PAK1 Regulates MEC-17 Acetyltransferase Activity and Microtubule Acetylation during Proplatelet Extension

 ,
, 
Abstract
1. Introduction
2. Results
2.1. PAK1 Colocalizes with Microtubules and Actin in Proplatelet Extensions
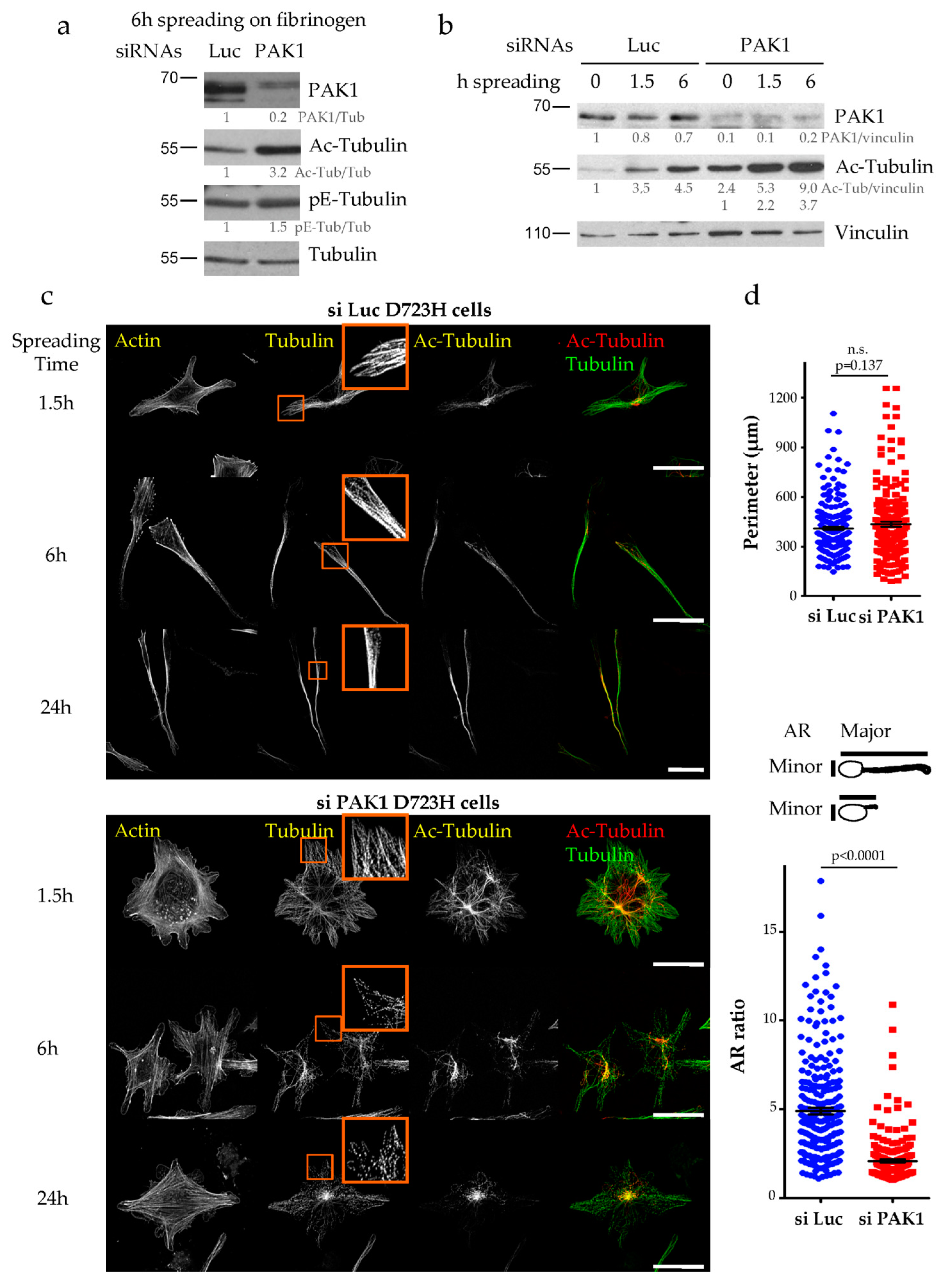
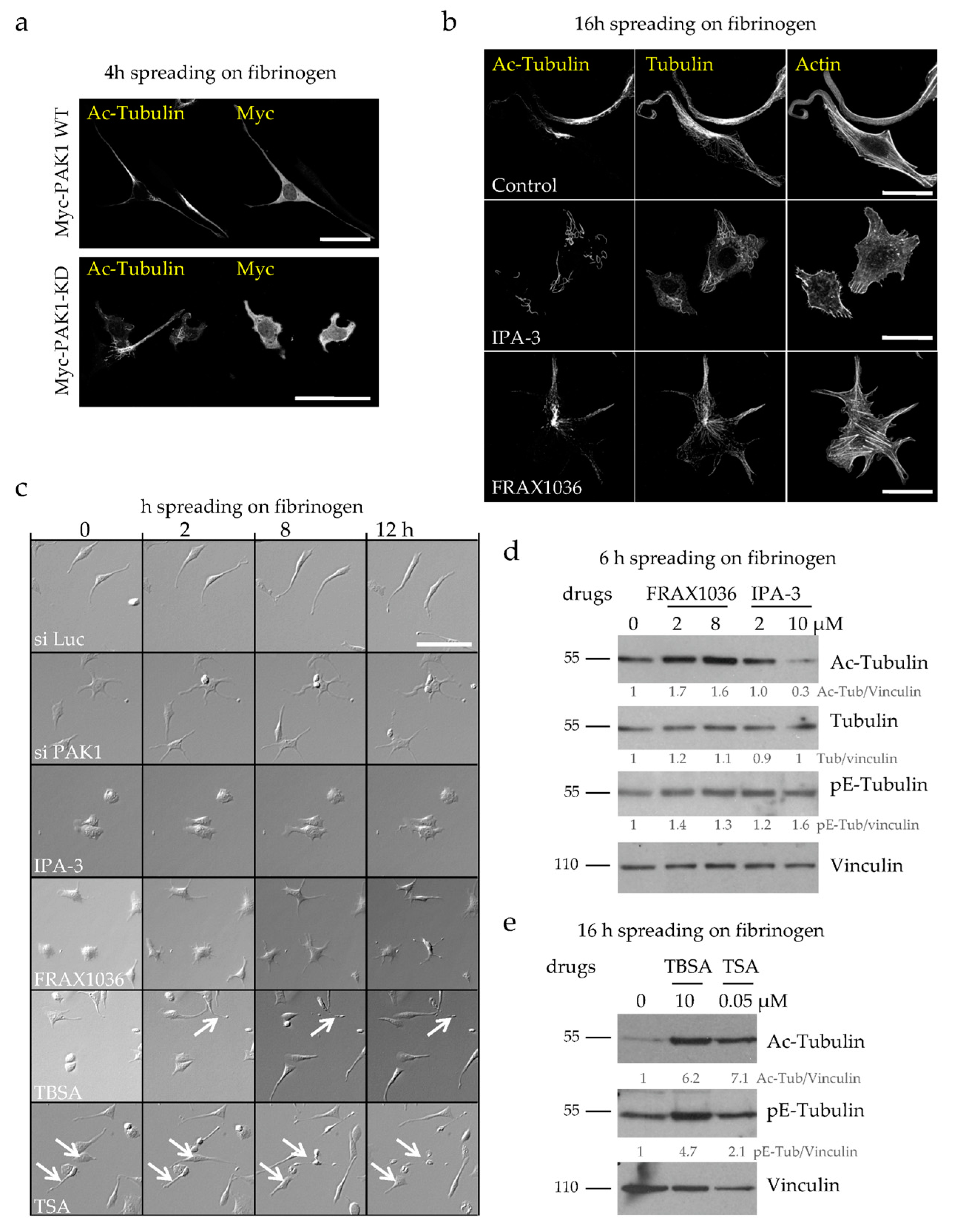
2.2. Loss of PAK1 Kinase Activity Induces the Formation of Many Short PPLLs, Increases MT Acetylation and Impairs MT Integrity
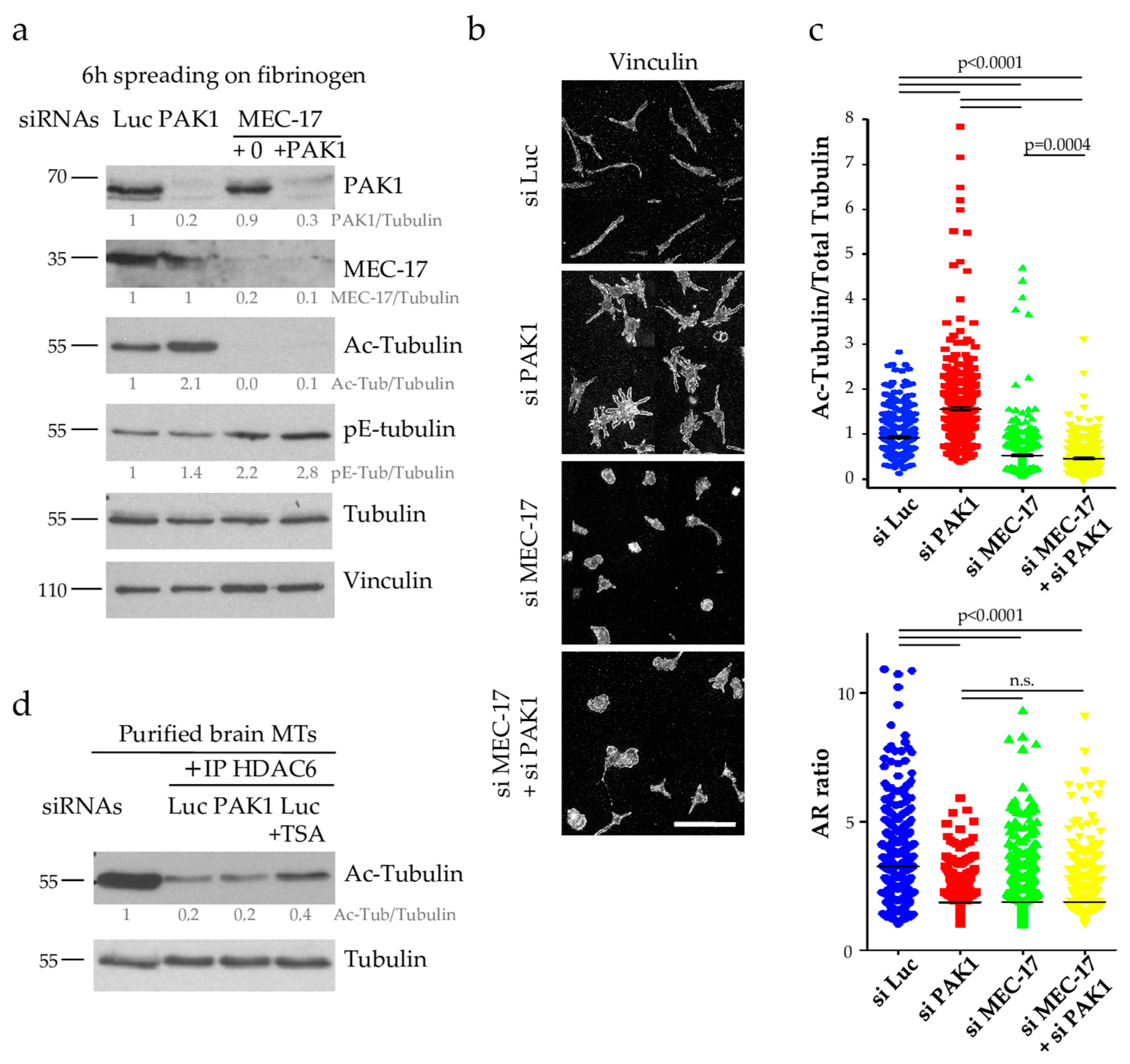
2.3. MT Hyperacetylation Does Not Increase the Number of PPLLs but Impairs MT Integrity
2.4. PAK1 Does Not Affect the Activity of the Tubulin Deacetylase HDAC6
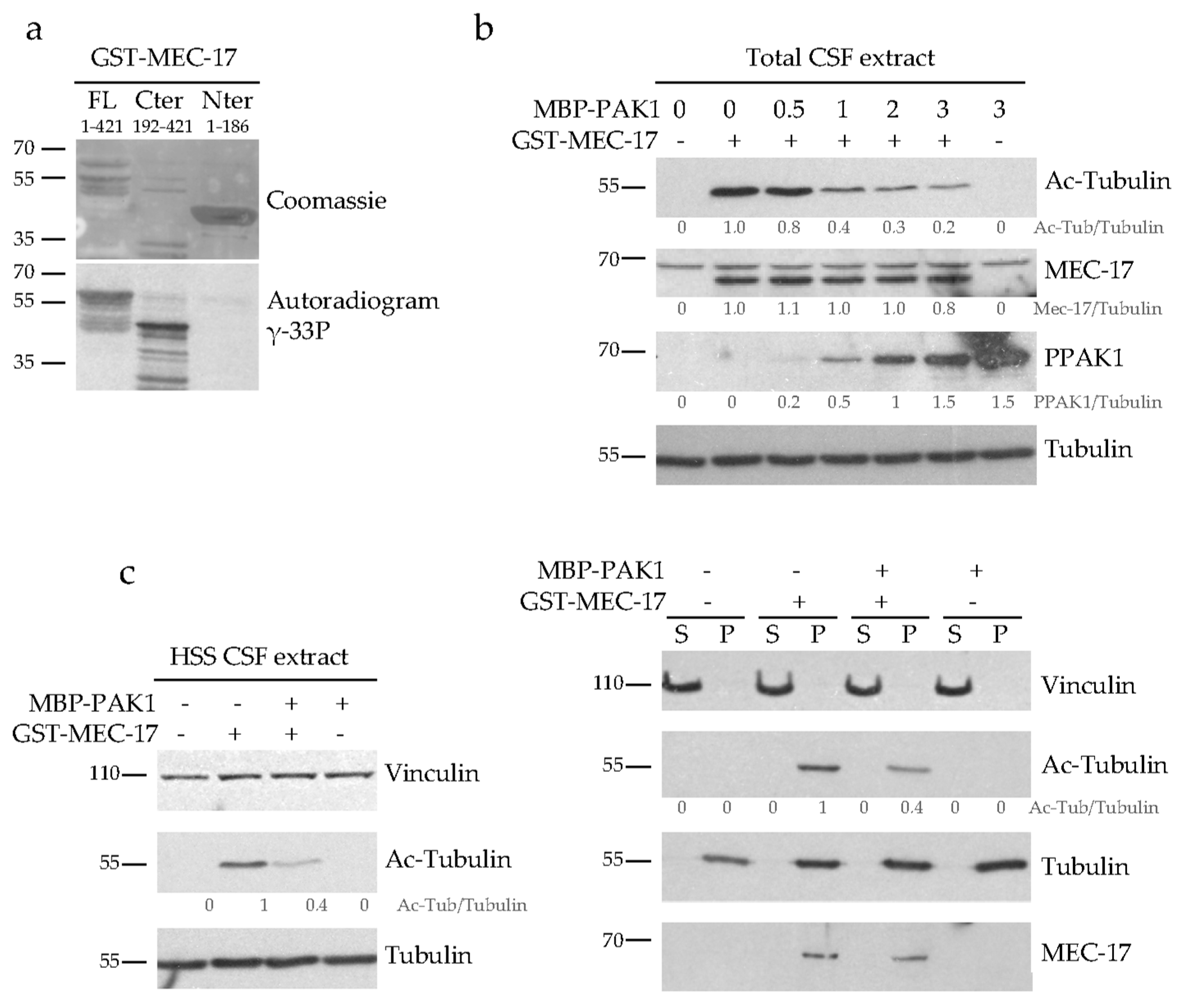
2.5. PAK1 Directly Phosphorylates MEC-17 and Inhibits Its Acetyltransferase Activity
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Megakaryocytes
4.3. Cell Lines, Cell Transfection and Drug Treatment
4.4. siRNA, Plasmids, Site Directed Mutagenesis
4.5. Immunofluorescence
4.6. Western Blot Quantification
4.7. Cell Imaging
4.8. Image Quantification and Statistical Analyses
4.9. HDAC6 Activity Assays
4.10. Recombinant Protein Purification
4.11. Xenopus CSF Extracts
4.12. In Vitro Kinase Reaction
4.13. MEC-17 Activity in CSF Extract
4.14. MEC-17 Binding to MTs
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ac-tubulin | Acetylated tubulin |
| BSA | Bovine serum albumin |
| CHO | Chinese hamster ovary |
| DMS | Demarcation membrane system |
| HDAC6 | Histone deacetylase 6 |
| LUC | Luciferase |
| MIP | Maximum intensity projection |
| MK | Megakaryocyte |
| MT | Microtubule |
| PAK1 | P21-activated kinase |
| pE-tubulin | Polyglutamylated tubulin |
| PPL | Proplatelet |
| PPLL | Proplatelet-like |
| SEM | Standard error of the mean |
| TBSA | Tubastatin A hydrochloride |
| TPO | Thrombopoietin |
| TSA | Trichostatin A |
References
- Schulze, H.; Korpal, M.; Hurov, J.; Kim, S.-W.; Zhang, J.; Cantley, L.C.; Graf, T.; Shivdasani, R.A. Characterization of the megakaryocyte demarcation membrane system and its role in thrombopoiesis. Blood 2006, 107, 3868–3875. [Google Scholar] [CrossRef] [PubMed]
- Junt, T.; Schulze, H.; Chen, Z.; Massberg, S.; Goerge, T.; Krueger, A.; Wagner, D.D.; Graf, T.; Italiano, J.E.; Shivdasani, R.A.; et al. Dynamic Visualization of Thrombopoiesis Within Bone Marrow. Science 2007, 317, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E.; Patel-Hett, S.; Hartwig, J.H. Mechanics of proplatelet elaboration: Mechanics of proplatelet elaboration. J. Thromb. Haemost. 2007, 5, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E.; Lecine, P.; Shivdasani, R.A.; Hartwig, J.H. Blood Platelets Are Assembled Principally at the Ends of Proplatelet Processes Produced by Differentiated Megakaryocytes. J. Cell Biol. 1999, 147, 1299–1312. [Google Scholar] [CrossRef]
- Patel, S.R.; Richardson, J.L.; Schulze, H.; Kahle, E.; Galjart, N.; Drabek, K.; Shivdasani, R.A.; Hartwig, J.H.; Italiano, J.E. Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood 2005, 106, 4076–4085. [Google Scholar] [CrossRef]
- Bender, M.; Thon, J.N.; Ehrlicher, A.J.; Wu, S.; Mazutis, L.; Deschmann, E.; Sola-Visner, M.; Italiano, J.E.; Hartwig, J.H. Microtubule sliding drives proplatelet elongation and is dependent on cytoplasmic dynein. Blood 2015, 125, 860–868. [Google Scholar] [CrossRef]
- Schwer, H.D.; Lecine, P.; Tiwari, S.; Italiano, J.E.; Hartwig, J.H.; Shivdasani, R.A. A lineage-restricted and divergent β-tubulin isoform is essential for the biogenesis, structure and function of blood platelets. Curr. Biol. 2001, 11, 579–586. [Google Scholar] [CrossRef]
- Kunishima, S.; Kobayashi, R.; Itoh, T.J.; Hamaguchi, M.; Saito, H. Mutation of the β1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood 2009, 113, 458–461. [Google Scholar] [CrossRef]
- Kunishima, S.; Nishimura, S.; Suzuki, H.; Imaizumi, M.; Saito, H. TUBB1 mutation disrupting microtubule assembly impairs proplatelet formation and results in congenital macrothrombocytopenia. Eur. J. Haematol. 2014, 92, 276–282. [Google Scholar] [CrossRef]
- Basciano, P.A.; Matakas, J.; Pecci, A.; Civaschi, E.; Cagioni, C.; Bompiani, N.; Burger, P.; Christos, P.; Snyder, J.P.; Bussel, J.; et al. β-1 tubulin R307H SNP alters microtubule dynamics and affects severity of a hereditary thrombocytopenia. J. Thromb. Haemost. 2015, 13, 651–659. [Google Scholar] [CrossRef]
- Stächele, J.; Bakchoul, T.; Najm, J.; Felbor, U.; Knöfler, R. Congenital macrothrombocytopenia associated with a combination of functional polymorphisms in the TUBB1 gene. Hamostaseologie 2015, 35 (Suppl. S1), S18–S21. [Google Scholar]
- van Dijk, J.; Bompard, G.; Cau, J.; Kunishima, S.; Rabeharivelo, G.; Mateos-Langerak, J.; Cazevieille, C.; Cavelier, P.; Boizet-Bonhoure, B.; Delsert, C.; et al. Microtubule polyglutamylation and acetylation drive microtubule dynamics critical for platelet formation. BMC Biol. 2018, 16, 116. [Google Scholar] [CrossRef]
- Tapon, N. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol. 1997, 9, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Pleines, I.; Dütting, S.; Cherpokova, D.; Eckly, A.; Meyer, I.; Morowski, M.; Krohne, G.; Schulze, H.; Gachet, C.; Debili, N.; et al. Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood 2013, 122, 3178–3187. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sanawar, R.; Li, X.; Li, F. Structure, biochemistry, and biology of PAK kinases. Gene 2017, 605, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Kosoff, R.E.; Aslan, J.E.; Kostyak, J.C.; Dulaimi, E.; Chow, H.Y.; Prudnikova, T.Y.; Radu, M.; Kunapuli, S.P.; McCarty, O.J.T.; Chernoff, J. Pak2 restrains endomitosis during megakaryopoiesis and alters cytoskeleton organization. Blood 2015, 125, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Meng, J. Abnormal Long-Lasting Synaptic Plasticity and Cognition in Mice Lacking the Mental Retardation Gene Pak3. J. Neurosci. 2005, 25, 6641–6650. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, M. The Pak1 Kinase: An Important Regulator of Neuronal Morphology and Function in the Developing Forebrain. Mol. Neurobiol. 2008, 37, 187–202. [Google Scholar] [CrossRef]
- Koth, A.P.; Oliveira, B.R.; Parfitt, G.M.; de Quadros Buonocore, J.; Barros, D.M. Participation of group I p21-activated kinases in neuroplasticity. J. Physiol. Paris 2014, 108, 270–277. [Google Scholar] [CrossRef]
- Asrar, S.; Meng, Y.; Zhou, Z.; Todorovski, Z.; Huang, W.W.; Jia, Z. Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 2009, 56, 73–80. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, Z.; Asrar, S.; Henkelman, M.; Xie, W.; Jia, Z. p21-Activated Kinases 1 and 3 Control Brain Size through Coordinating Neuronal Complexity and Synaptic Properties. Mol. Cell. Biol. 2011, 31, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An Isoform-Selective, Small-Molecule Inhibitor Targets the Autoregulatory Mechanism of p21-Activated Kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.C.; Gierke, S.; Pitt, C.; Sagolla, M.; Cheng, C.K.; Zhou, W.; Jubb, A.M.; Strickland, L.; Schmidt, M.; Duron, S.G.; et al. Small molecule inhibition of group I p21-activated kinases in breast cancer induces apoptosis and potentiates the activity of microtubule stabilizing agents. Breast Cancer Res. 2015, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Song, S.W.; Sun, J.; Bruner, J.M.; Fuller, G.N.; Zhang, W. IIp45 Inhibits Cell Migration through Inhibition of HDAC6. J. Biol. Chem. 2010, 285, 3554–3560. [Google Scholar] [CrossRef]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.-T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 Interacts with HDAC6 and Regulates Deacetylase Activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef]
- Deakin, N.O.; Turner, C.E. Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J. Cell Biol. 2014, 206, 395–413. [Google Scholar] [CrossRef]
- Salemi, L.M.; Maitland, M.E.R.; Yefet, E.R.; Schild-Poulter, C. Inhibition of HDAC6 activity through interaction with RanBPM and its associated CTLH complex. BMC Cancer 2017, 17, 460. [Google Scholar] [CrossRef]
- Balabanian, L.; Berger, C.L.; Hendricks, A.G. Acetylated Microtubules Are Preferentially Bundled Leading to Enhanced Kinesin-1 Motility. Biophys. J. 2017, 113, 1551–1560. [Google Scholar] [CrossRef]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone Deacetylase 6 Inhibition Compensates for the Transport Deficit in Huntington’s Disease by Increasing Tubulin Acetylation. J. Neuroscience 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Alper, J.D.; Decker, F.; Agana, B.; Howard, J. The Motility of Axonemal Dynein Is Regulated by the Tubulin Code. Biophys. J. 2014, 107, 2872–2880. [Google Scholar] [CrossRef] [PubMed]
- Sudo, H.; Baas, P.W. Acetylation of Microtubules Influences Their Sensitivity to Severing by Katanin in Neurons and Fibroblasts. J. Neurosci. 2010, 30, 7215–7226. [Google Scholar] [CrossRef]
- Mao, C.-X.; Xiong, Y.; Xiong, Z.; Wang, Q.; Zhang, Y.Q.; Jin, S. Microtubule-severing protein Katanin regulates neuromuscular junction development and dendritic elaboration in Drosophila. Development 2014, 141, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Ververis, A.; Christodoulou, A.; Christoforou, M.; Kamilari, C.; Lederer, C.W.; Santama, N. A novel family of katanin-like 2 protein isoforms (KATNAL2), interacting with nucleotide-binding proteins Nubp1 and Nubp2, are key regulators of different MT-based processes in mammalian cells. Cell. Mol. Life Sci. 2016, 73, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Sudo, H. Microtubule Hyperacetylation Enhances KL1-Dependent Micronucleation under a Tau Deficiency in Mammary Epithelial Cells. IJMS 2018, 19, 2488. [Google Scholar] [CrossRef] [PubMed]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef]
- Eshun-Wilson, L.; Zhang, R.; Portran, D.; Nachury, M.V.; Toso, D.B.; Löhr, T.; Vendruscolo, M.; Bonomi, M.; Fraser, J.S.; Nogales, E. Effects of α-tubulin acetylation on microtubule structure and stability. Proc. Natl. Acad. Sci. USA 2019, 116, 10366–10371. [Google Scholar] [CrossRef]
- Davenport, A.M.; Collins, L.N.; Chiu, H.; Minor, P.J.; Sternberg, P.W.; Hoelz, A. Structural and Functional Characterization of the α-Tubulin Acetyltransferase MEC-17. J. Mol. Biol. 2014, 426, 2605–2616. [Google Scholar] [CrossRef]
- Pakala, S.B.; Nair, V.S.; Reddy, S.D.; Kumar, R. Signaling-dependent Phosphorylation of Mitotic Centromere-associated Kinesin Regulates Microtubule Depolymerization and Its Centrosomal Localization. J. Biol. Chem. 2012, 287, 40560–40569. [Google Scholar] [CrossRef]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of Microtubule Destabilizing Activity of Op18/Stathmin Downstream of Rac1. J. Biol. Chem. 2004, 279, 6196–6203. [Google Scholar] [CrossRef]
- Rane, C.K.; Minden, A. P21 activated kinase signaling in cancer. Semin. Cancer Biol. 2019, 54, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Li, C.; Rajoka, M.S.R.; He, Z.; Huang, J.; Wang, J.; Zhang, J. P21-Activated Kinase 1: Emerging biological functions and potential therapeutic targets in Cancer. Theranostics 2020, 10, 9741–9766. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H. Culture of Murine Megakaryocytes and Platelets from Fetal Liver and Bone Marrow. In Platelets and Megakaryocytes; Gibbins, J.M., Mahaut-Smith, M.P., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2012; Volume 788, pp. 193–203. ISBN 978-1-61779-306-6. [Google Scholar]
- Faure, S. A member of the Ste20/PAK family of protein kinases is involved in both arrest of Xenopus oocytes at G2/prophase of the first meiotic cell cycle and in prevention of apoptosis. EMBO J. 1997, 16, 5550–5561. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]
- Mackeh, R.; Lorin, S.; Ratier, A.; Mejdoubi-Charef, N.; Baillet, A.; Bruneel, A.; Hamaï, A.; Codogno, P.; Poüs, C.; Perdiz, D. Reactive Oxygen Species, AMP-activated Protein Kinase, and the Transcription Cofactor p300 Regulate α-Tubulin Acetyltransferase-1 (αTAT-1/MEC-17)-dependent Microtubule Hyperacetylation during Cell Stress. J. Biol. Chem. 2014, 289, 11816–11828. [Google Scholar] [CrossRef]
- Castoldi, M.; Popov, A.V. Purification of brain tubulin through two cycles of polymerization–depolymerization in a high-molarity buffer. Protein Expr. Purif. 2003, 32, 83–88. [Google Scholar] [CrossRef]
- Cau, J.; Faure, S.; Vigneron, S.; Labbé, J.C.; Delsert, C.; Morin, N. Regulation of Xenopus p21-activated Kinase (X-PAK2) by Cdc42 and Maturation-promoting Factor Controls Xenopus Oocyte Maturation. J. Biol. Chem. 2000, 275, 2367–2375. [Google Scholar] [CrossRef]
- Bompard, G.; Rabeharivelo, G.; Frank, M.; Cau, J.; Delsert, C.; Morin, N. Subgroup II PAK-mediated phosphorylation regulates Ran activity during mitosis. J. Cell Biol. 2010, 190, 807–822. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MEC-17 Cloning and Mutagenesis Primers | 5′ → 3′ Sequence |
|---|---|
| Forward primer for cloning Mec-17 Full Length and N-ter | ccgcggatccgagttcccgttcg |
| Reverse primer for cloning Mec-17 Full Length and C-ter | tccgctcgagttaccaaggcctggtgctgc |
| Forward primer for cloning Mec-17 C-ter | ccgcggatcccatcagcaccggccccc |
| Reverse primer for cloning Mec-17 N-ter | tccgctcgagttaaaagatgacaaagttgttcacc |
| Forward primer T260A | ggcccctcggcgtgccGCAcctccagc |
| Reverse primer T260A | ggtgggtgggctggaggTGCggcacgc |
| Forward primer S270A | cccacccacctccacgtGCTagcagcctgg |
| Reverse primer S270A | gagttgcccaggctgctAGCacgtggagg |
| Forward primer S271A | cccacctccacgttctGCCagcctgggc |
| Reverse primer S271A | ggtgagttgcccaggctGGCagaacgtgg |
| Forward primer S272A | ccacctccacgttctagcGGCctgggc |
| Reverse primer S272A | ccggtgagttgcccagGCCgctagaacg |
| Forward primer S276A | ctagcagcctgggcaacGCAccggatcg |
| Reverse primer S276A | gggaccccgatccggTGCgttgcccagg |
| Forward primer S315A | ccactgaccctggaggcGCCccagccc |
| Reverse primer S315A | cgtctgcgctgggctggGGCgcctccagg |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Dijk, J.; Bompard, G.; Rabeharivelo, G.; Cau, J.; Delsert, C.; Morin, N. PAK1 Regulates MEC-17 Acetyltransferase Activity and Microtubule Acetylation during Proplatelet Extension. Int. J. Mol. Sci. 2020, 21, 7531. https://doi.org/10.3390/ijms21207531
van Dijk J, Bompard G, Rabeharivelo G, Cau J, Delsert C, Morin N. PAK1 Regulates MEC-17 Acetyltransferase Activity and Microtubule Acetylation during Proplatelet Extension. International Journal of Molecular Sciences. 2020; 21(20):7531. https://doi.org/10.3390/ijms21207531
Chicago/Turabian Stylevan Dijk, Juliette, Guillaume Bompard, Gabriel Rabeharivelo, Julien Cau, Claude Delsert, and Nathalie Morin. 2020. "PAK1 Regulates MEC-17 Acetyltransferase Activity and Microtubule Acetylation during Proplatelet Extension" International Journal of Molecular Sciences 21, no. 20: 7531. https://doi.org/10.3390/ijms21207531
APA Stylevan Dijk, J., Bompard, G., Rabeharivelo, G., Cau, J., Delsert, C., & Morin, N. (2020). PAK1 Regulates MEC-17 Acetyltransferase Activity and Microtubule Acetylation during Proplatelet Extension. International Journal of Molecular Sciences, 21(20), 7531. https://doi.org/10.3390/ijms21207531