1. Introduction
Amino acids are essential biomolecules, which are involved in cellular processes ranging from energy production to protein synthesis and signaling. Membrane proteins belonging to different solute carrier (SLC) families mediate the transport of amino acids and their derivatives across biological membranes [
1]. Among these transporter families, the SLC7 family consists of 15 genes [
2], which encode amino acid transporters that belong to the amino acid, polyamine and organocation (APC) superfamily of transporters (transport classification (TC) system No. 2.A.3;
http://www.tcdb.org) [
3]. The SLC7 family consists of two subgroups: the cationic amino acid transporters (CATs;
SLC7A1-A4 and
SLC7A14) and the glycoprotein-associated L-type amino acid transporters (LATs;
SLC7A5-A11, Slc7a12, SLC7A13, and
Slc7a15) [
2]. CATs are N-glycosylated, while LATs are not. In contrast to CATs, LATs associate with type-II membrane N-glycoproteins, which belong to the SLC3 family, e.g., 4F2hc (SLC3A2; CD98) and rBAT (SLC3A1), to form heterodimeric amino acid transporters (HATs). These ancillary proteins, also called heavy chains, contain an intracellular, N-terminal domain, a single transmembrane α-helix, and a large extracellular C-terminal domain. The atomic structure of the extracellular domain of the heavy chain 4F2hc is similar to the structure of bacterial glucosidases, but does not possess glucosidase activity [
4]. The ancillary glycoproteins 4F2hc and rBAT, which are covalently linked to corresponding LATs, i.e., the light chains of HATs, by conserved disulfide bridges are essential for the correct trafficking of the heterodimer to the plasma membrane in mammalian cells [
2,
5,
6].
HATs have major impacts on human health being implicated in several human diseases such as aminoacidurias (cystinuria and lysinuric protein intolerance), tumor cell growth, glioma invasion, Kaposi’s sarcoma-associated herpesvirus infection, and cocaine relapse [
2,
5,
7]. 4F2hc-LAT1 and 4F2hc-LAT2 are expressed in different tissues, e.g., LAT1 (SLC7A5) in brain, ovary, testis, placenta, spleen, colon, blood-brain barrier, fetal liver, activated lymphocytes, tumor cells and LAT2 (SLC7A8) in small intestine, kidney, lung, heart, spleen, liver, brain, placenta, prostate, ovary, fetal liver, testis, skeletal muscle [
2]. They mediate sodium-independent obligatory exchange of substrates across cell membranes with a 1:1 stoichiometry [
2,
5]. Predominant substrates of 4F2hc-LAT1 are large neutral L-amino acids [
8], L-DOPA [
9], and the thyroid hormones T
3 and T
4 [
10,
11]. 4F2hc-LAT2 has specificity toward neutral L-amino acids including small ones [
8,
12,
13], and T
3 and T
4 [
11]. 4F2hc-LAT1 is a target for cancer diagnosis and treatment [
1,
2,
14,
15,
16]. In several cancer cells, 4F2hc-LAT1 is overexpressed mediating increased uptake of L-leucine. Relative high concentrations in L-leucine result in increased mammalian target of rapamycin (mTOR) activation [
17], which supports growth and survival of cancer cells [
18]. Because of its localization in the blood-brain barrier, 4F2hc-LAT1 is a promising transport system, which is also utilized to shuttle drugs and prodrugs into the brain [
19]. Recently, it was suggested that lack or defects in LAT2 are implicated in age-related hearing loss [
20] and cataract formation [
21].
Cryo- and negative stain-electron microscopy (EM) elucidated the supramolecular organization and structures of selected HATs, i.e., of 4F2hc-LAT1 [
22,
23,
24], 4F2hc-LAT2 [
25,
26,
27,
28], and rBAT-b
0,+AT [
29]. Furthermore, the high-resolution cryo-EM structures of 4F2hc-LAT1 [
22,
23] and rBAT-b
0,+AT [
29] provided detailed insights into the interactions between the ancillary glycoproteins and the corresponding membrane transporters at the molecular level.
The methylotrophic yeast
Pichia pastoris has been successfully used for the overexpression of eukaryotic membrane proteins [
30,
31] and for large-scale production of recombinant soluble and membrane proteins at high cell densities [
32]. In an overexpression screening campaign using
P. pastoris, the human HAT 4F2hc-LAT2 was successfully identified as a promising candidate for milligram protein production [
33]. Expression in
P. pastoris resulted in functional recombinant human 4F2hc-LAT2 containing the conserved disulfide bridge between light and heavy chain [
25,
26,
33]. Importantly, when LAT2 is expressed in
P. Pastoris, the recombinant protein is properly folded, correctly trafficked to the plasma membrane and functional even in the absence of co-expressed 4F2hc [
25]. In contrast, surface expression of LAT2 in the absence of its ancillary glycoprotein is severely impaired in mammalian cells [
6]. This important difference makes the Pichia expression system special, and gives the opportunity to study LATs alone, e.g., LAT1 and LAT2, and to explore possible effects of the ancillary glycoprotein 4F2hc on their transport function. To this aim, we expressed the HATs 4F2hc-LAT1 and 4F2hc-LAT2, and the LATs LAT1 and LAT2 in the methylotrophic yeast
P. pastoris, and characterized and compared their L-leucine transport kinetics and amino acid specificities using [
3H]L-leucine-based uptake and competition assays. We found that the heavy chain 4F2hc modulates the substrate affinity and specificity of LATs. In addition, the presented data confirm that the light chains LAT1 and LAT2 constitute the substrate-transporting components of the HATs and that these two light chains are also functional in the absence of 4F2hc. Thus,
P. pastoris has proven useful as eukaryotic system to express and characterize the transport function of the human light chains LAT1 and LAT2 in the absence of co-expressed heavy chain/ancillary protein 4F2hc.
2. Results and Discussion
The human HATs: 4F2hc-LAT1 and 4F2hc-LAT2, and LATs: LAT1 and LAT2 were expressed in the methylotrophic yeast
Pichia pastoris. Western blot analysis indicated expression of the corresponding HATs and LATs (
Figure S1). Transport activities were determined by measuring the uptake of [
3H]L-leucine into
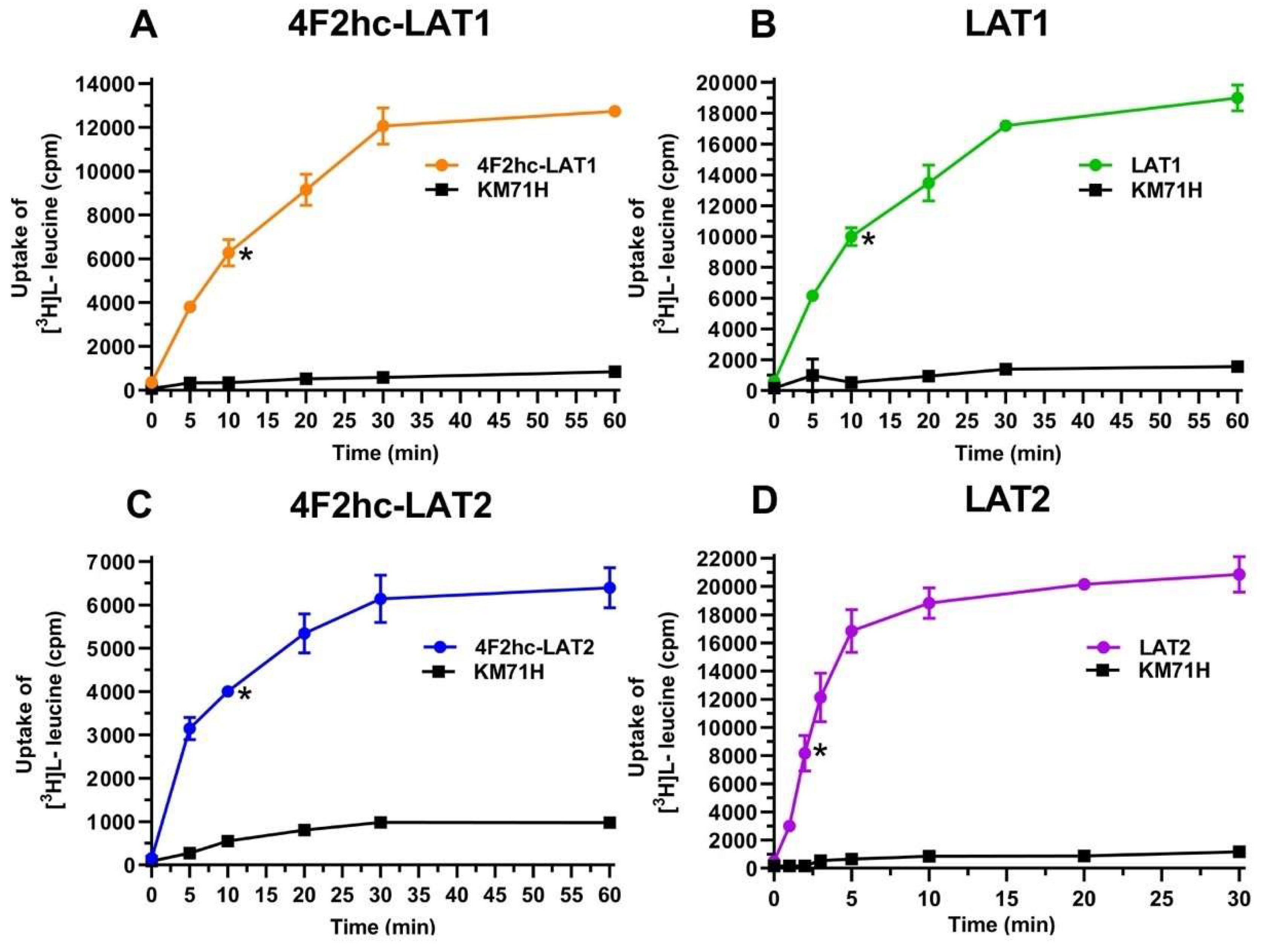
P. pastoris cells expressing the corresponding HAT or LAT. Time-course experiments showed clear HAT- and LAT-specific transport activities, which were much higher than the [
3H]L-leucine uptake into untransformed host cells (
Figure 1).
We determined the half maximal inhibitory concentrations (IC
50s) of L-leucine by homologues competition for all constructs using the obtained time points, i.e., 10 min (4F2hc-LAT1, LAT1, 4F2hc-LAT2) and 2 min (LAT2) (
Figure S2). These IC
50s (
Figure S2) gave first impressions of the affinities of the HATs and LATs for L-leucine. HAT- and LAT-mediated transport of [
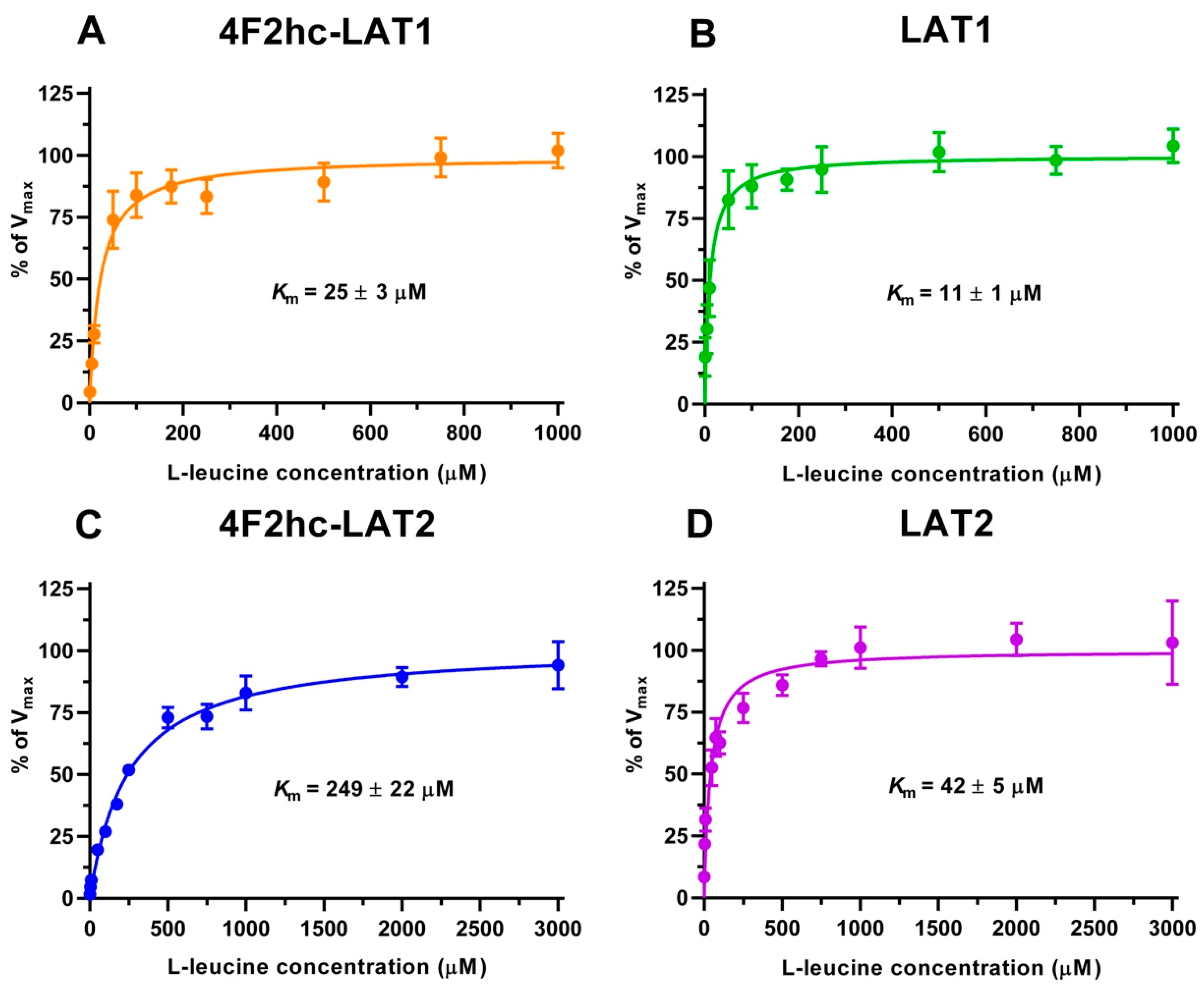
3H]L-leucine was saturable and followed Michaelis–Menten kinetics with
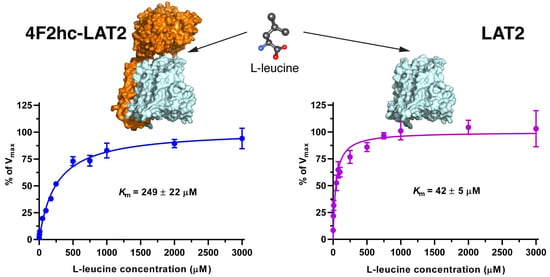
Km values of 25 µM (4F2hc-LAT1), 11 µM (LAT1), 249 µM (4F2hc-LAT2), and 42 µM (LAT2) (
Figure 2). As reflected from this data, the impact of the heavy chain 4F2hc on the affinity of the light chain for L-leucine was most pronounced for LAT2, where the
Km value increases almost six-fold upon association with 4F2hc. The measured
Kms of L-leucine for the two HATs were comparable with values from previous publications, i.e., 18 µM [
34] and 20 µM [
35] for 4F2hc-LAT1, and 220 µM for 4F2hc-LAT2 [
13]. Recently, a study using proteoliposomes showed that human LAT1 has no transport activity in the absence of 4F2hc and concluded that the ancillary protein is essential for the transport activity of the complex [
23]. In contrast, we demonstrate that LAT1 and LAT2 are able to transport [
3H]L-leucine in the absence of the heavy chain 4F2hc. This finding is further supported by studies, in which LAT1 was reconstituted into liposomes, and transport activity of this light chain was shown in the absence of the heavy chain 4F2hc [
36,
37].
The substrate specificities of 4F2hc-LAT1 and 4F2hc-LAT2, and LAT1 and LAT2 were determined by measuring the ability of proteinogenic amino acids and D-leucine at concentrations of about ten times
Km to compete with [
3H]L-leucine uptake (
Figure 3). 4F2hc-LAT1 showed a relatively broad substrate specificity with highest for L-leucine and L-histidine, in line with previous reports [
34,
35,
36]. The specificity for D-leucine was significantly lower compared to its L-isomer indicating stereospecificity of 4F2hc-LAT1 (
Figure 3). Having established an [
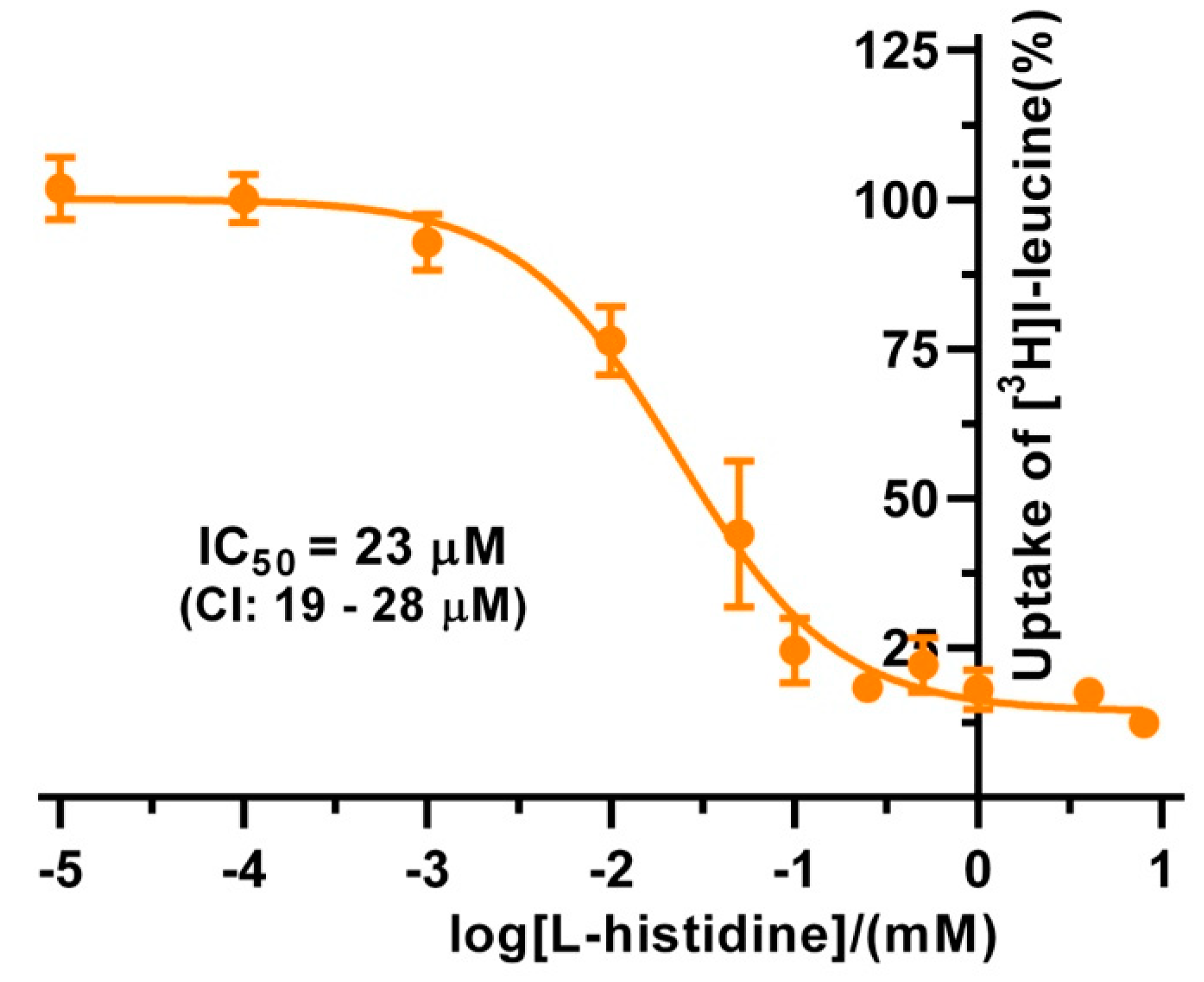
3H]L-leucine-based uptake assay and for comparison with kinetic values in the literature, we determined the half maximal inhibition concentration (IC
50) for L-histidine to 23 µM using
P. pastoris cells expressing human 4F2hc-LAT1 (
Figure 4). This IC
50 is comparable to previously reported
Km values for L-histidine, i.e., 12.7 µM [
35] and 24.6 µM [
36]. Competition data clearly showed that this HAT, 4F2hc-LAT1, has no considerable affinity (i.e., more than 50% residual [
3H]L-leucine uptake;
Figure 3A) for glycine, L-proline, L-serine and the negatively charged amino acids L-aspartate and L-glutamate. The specificity for most tested amino acids decreased when [
3H]L-leucine transport competition was studied for the light chain LAT1 alone (
Figure 3B). The stereospecificity with respect to L-leucine increased significantly compared to 4F2hc-LAT1, i.e., almost no observed inhibition of [
3H]L-leucine transport by D-leucine (
Figure 3B). L-alanine, which was competing with HAT-mediated L-leucine transport, was not recognized by LAT1 at all. In summary, LAT1 is highly specific for L-leucine and has a modest affinity for other amino acids (
Figure 3B). Interestingly, the effect of competition for L-histidine in LAT1 is significantly decreased in the absence of the heavy chain 4F2hc (
Figure 3A,B), which suggests a markedly lower affinity for this amino acid compared to the heterodimer. As observed for 4F2hc-LAT1, the HAT 4F2hc-LAT2 has also a relatively broad substrate specificity (
Figure 3C). Most amino acids with the exception of L-proline, L-serine, L-glutamate, and the positively charged amino acids L-lysine, L-arginine, and L-histidine reduced the residual [
3H]L-leucine uptake below 45%.
In contrast to 4F2hc-LAT1, specificities of 4F2hc-LAT2 for the branched chain amino acids (BCAA) L-leucine, L-isoleucine, and L-valine were comparable (
Figure 3C). Again, in the absence of the heavy chain 4F2hc the competition pattern changed significantly (
Figure 3D). As observed for LAT1, L-leucine showed the strongest reduction of LAT2-mediated [
3H]L-leucine uptake in contrast to its D-isomer, reflecting stereospecificity of the transporter (
Figure 3D). Only L-isoleucine and L-valine also reduced the residual [
3H]L-leucine uptake below 35%, which shows that LAT2 has a preferred affinity for BCAA. The strongest increase in competition associated with co-expression of the heavy chain 4F2hc is observed for L-tyrosine, L-cysteine, L-threonine, L-asparagine, L-glutamine, and L-aspartate. In general, association of the light chain LAT2 with the heavy chain 4F2hc expands the substrate specificity of the HAT compared to the BCAA-specific light chain LAT2 alone. Interestingly, the stereospecificity for leucine of the light chains LAT1 and LAT2 is more pronounced in the absence of the heavy chain 4F2hc (
Figure 4).
4. Materials and Methods
4.1. Cloning of Human 4F2hc-LAT1, 4F2hc-LAT2, LAT1, and LAT2
The making of the 4F2hc-LAT2 and LAT2 expression constructs, i.e., pPICZB-4F2hc-LAT2 and pPICZB-LAT2 in the pPICZB vector (Thermo Fisher Scientific, Waltham, MA, USA), and of the
Pichia pastoris clones expressing human 4F2hc-LAT2 or LAT2 was described in detail previously [
33]. These same two Pichia clones expressing 4F2hc-LAT2 and LAT2 were used in the here-presented study. We generated a pPICZB-based expression construct for human 4F2hc-LAT1 (pPICZB-4F2hc-LAT1) as described for 4F2hc-LAT2 [
33], but using the cDNA of the light chain LAT1 (UniProt ID code Q01650) instead of LAT2. For the LAT1 construct, the human gene (UniProt ID code Q01650) was synthesized codon-optimized for expression in the methylotrophic yeast
Pichia pastoris with 5′-HindIII and 3′-XhoI restriction sites (GenScript). In contrast to this codon-optimized
LAT1 gene, the previously mentioned constructs were generated from cDNA. The
LAT1 gene was ligated into the vector pZUDFPICZ-10His3C using 5′-HindIII and 3′-XhoI restriction sites yielding the construct pZUDFPICZ-10His3C-LAT1. pZUDFPICZ-10His3C is a modified version of the pPICZB plasmid (Thermo Fisher Scientific), which has been modified as follows. First, the single HindIII restriction site of pPICZB was removed by site-directed mutagenesis using the primer (5′-3′) TGG TTC CAA TTG ACA AAC TTT TGA TTT TAA CGA. Then, an XbaI restriction site was introduced after the polyhistidine tag of the modified pPICZB plasmid using the primer (5′-3′) ATC ATC ATC ATC ATC ATT CTA GAT GAG TTT GTA GCC TTA GA. Both mutagenesis reactions were performed using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). Finally, the region of the modified plasmid between the unique EcoRI restriction site and the newly created XbaI restriction site was replaced by the synthetic polynucleotide (5′-3′) GAA TTC
ACC ATG GCA CAT CAT CAT CAT CAT CAT CAT CAT CAC CAC GAG CTC CTT GAG GTC CTT TTT CAG GGT CCT AAG CTT GCG GCC GCC CTC GAG TCT AGA. This results in the new expression plasmid pZUDFPICZ-10His3C with a Kozak sequence (in bold), an N-terminal decahistidine-tag (His-tag) followed by a human rhinovirus 3C (HRV3C) protease cleavage site and a multicloning site (HindIII, NotI, XhoI, and XbaI).
4.2. Pichia Pastoris Culture and Expression
Electrocompetent P. pastoris strain KM71H cells (Thermo Fisher Scientific) were transformed with PmeI-linearized human 4F2hc-LAT1 and LAT1 plasmids by electroporation using a Gene Pulser II system (Bio-Rad, Hercules, CA, USA) and the settings 1.5 kV, 200 Ω, and 25 µF. To select for plasmid integration, transformed P. pastoris cells were plated on YPD-agar plates (1% (v/v) bacto yeast extract (BD Biosciences, Franklin Lakes, NJ, USA), 2% (w/v) peptone (Condalab, Madrid, Spain), 2% (w/v) dextrose (Sigma, St. Louis, MO, USA), 2% (w/v) agar (BD Biosciences) supplemented with 200 µg/mL zeocin (InvivoGen, San Diego, CA, USA) and incubated for 2–3 days at 30°C. Colonies, which grew in the presence of 200 µg/mL zeocin, were streaked on new YPD-agar plates containing increasing zeocin concentrations (500, 1000, 2000, and 4000 µg/mL). Each plate was incubated for 2–3 days at 30 °C. To screen for clones, which grew in the presence of 4000 µg/mL zeocin with high transport activity, individual colonies were cultivated under expression conditions and their transport activities were assessed. Clones of corresponding constructs showing the highest uptake of [3H]L-leucine were selected for further experiments and verified for correct integration by PCR. Selected clones and untransformed P. pastoris KM71H cells were initially inoculated in 10 mL of YPD media as a seed culture in 50 mL culture tubes and grown for 24 h at 30 °C and 300 rpm in an incubator shaker (Multitron, Infors HT, Bottmingen, Switzerland). From seed cultures, 5 mL of inoculum were added to 500 mL buffered glycerol-complex medium (BMGY; 1% (v/v) glycerol, 1% (w/v) bacto yeast extract (BD Biosciences), 2% (w/v) peptone (Condalab), 100 mM potassium phosphate pH 6.0, 1.34% (w/v) yeast nitrogen base YNB (Condalab), 4 × 10−5% (w/v) biotin) and grown overnight (12–14 h) to OD600 4–6 at 30 °C and 200 rpm using 2 L Erlenmeyer culture flasks in an incubator shaker (Multitron, Infors HT). For protein expression, cells were pelleted by centrifugation (3000× g, 15 min, room temperature) and resuspended in 1/5 to 1/10 of the original culture volume (as described in the manufacturer’s manual for Muts strains; Thermo Fisher Scientific) in buffered methanol-complex medium (BMMY; buffered-complex medium with the same composition as BMGY except having a final concentration of 1% (v/v) methanol instead of 1% (v/v) glycerol) to a final cell density of OD600 40. Resuspended cells (50–75 mL) were grown in 1 L Erlenmeyer culture flasks at 30 °C and 300 rpm in an incubator shaker (Multitron, Infors HT). Conditions for induction were maintained by supplementing the expression culture with methanol after 20 and 24 h to a final concentration of 1% (v/v). Cells were harvested 28 h post induction by centrifugation (3000× g, 15 min, room temperature). The resulting pellet was resuspended in transport buffer (150 mM choline chloride (ChCl), 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES and 10 mM Tris, pH 7.4) containing 50% (v/v) glycerol, the OD600 was adjusted to 40, and the cells were finally stored at −18 °C.
4.3. Western Blot Analysis
Immunoblotting experiments were performed using P. pastoris cells expressing the corresponding HAT or LAT used in the here-presented functional studies. For Western blots, ~15 mg Pichia cells expressing HATs or LATs were suspended in 1 mL of 50 mM potassium phosphate pH 7.4, 1 mM EDTA, 5% (v/v) glycerol, 2% (w/v) SDS, 5 mM oxidized glutathione and protease inhibitor (SigmaFASTTM Protease Inhibitor Cocktail Tablet, EDTA Free, Sigma-Aldrich, St. Louis, MO, USA). Cells were lysed at room temperature by homogenization with 20 strokes using a glass Teflon homogenizer. This lysis procedure was repeated four more times after 30 min incubation between each pass. Insolubilized cells and debris were separated by centrifugation at 12,000× g (10 min, 4 °C). The supernatant was mixed with 5x non-reducing sample buffer (60 mM Tris-HCl, pH 6.8, 10% (v/v) glycerol, 2% (w/v) SDS, 0.01% (w/v) bromophenol blue) to a final concentration of 2.5x non-reducing sample buffer and separated on a 10% SDS-PAGE gel. The His-tagged transporter LAT1 was detected using a mouse anti-His5 primary antibody (Qiagen, catalog number 34660, Hilden, Germany) at a dilution of 1:3000 and a goat anti mouse IgG (H+L) horse radish peroxidase (HRP) conjugated secondary antibody (Bio-Rad, catalog number 172-1011) at a dilution of 1:3000. Strep-tagged transporters (4F2hc-LAT1, 4F2hc-LAT1, and LAT2) were detected with HRP-conjugated streptavidin (StrepMAB-Classic-HRP, IBA Lifesciences, catalog number 2-1509-001) at a dilution of 1:30,000.
4.4. [3H]L-Leucine Radioligand Transport Assay
For transport experiments, 3 mL P. pastoris cells at OD600 40 expressing the corresponding transporter were thawed, diluted 1:50 in transport buffer, and pelleted by centrifugation (3000× g, 15 min, room temperature). Subsequently, the pellet was washed by resuspending in 50 mL transport buffer and pelleted again. The washing step was repeated twice. Finally, the cell pellet was resuspended in 4 mL of transport buffer and incubated for 20 min at 30 °C under agitation (300 rpm, Multitron, Infors HT). The density of the yeast suspension was adjusted with transport buffer to OD600 25 (LAT1, 4F2hc-LAT2 or 4F2hc-LAT1) and 7.5 (LAT2). All transport experiments were performed in a reaction volume of 100 µL. For time course experiments, the reaction mixture contained 40 µL cell suspension and 60 µL substrate master mix (167 nM L-leucine spiked with [3H]L-leucine (American Radiolabeled Chemicals)) to a specific activity of 20 Ci/mmol resulting in a final L-leucine concentration of 100 nM. For the determination of the Michaelis–Menten constant (Km), the reaction mixture contained 40 µL cell suspension and 60 µL of L-leucine solution yielding final concentrations ranging from 1–3000 µM (4F2hc-LAT2 and LAT2) and 1–1000 µM (4F2hc-LAT1 and LAT1), which were spiked with [3H]L-leucine to a specific activity of 0.033 Ci/mmol. For L-leucine and L-histidine IC50 experiments, the reaction mixture contained 40 µL cell suspension, 50 µL of competitor solution at different concentrations, i.e., 0.01–8000 µM (L-histidine, 4F2hc-LAT1), 0.01–10,000 µM (L-leucine, 4F2hc-LAT1, and LAT1) and 1–10,000 µM (L-leucine, 4F2hc-LAT2, and LAT2), and 10 µL substrate master mix (1 µM L-leucine spiked with [3H]L-leucine (American Radiolabeled Chemicals, St. Louis, MO, USA)) to a specific activity of 20 Ci/mmol resulting in a final L-leucine concentration of 100 nM. For substrate profiling (i.e., competition experiments), the reaction mixture contained 40 µL cell suspension, 50 µL of competitor solution (i.e., the final competitor concentration was about 10× higher than the L-leucine Km value of the respective transporter), and 10 µL substrate master mix (1 µM L-leucine spiked with [3H]L-leucine (American Radiolabeled Chemicals)) to a specific activity of 20 Ci/mmol resulting in a final L-leucine concentration of 100 nM. Competitors were prepared in 10% (v/v) DMSO yielding a final concentration of 0.5% in the assay. Control samples contained the same concentration of DMSO. Final OD600 values in uptake experiments were 10 for 4F2hc-LAT1, 4F2hc-LAT2, and LAT1, and 3 for LAT2. All transport reactions were done in 2 mL reaction tubes (Eppendorf) at 25 °C under agitation (1000 rpm, Thermomixer compact, Eppendorf, Hamburg, Germany). Transport was terminated after 10 min for 4F2hc-LAT1, 4F2hc-LAT2, or LAT, and 2 min for LAT2 by addition of 600 µL of pre-chilled transport buffer. Cells were rapidly separated from the buffer by transferring the stopped reactions on a 96-well 0.66 mm glass fiber filter plate (Corning FiltrEX, Corning, NY, USA) and vacuum filtration. Each well was washed with 2 mL of ice-cold transport buffer to remove free radioligand. The plate was then dried overnight at 37 °C and the backside was sealed with back seal (PerkinElmer, Waltham, MA, USA). The trapped radioligand was released by addition of 200 µL scintillation cocktail (MicroScint 40, PerkinElmer) to each well and the plate topside was sealed with TopsealTM-A Plus (PerkinElmer), followed by incubation for 30 min at 25 °C and 1000 rpm (Thermomixer compact, Eppendorf, Hamburg, Germany). Counts were measured in each well for 2 min with a scintillation counter (TopCount NXT, PerkinElmer).
4.5. Statistics
Experimental data points were performed at least in triplicate. For data analysis, the signal of the untransformed P. pastoris cells was subtracted from the transporter signal. Michaelis–Menten saturation curves were fitted into data points of independent experiments. Data points were then individually normalized using the corresponding Vmax values (i.e., the fitted upper plateau value corresponds to 100%). Data points from corresponding concentrations were averaged and SD obtained. Finally, Michaelis–Menten saturation curves were fitted to the averaged data yielding Km values. To determine the half maximal inhibitory concentration (IC50) values of heterologous (i.e., L-histidine) L-leucine transport competition, a sigmoidal model curve was fitted to the net transport signals of independent experiments. Every experimental data point was individually normalized using the corresponding upper plateau values (i.e., the fitted upper plateau value corresponds to 100%). Data points from corresponding concentrations were averaged and a sigmoidal model curve was fitted to the data in order to obtain the IC50 value. Prism6 (GraphPad Software) was used for data analysis.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



