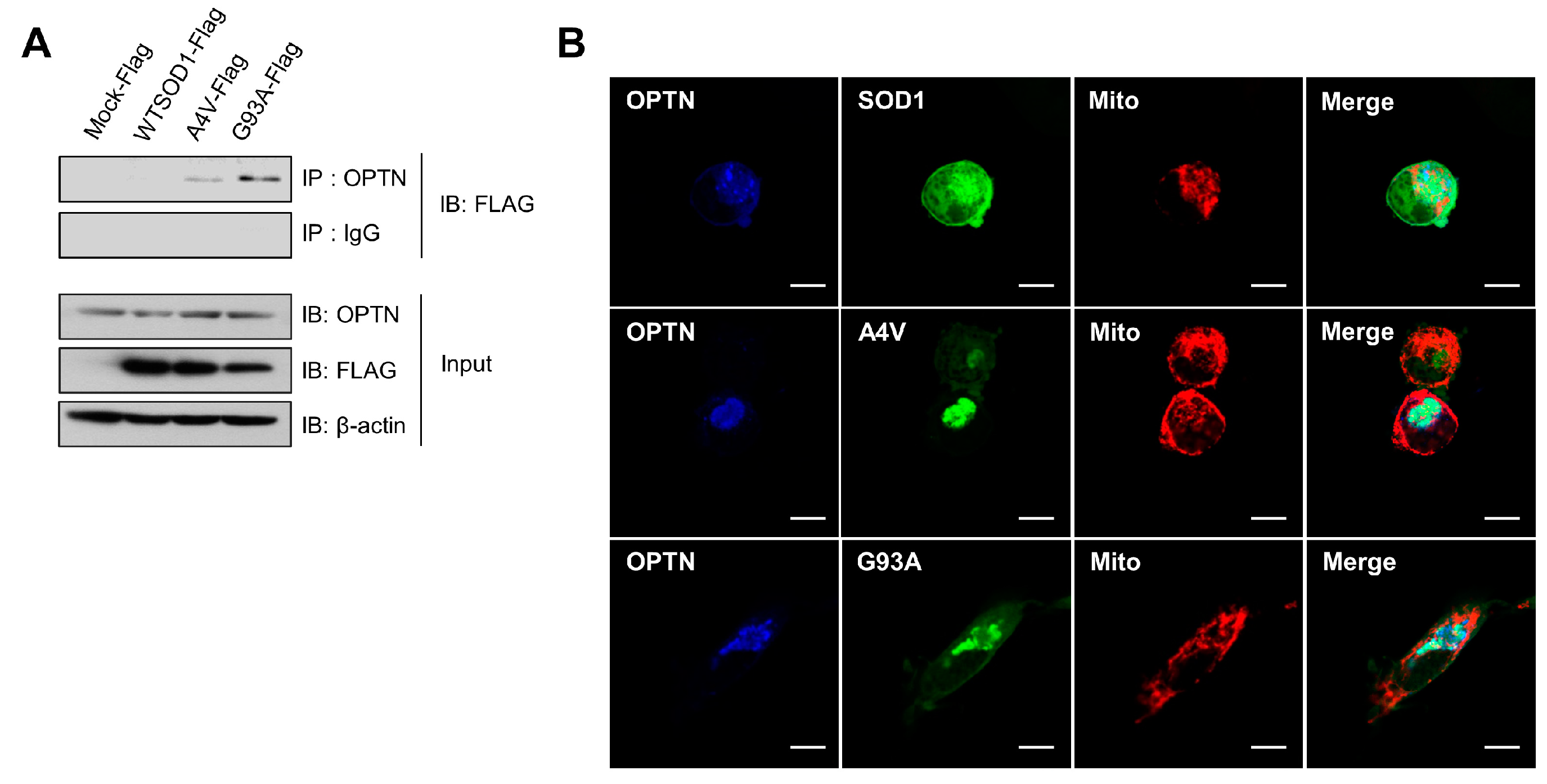
2.1. Optineurin Associates with Mutant SOD1 Proteins, A4V and G93A
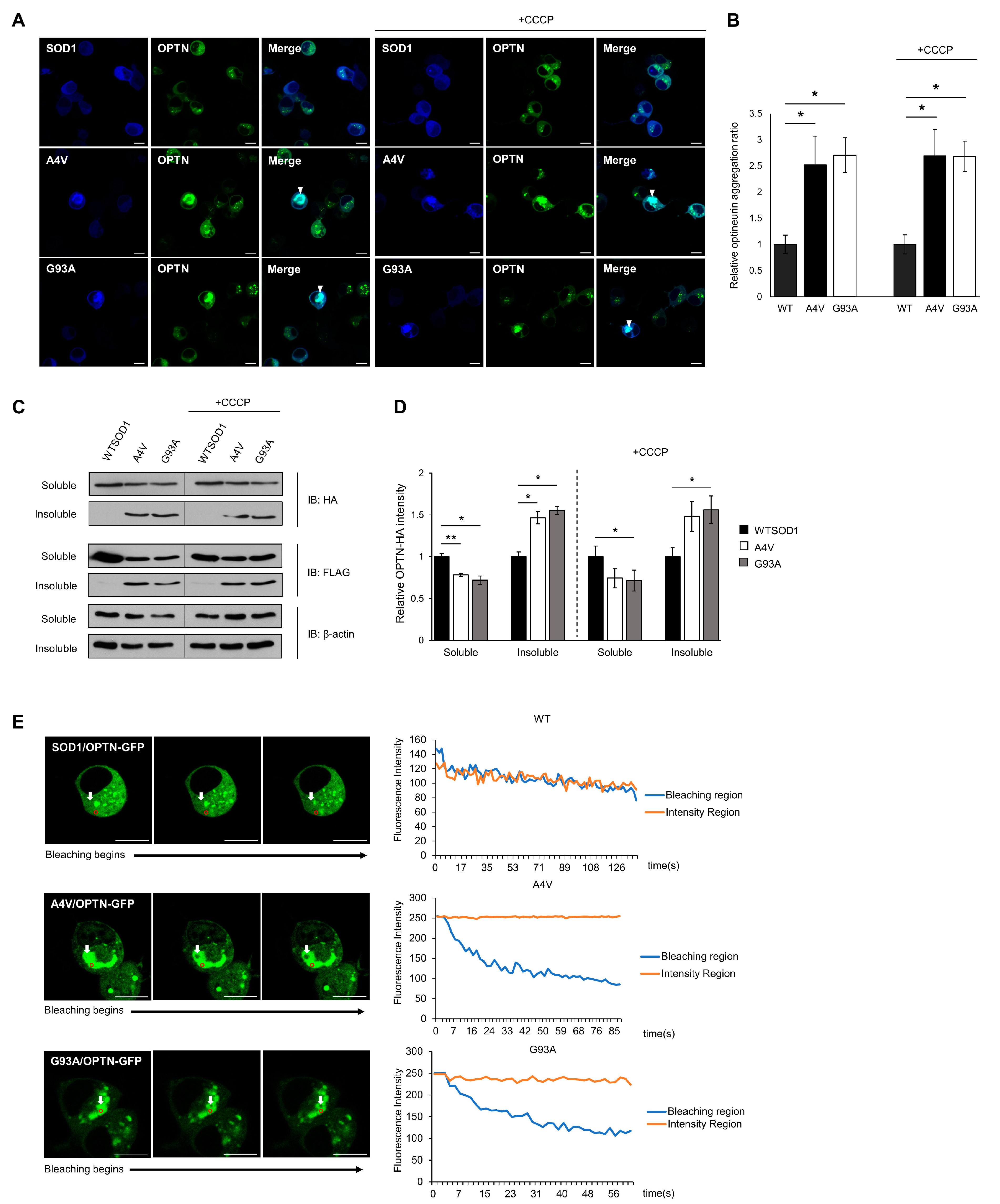
2.2. Insoluble Optineurin Significantly Increases in N2a Cells Containing Mutant SOD1 Aggregates
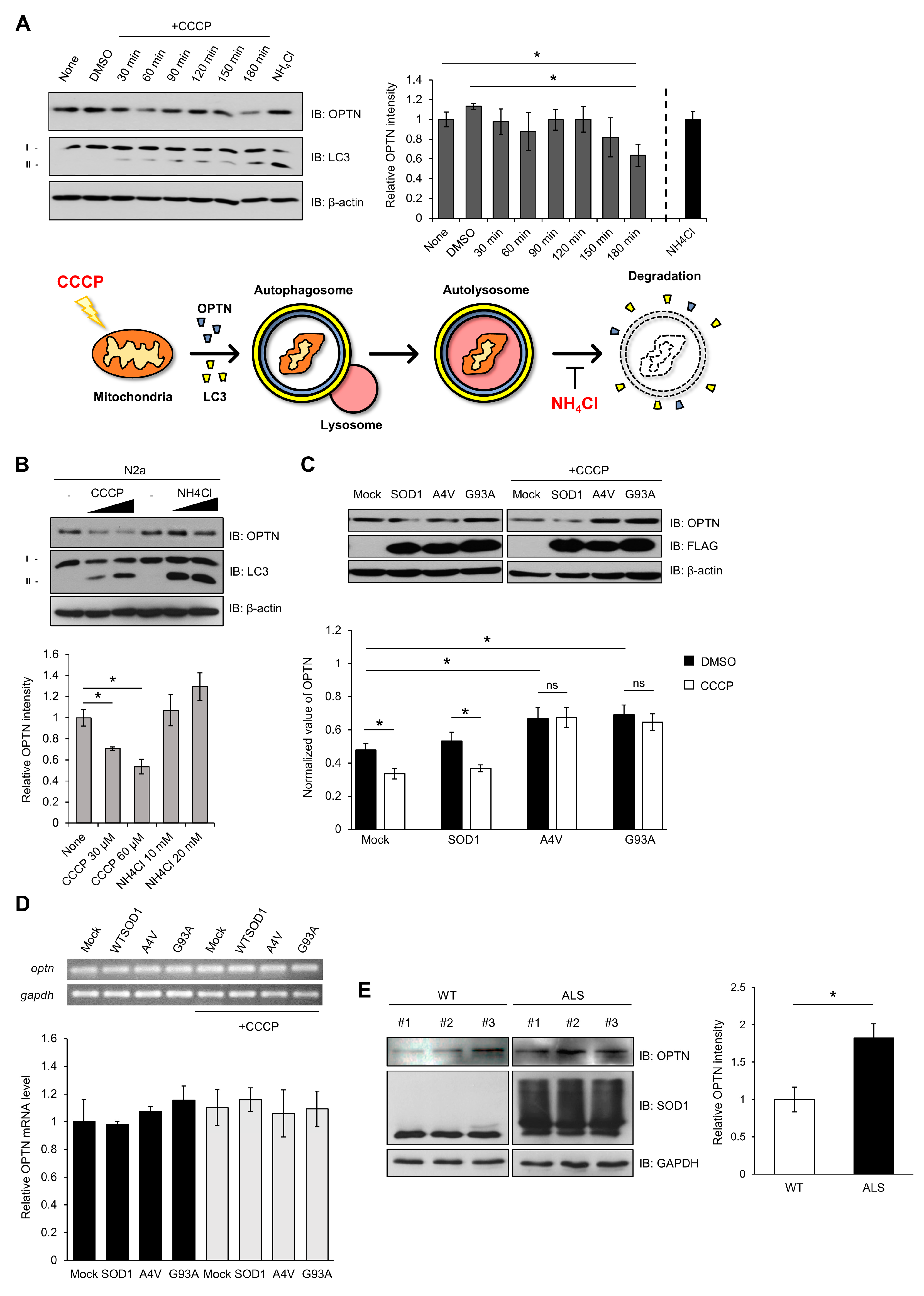
2.3. Mutant SOD1 Aggregates Sequester OPTN and Affect the Mitophagy Process
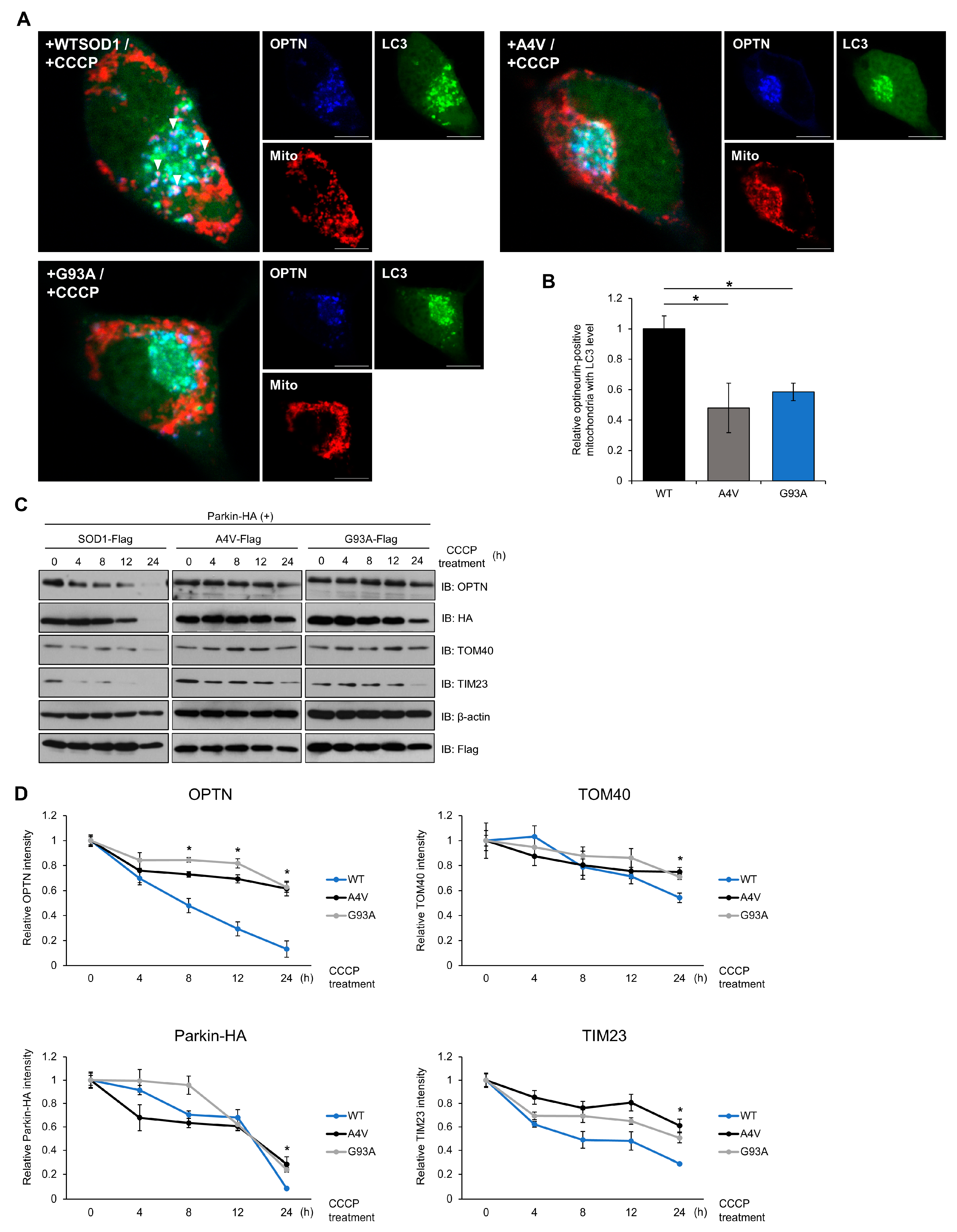
2.4. Mutant SOD1 Aggregates Inhibit Mitophagosome Formation and Mitophagy Flux
2.5. Over-Expression of OPTN Reduces Cytotoxicity Induced by Mutant SOD1
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Plasmid Constructions
4.3. Reagents and Antibodies
4.4. Immunoblotting Assay
4.5. Co-Immunoprecipitation (Co-IP)
4.6. Animals
4.7. Cell Viability Assay
4.8. DCF-DA Assay
4.9. Soluble/Insoluble Fraction
4.10. FLIP
4.11. Immunofluorescence Assay (IFA)
4.12. qRT-PCR
4.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| SOD1 | Superoxide dismutase 1 |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| HD | Huntington’s disease |
| OPTN | Optineurin |
| UBD | Ubiquitin binding domain |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| TDP-43 | TAR DNA binding protein 43 |
| CCCP | Carbonyl cyanide m-chlorophenyl hydrazone |
| FLIP | Fluorescence loss in photobleaching |
| ROS | Reactive oxygen species |
| CHX | Cycloheximide |
| TIM23 | Translocase of the inner mitochondrial membrane 23 |
| TOM40 | Translocase of the outer mitochondrial membrane 40 |
| PARP | Poly (ADP-ribose) polymerase |
| DCF | Dichlorofluorescein |
| PI | Propidium iodide |
| PINK1 | PTEN-induced kinase 1 |
| TBK1 | TANK-binding kinase 1 |
References
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurosci. 2015, 11, 266–279. [Google Scholar] [CrossRef]
- Cook, C.; Petrucelli, L. Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 2019, 101, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Valentine, J.S.; Doucette, P.A.; Zittin Potter, S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Ann. Rev. Biochem. 2005, 74, 563–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munch, C.; Bertolotti, A. Exposure of hydrophobic surfaces initiates aggregation of diverse ALS-causing superoxide dismutase-1 mutants. J. Mol. Biol. 2010, 399, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Vassall, K.A.; Stubbs, H.R.; Primmer, H.A.; Tong, M.S.; Sullivan, S.M.; Sobering, R.; Srinivasan, S.; Briere, L.A.; Dunn, S.D.; Colon, W.; et al. Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc. Natl. Acad. Sci. USA 2011, 108, 2210–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banci, L.; Bertini, I.; Boca, M.; Calderone, V.; Cantini, F.; Girotto, S.; Vieru, M. Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc. Natl. Acad. Sci. USA 2009, 106, 6980–6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urushitani, M.; Kurisu, J.; Tsukita, K.; Takahashi, R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 83, 1030–1042. [Google Scholar] [CrossRef]
- Park, J.H.; Jang, H.R.; Lee, I.Y.; Oh, H.K.; Choi, E.J.; Rhim, H.; Kang, S. Amyotrophic lateral sclerosis-related mutant superoxide dismutase 1 aggregates inhibit 14–3–3-mediated cell survival by sequestration into the JUNQ compartment. Hum. Mol. Genet. 2017, 26, 3615–3629. [Google Scholar] [CrossRef]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Sundaramoorthy, V.; Walker, A.K.; Tan, V.; Fifita, J.A.; McCann, E.P.; Williams, K.L.; Blair, I.P.; Guillemin, G.J.; Farg, M.A.; Atkin, J.D. Defects in optineurin-and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 3830–3846. [Google Scholar] [CrossRef] [Green Version]
- Akizuki, M.; Yamashita, H.; Uemura, K.; Maruyama, H.; Kawakami, H.; Ito, H.; Takahashi, R. Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J. Neurochem. 2013, 126, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A coordinator of membrane-associated cargo trafficking and autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodolfo, C.; Campello, S.; Cecconi, F. Mitophagy in neurodegenerative diseases. Neurochem. Int. 2018, 117, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genet. Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [Green Version]
- Pedrini, S.; Sau, D.; Guareschi, S.; Bogush, M.; Brown, R.H., Jr.; Naniche, N.; Kia, A.; Trotti, D.; Pasinelli, P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum. Mol. Genet. 2010, 19, 2974–2986. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef] [Green Version]
- Leadsham, J.E.; Sanders, G.; Giannaki, S.; Bastow, E.L.; Hutton, R.; Naeimi, W.R.; Breitenbach, M.; Gourlay, C.W. Loss of cytochrome c oxidase promotes RAS-dependent ROS production from the ER resident NADPH oxidase, Yno1p, in yeast. Cell Metab. 2013, 18, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H., Jr.; Julien, J.P.; Arbour, N.; Vande Velde, C. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Osawa, T.; Mizuno, Y.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Optineurin in neurodegenerative diseases. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2011, 31, 569–574. [Google Scholar] [CrossRef]
- Tu, P.H.; Raju, P.; Robinson, K.A.; Gurney, M.E.; Trojanowski, J.Q.; Lee, V.M. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. USA 1996, 93, 3155–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Magrane, J.; Hervias, I.; Henning, M.S.; Damiano, M.; Kawamata, H.; Manfredi, G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet. 2009, 18, 4552–4564. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Gal, J.; Kuang, L.; Barnett, K.R.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.V.; Hayward, L.J.; Kasarskis, E.J.; Zhu, H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016, 132, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, N.H.; Tak, Y.J.; Rhim, H.; Kang, S. Hip2 ubiquitin-conjugating enzyme has a role in UV-induced G1/S arrest and re-entry. Genet. Genom. 2019, 41, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.J.; Park, H.J.; Kim, G.Y.; Cho, H.M.; Choi, J.H.; Park, H.Y.; Jang, J.Y.; Rhim, H.S.; Kang, S.M. Intracellular amyloid beta interacts with SOD1 and impairs the enzymatic activity of SOD1: Implications for the pathogenesis of amyotrophic lateral sclerosis. Exp. Mol. Med. 2009, 41, 611–617. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.H.; Jang, J.Y.; Rhim, H.; Kang, S. Characterization and Hsp104-induced artificial clearance of familial ALS-related SOD1 aggregates. Biochem. Biophys. Res. Commun. 2013, 434, 521–526. [Google Scholar] [CrossRef]
- Park, H.M.; Kim, G.Y.; Nam, M.K.; Seong, G.H.; Han, C.; Chung, K.C.; Kang, S.; Rhim, H. The serine protease HtrA2/Omi cleaves Parkin and irreversibly inactivates its E3 ubiquitin ligase activity. Biochem. Biophys. Res. Commun. 2009, 387, 537–542. [Google Scholar] [CrossRef]
- Jang, J.Y.; Cho, H.; Park, H.Y.; Rhim, H.; Kang, S. ALS-linked mutant SOD1 proteins promote Aβ aggregates in ALS through direct interaction with Aβ. Biochem. Biophys. Res. Commun. 2017, 493, 697–707. [Google Scholar] [CrossRef]
- Leyton-Jaimes, M.F.; Benaim, C.; Abu-Hamad, S.; Kahn, J.; Guetta, A.; Bucala, R.; Israelson, A. Endogenous macrophage migration inhibitory factor reduces the accumulation and toxicity of misfolded SOD1 in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2016, 113, 10198–10203. [Google Scholar] [CrossRef] [Green Version]
- De Marco, N.; Buono, M.; Troise, F.; Diez-Roux, G. Optineurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J. Biol. Chem. 2006, 281, 16147–16156. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H. Ethyl acetate fraction of adenophora triphylla var. japonica inhibits migration of lewis lung carcinoma cells by suppressing macrophage polarization toward an M2 phenotype. J. Pharmacopunct. 2019, 22, 253–259. [Google Scholar]
- Park, J.H.; Park, H.S.; Hong, S.; Kang, S. Motor neurons derived from ALS-related mouse iPS cells recapitulate pathological features of ALS. Exp. Mol. Med. 2016, 48, e276. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tak, Y.J.; Park, J.-H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. https://doi.org/10.3390/ijms21207525
Tak YJ, Park J-H, Rhim H, Kang S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. International Journal of Molecular Sciences. 2020; 21(20):7525. https://doi.org/10.3390/ijms21207525
Chicago/Turabian StyleTak, Yeong Jin, Ju-Hwang Park, Hyangshuk Rhim, and Seongman Kang. 2020. "ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin" International Journal of Molecular Sciences 21, no. 20: 7525. https://doi.org/10.3390/ijms21207525
APA StyleTak, Y. J., Park, J.-H., Rhim, H., & Kang, S. (2020). ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. International Journal of Molecular Sciences, 21(20), 7525. https://doi.org/10.3390/ijms21207525