Intimate Relations—Mitochondria and Ageing

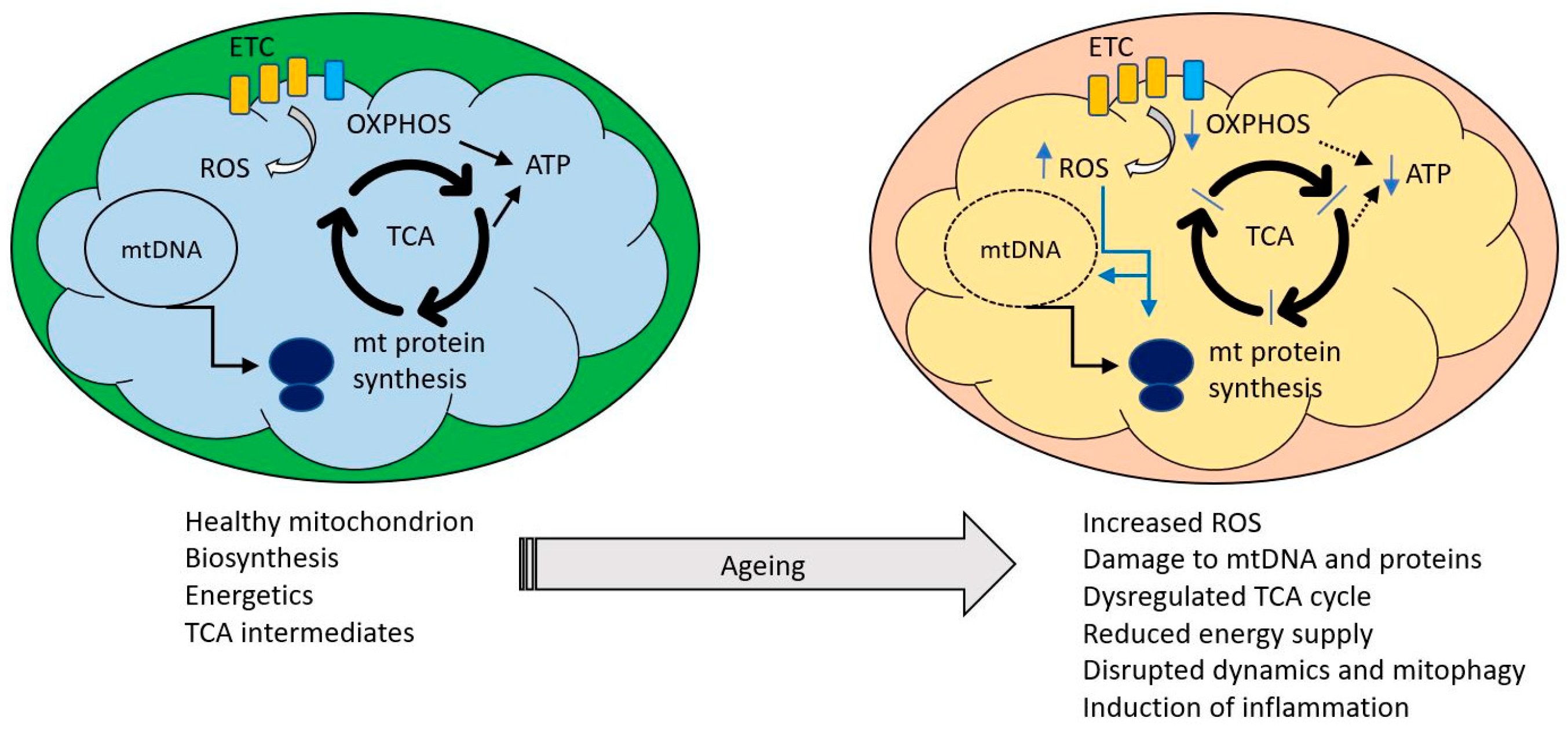{kind=link}
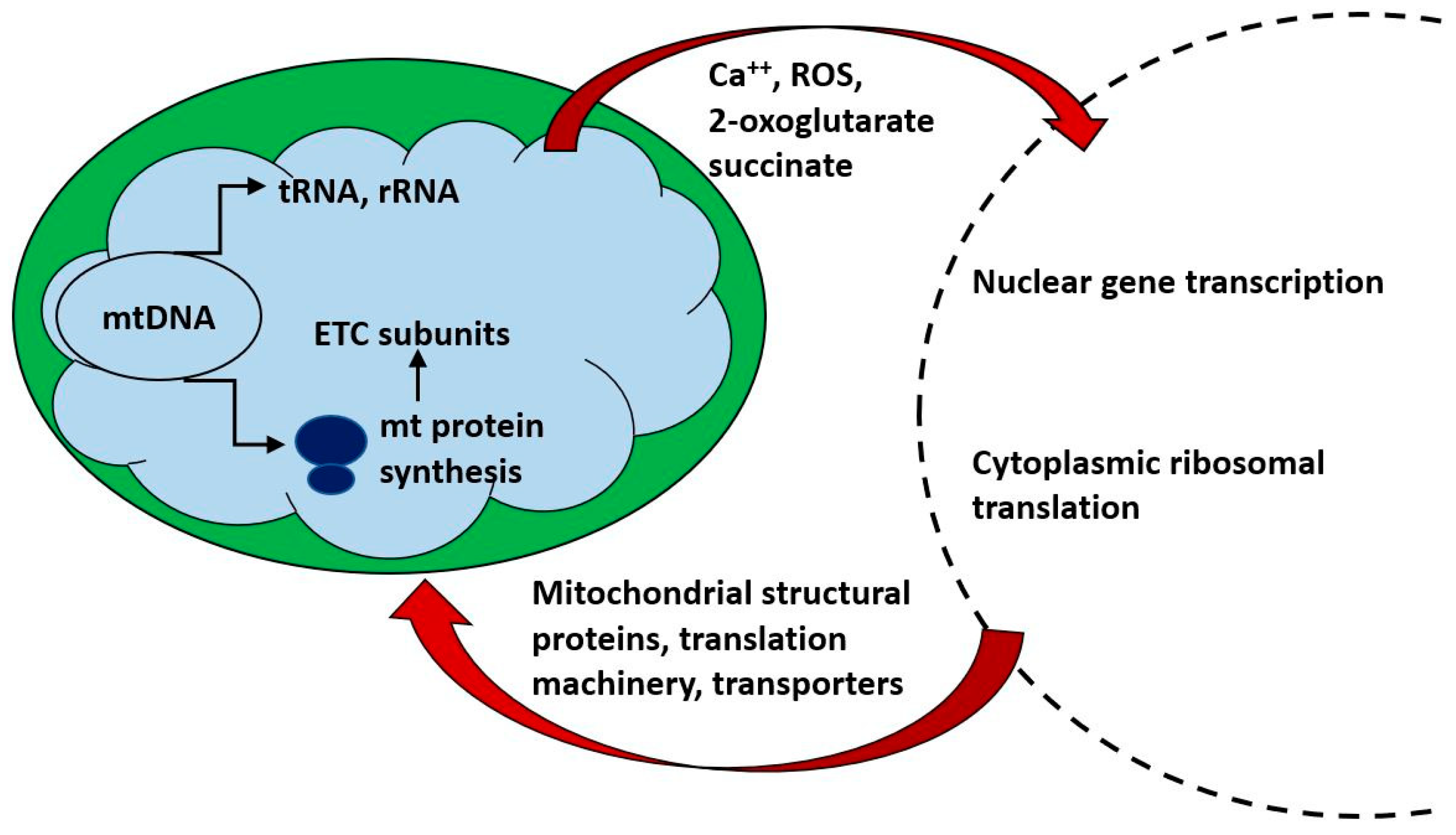{kind=link}
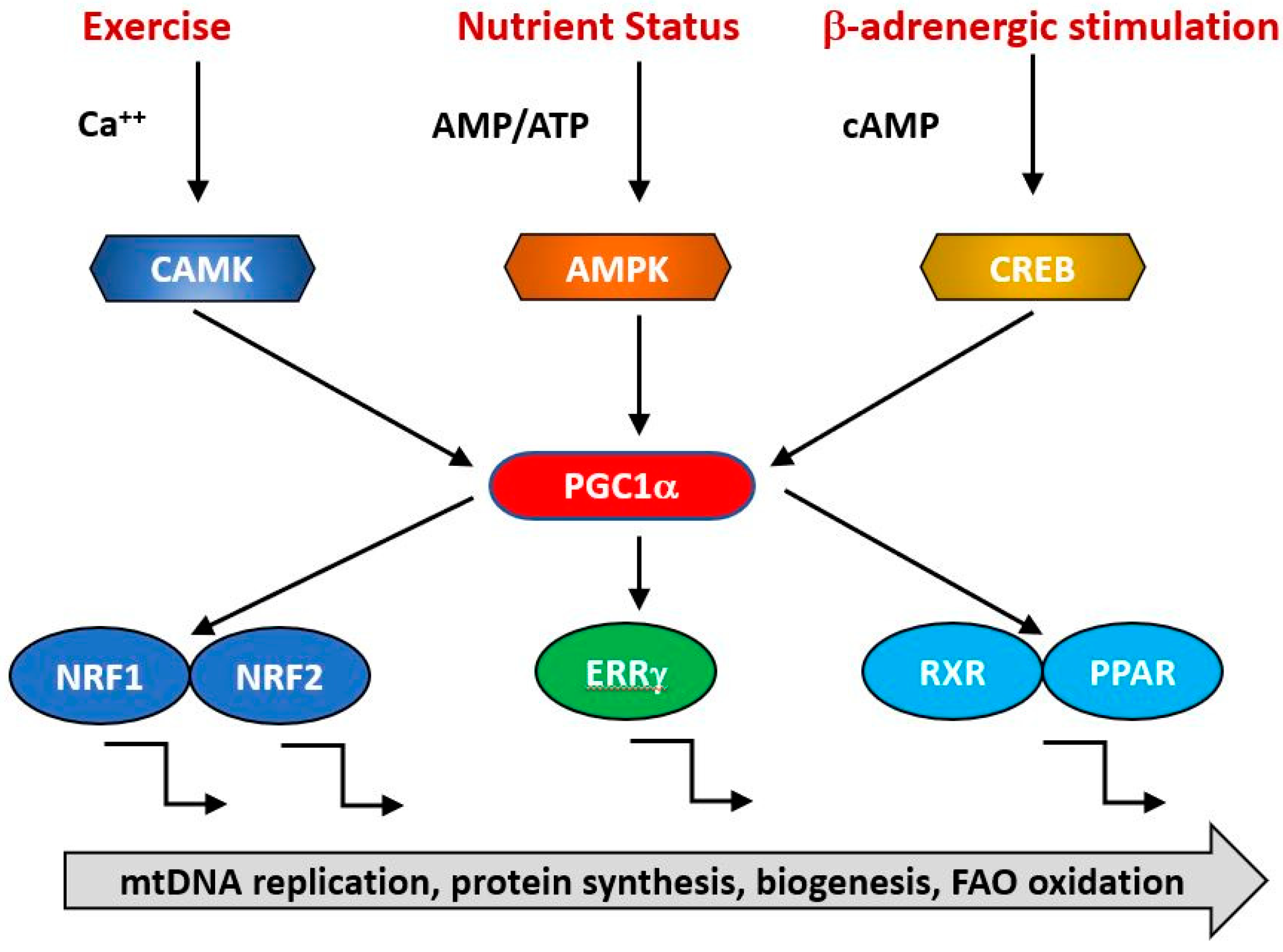{kind=link}
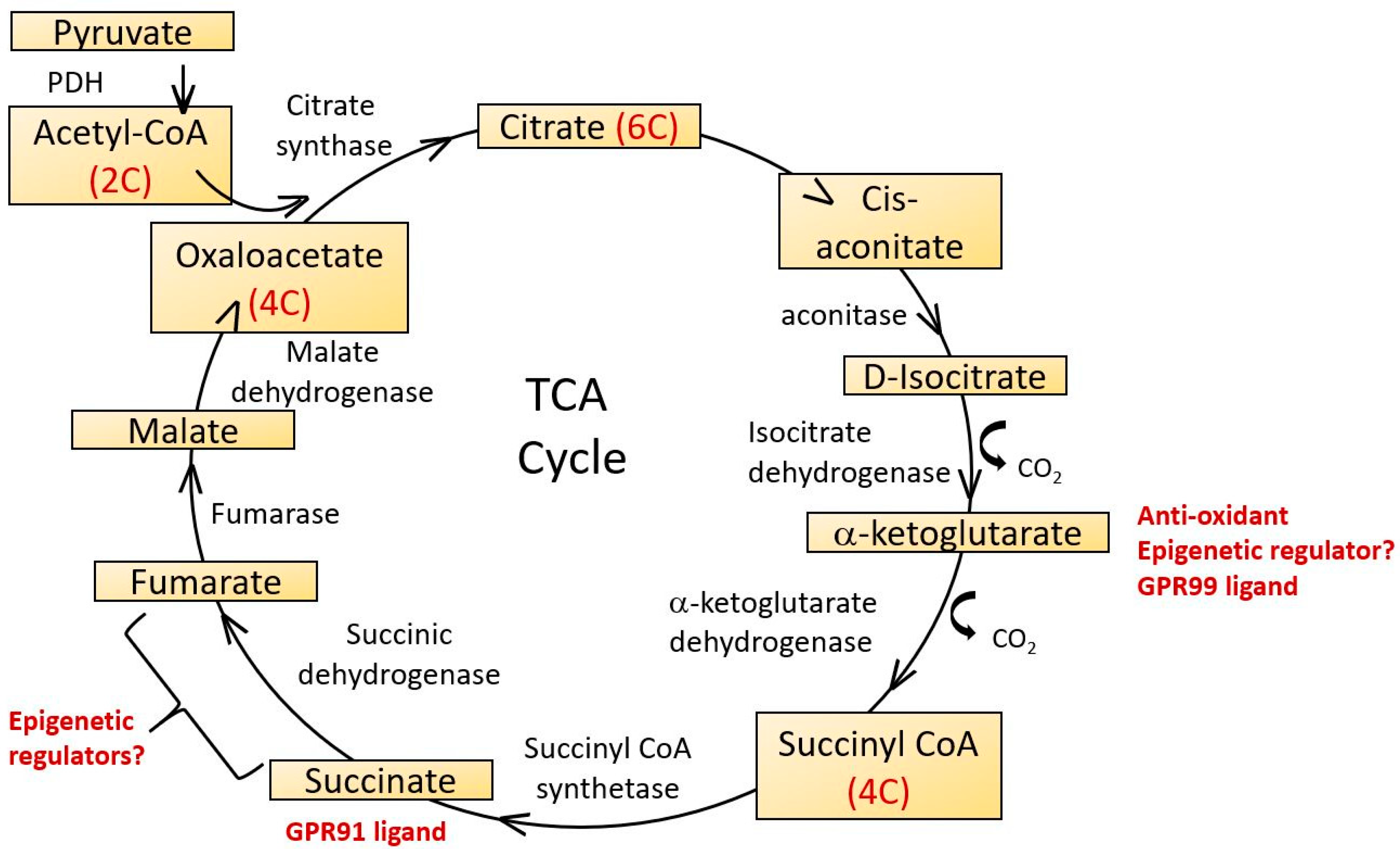{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Biology of mtDNA
3. Regulation of Mitochondrial Gene Expression and Age-Related Changes in Mitochondrially Encoded Proteins
4. Mitochondrial Biogenesis
5. Biochemistry of Mitochondrial Ageing
5.1. Small Metabolites
5.2. Mitochondrial Proteases
5.3. Cardiolipin
6. Genetics of Mitochondrial DNA
Association of mtDNA Mutations with Disease and Ageing
7. Mitochondrial Mutations and the Oxidative Damage Theory
8. Cellular Programs and Aging–Mitochondria in Stem Cells and Senescence
9. Mitochondrial Quality Control
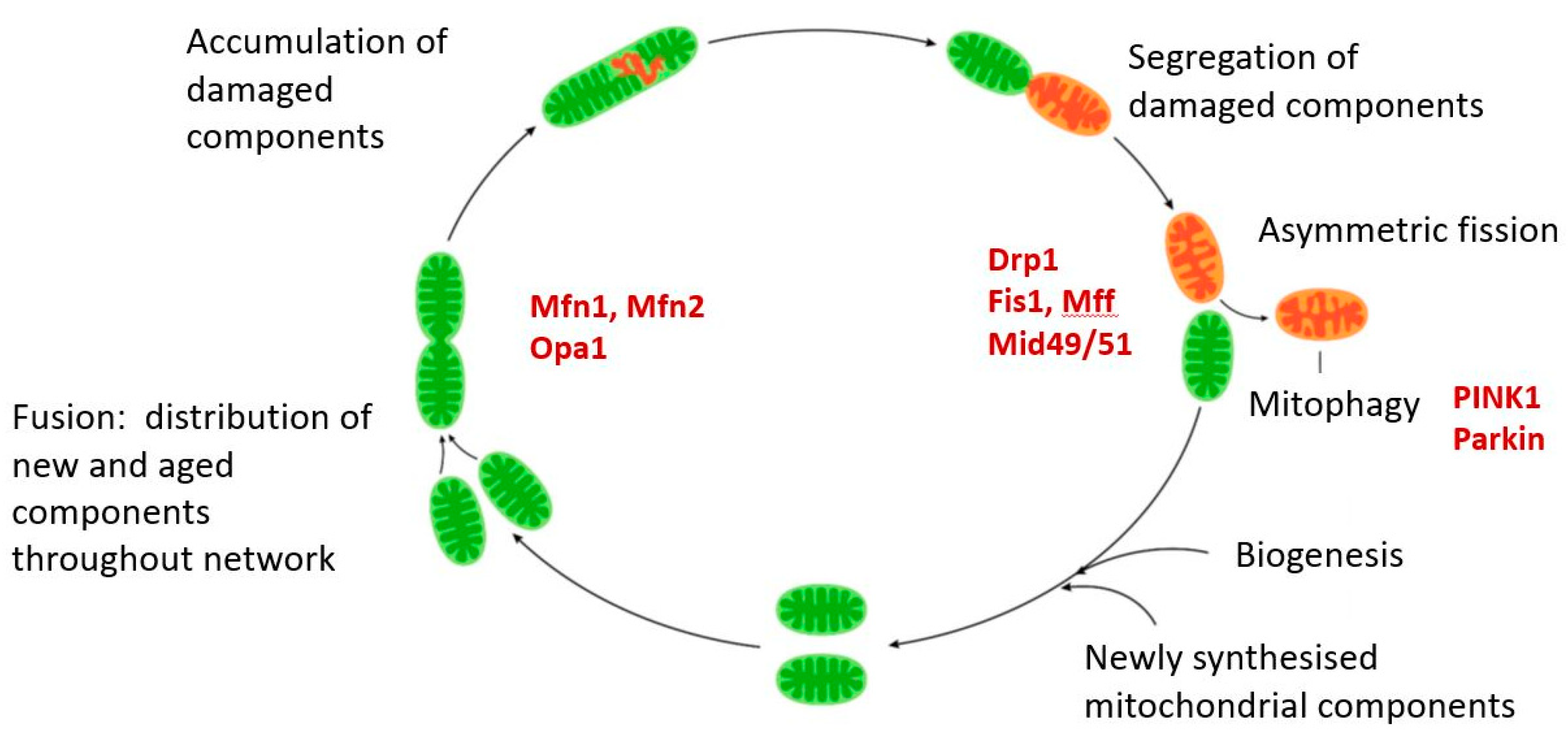
9.1. Mitophagy
9.2. Fission/Fusion
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2, REVIEWS1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, T.C.; Chen, Y.S.; King, K.L.; Yeh, S.H.; Wei, Y.H. Liver mitochondrial respiratory functions decline with age. Biochem. Biophys. Res. Commun. 1989, 165, 944–1003. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [Green Version]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Mitochondrial decay in aging. Biochim. Biophys. Acta 1995, 1271, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Young, I.G.; Anderson, S. The genetic code in bovine mitochondria: Sequence of genes for the cytochrome oxidase subunit II and two tRNAs. Gene 1980, 12, 257–265. [Google Scholar] [CrossRef]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, 6296. [Google Scholar] [CrossRef] [Green Version]
- Hensen, F.; Potter, A.; van Esveld, S.L.; Tarres-Sole, A.; Chakraborty, A.; Sola, M.; Spelbrink, J.N. Mitochondrial RNA granules are critically dependent on mtDNA replication factors Twinkle and mtSSB. Nucleic Acids Res. 2019, 47, 3680–3698. [Google Scholar] [CrossRef] [Green Version]
- Beckman, K.B.; Ames, B.N. Endogenous oxidative damage of mtDNA. Mutat. Res. 1999, 424, 51–58. [Google Scholar] [CrossRef]
- Radzvilavicius, A.L.; Hadjivasiliou, Z.; Pomiankowski, A.; Lane, N. Selection for Mitochondrial Quality Drives Evolution of the Germline. PLoS Biol. 2016, 14, e2000410. [Google Scholar] [CrossRef] [PubMed]
- Ziada, A.S.; Lu, M.Y.; Ignas-Menzies, J.; Paintsil, E.; Li, M.; Ogbuagu, O.; Saberi, S.; Hsieh, A.Y.Y.; Sattha, B.; Harrigan, P.R.; et al. Mitochondrial DNA somatic mutation burden and heteroplasmy are associated with chronological age, smoking, and HIV infection. Aging Cell 2019, 18, e13018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, M.J.; Nguyen, D.; Kunkel, T.A.; Copeland, W.C. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 2001, 276, 38555–38562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spelbrink, J.N.; Toivonen, J.M.; Hakkaart, G.A.; Kurkela, J.M.; Cooper, H.M.; Lehtinen, S.K.; Lecrenier, N.; Back, J.W.; Speijer, D.; Foury, F.; et al. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J. Biol. Chem. 2000, 275, 24818–24828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-hydroxy-2’-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar] [CrossRef] [Green Version]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [Green Version]
- Monti, C.; Lane, L.; Fasano, M.; Alberio, T. Update of the Functional Mitochondrial Human Proteome Network. J. Proteome Res. 2018, 17, 4297–4306. [Google Scholar] [CrossRef]
- Palmfeldt, J.; Bross, P. Proteomics of human mitochondria. Mitochondrion 2017, 33, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Grabowski, P.; Schrader, T.A.; Passmore, J.B.; Schrader, M.; Rappsilber, J. Co-regulation map of the human proteome enables identification of protein functions. Nat. Biotechnol. 2019, 37, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–15623. [Google Scholar] [CrossRef] [Green Version]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging (Albany NY) 2014, 6, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Zahn, J.M.; Sonu, R.; Vogel, H.; Crane, E.; Mazan-Mamczarz, K.; Rabkin, R.; Davis, R.W.; Becker, K.G.; Owen, A.B.; Kim, S.K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006, 2, e115. [Google Scholar] [CrossRef]
- Terman, A. Catabolic insufficiency and aging. Ann. N. Y. Acad. Sci. 2006, 1067, 27–36. [Google Scholar] [CrossRef]
- Byun, H.O.; Jung, H.J.; Seo, Y.H.; Lee, Y.K.; Hwang, S.C.; Hwang, E.S.; Yoon, G. GSK3 inactivation is involved in mitochondrial complex IV defect in transforming growth factor (TGF) beta1-induced senescence. Exp. Cell Res. 2012, 318, 1808–1819. [Google Scholar] [CrossRef]
- Lafargue, A.; Degorre, C.; Corre, I.; Alves-Guerra, M.C.; Gaugler, M.H.; Vallette, F.; Pecqueur, C.; Paris, F. Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic. Biol. Med. 2017, 108, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Victorelli, S.; Passos, J.F. Reactive Oxygen Species Detection in Senescent Cells. Methods Mol. Biol. 2019, 1896, 21–29. [Google Scholar]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Fayet, G.; Jansson, M.; Sternberg, D.; Moslemi, A.R.; Blondy, P.; Lombes, A.; Fardeau, M.; Oldfors, A. Ageing muscle: Clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul. Disord. 2002, 12, 484–493. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef]
- Lin, M.T.; Simon, D.K.; Ahn, C.H.; Kim, L.M.; Beal, M.F. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum. Mol. Genet. 2002, 11, 133–145. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [Green Version]
- Stenton, S.L.; Prokisch, H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018, 62, 399–408. [Google Scholar]
- Singh, R.K.; Srivastava, A.; Kalaiarasan, P.; Manvati, S.; Chopra, R.; Bamezai, R.N. mtDNA germ line variation mediated ROS generates retrograde signaling and induces pro-cancerous metabolic features. Sci. Rep. 2014, 4, 6571. [Google Scholar] [CrossRef] [Green Version]
- Molina-Heredia, F.P.; Houée-Levin, C.; Berthomieu, C.; Touati, D.; Tremey, E.; Favaudon, V.; Adam, V.; Nivière, V. Detoxification of superoxide without production of H2O2: Antioxidant activity of superoxide reductase complexed with ferrocyanide. Proc. Natl. Acad. Sci. USA 2006, 103, 14750–14755. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef]
- Beretta, S.; Mattavelli, L.; Sala, G.; Tremolizzo, L.; Schapira, A.H.; Martinuzzi, A.; Carelli, V.; Ferrarese, C. Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines. Brain 2004, 127, 2183–2192. [Google Scholar] [CrossRef]
- Carreño-Gago, L.; Gamez, J.; Camara, Y.; García-Arumí, E.; Aller-Alvarez, J.S.; Moncho, D.; Salvado, M.; Galan, A.; De La Cruz, X.; Pinós, T.; et al. Identification and characterization of the novel point mutation m.3634A > G in the mitochondrial MT—ND1 gene associated with LHON syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 182–187. [Google Scholar] [CrossRef]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise Intolerance Due to Mutations in the CytochromebGene of Mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef]
- Reutzel, M.; Grewal, R.; Dilberger, B.; Silaidos, C.; Joppe, A.; Eckert, G.P. Cerebral Mitochondrial Function and Cognitive Performance during Aging: A Longitudinal Study in NMRI Mice. Oxid. Med. Cell Longev. 2020, 4060769. [Google Scholar] [CrossRef] [Green Version]
- Bowling, A.C.; Mutisya, E.M.; Walker, L.C.; Price, D.L.; Cork, L.C.; Beal, M.F. Age-dependent impairment of mitochondrial function in primate brain. J. Neurochem. 1993, 60, 1964–1967. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef]
- Kwong, L.K.; Sohal, R.S. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys. 2000, 373, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.M.; Leeuwenburgh, C.; Lezza, A.M. Aging and calorie restriction oppositely affect mitochondrial biogenesis through TFAM binding at both origins of mitochondrial DNA replication in rat liver. PLoS ONE 2013, 8, e74644. [Google Scholar] [CrossRef] [Green Version]
- Rafalski, V.A.; Mancini, E.; Brunet, A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 2012, 125, 5597–5608. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Inoue, S.; Noda, S.; Kashima, K.; Nakada, K.; Hayashi, J.; Miyoshi, H. Mitochondrial respiration defects modulate differentiation but not proliferation ofhematopoietic stem and progenitor cells. FEBS Lett. 2010, 584, 3402–3409. [Google Scholar] [CrossRef] [Green Version]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Lobo-Jarne, T.; Ugalde, C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell Dev. Biol. 2018, 76, 179–190. [Google Scholar] [CrossRef]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 2016, 167, 1598–1609. [Google Scholar] [CrossRef] [Green Version]
- Rathore, S.; Berndtsson, J.; Marin-Buera, L.; Conrad, J.; Carroni, M.; Brzezinski, P.; Ott, M. Cryo-EM structure of the yeast respiratory supercomplex. Nat. Struct. Mol. Biol. 2019, 26, 50–57. [Google Scholar] [CrossRef]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 2014, 1837, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef]
- Gomez, L.A.; Monette, J.S.; Chavez, J.D.; Maier, C.S.; Hagen, T.M. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch. Biochem. Biophys. 2009, 490, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Frenzel, M.; Rommelspacher, H.; Sugawa, M.D.; Dencher, N.A. Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex. Exp. Gerontol. 2010, 45, 563–572. [Google Scholar] [CrossRef]
- Lombardi, A.; Silvestri, E.; Cioffi, F.; Senese, R.; Lanni, A.; Goglia, F.; De Lange, P.; Moreno, M. Defining the transcriptomic and proteomic profiles of rat ageing skeletal muscle by the use of a cDNA array, 2D- and Blue native-PAGE approach. J. Proteom. 2009, 72, 708–721. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, J.F.; Patel, H.V.; Naples, C.J.; Fujioka, H.; Hoppel, C.L. Decreased cytochrome c mediates an age-related decline of oxidative phosphorylation in rat kidney mitochondria. Biochem. J. 2010, 427, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genova, M.L.; Lenaz, G. The Interplay Between Respiratory Supercomplexes and ROS in Aging. Antioxid. Redox Signal. 2015, 23, 208–238. [Google Scholar] [CrossRef]
- Rieger, B.; Krajčová, A.; Duwe, P.; Busch, K.B. ALCAT1 Overexpression Affects Supercomplex Formation and Increases ROS in Respiring Mitochondria. Oxid. Med. Cell Longev. 2019, 2019, 9186469. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Adey, D.B.; Ades, P.A.; Nair, K.S. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc. Natl. Acad. Sci. USA 1996, 93, 15364–15369. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.F.; Robinson, M.M.; Bruss, M.D.; Hellerstein, M.; Hamilton, K.L. A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 2012, 11, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Couvillion, M.T.; Soto, C.; Shipkovenska, G.; Churchman, L.S. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Shekar, K.C.; Li, L.; Dabkowski, E.R.; Xu, W.; Ribeiro, R.F., Jr.; Hecker, P.A.; Recchia, F.A.; Sadygov, R.G.; Willard, B.; Kasumov, T.; et al. Cardiac mitochondrial proteome dynamics with heavy water reveals stable rate of mitochondrial protein synthesis in heart failure despite decline in mitochondrial oxidative capacity. J. Mol. Cell Cardiol. 2014, 75, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Wilkening, A.; Rub, C.; Sylvester, M.; Voos, W. Analysis of heat-induced protein aggregation in human mitochondria. J. Biol. Chem. 2018, 293, 11537–11552. [Google Scholar] [CrossRef] [Green Version]
- Suhm, T.; Kaimal, J.M.; Dawitz, H.; Peselj, C.; Masser, A.E.; Hanzen, S.; Ambrozic, M.; Smialowska, A.; Bjorck, M.L.; Brzezinski, P.; et al. Mitochondrial Translation Efficiency Controls Cytoplasmic Protein Homeostasis. Cell Metab. 2018, 27, 1309–1322.e6. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Perks, K.L.; Rossetti, G.; Rudler, D.L.; Hughes, L.A.; Ermer, J.A.; Scott, L.H.; Kuznetsova, I.; Richman, T.R.; Narayana, V.K.; et al. Stress signaling and cellular proliferation reverse the effects of mitochondrial mistranslation. EMBO J. 2019, 38, e102155. [Google Scholar] [CrossRef] [PubMed]
- Sharpley, M.S.; Marciniak, C.; Eckel-Mahan, K.; McManus, M.; Crimi, M.; Waymire, K.; Lin, C.S.; Masubuchi, S.; Friend, N.; Koike, M.; et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 2012, 151, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.; Aivio, S.; Knobel, P.A.; Bailey, L.J.; Casali, A.; Vinaixa, M.; Garcia-Cao, I.; Coyaud, E.; Jourdain, A.; Perez-Ferreros, P.; et al. EXD2 governs germ stem cell homeostasis and lifespan by promoting mitoribosome integrity and translation. Nat. Cell Biol. 2018, 20, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, T.; Kido, K.; Suga, T.; Isaka, T.; Hayashi, T.; Fujita, S. Exercise-induced mitochondrial biogenesis coincides with the expression of mitochondrial translation factors in murine skeletal muscle. Physiol. Rep. 2018, 6, e13893. [Google Scholar] [CrossRef]
- Stoldt, S.; Wenzel, D.; Kehrein, K.; Riedel, D.; Ott, M.; Jakobs, S. Spatial orchestration of mitochondrial translation and OXPHOS complex assembly. Nat. Cell Biol. 2018, 20, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS ONE 2014, 9, e96820. [Google Scholar] [CrossRef] [Green Version]
- Carden, T.; Singh, B.; Mooga, V.; Bajpai, P.; Singh, K.K. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J. Biol. Chem. 2017, 292, 20694–20706. [Google Scholar] [CrossRef] [Green Version]
- Grover, R.; Burse, S.A.; Shankrit, S.; Aggarwal, A.; Kirty, K.; Narta, K.; Srivastav, R.; Ray, A.K.; Malik, G.; Vats, A.; et al. Myg1 exonuclease couples the nuclear and mitochondrial translational programs through RNA processing. Nucleic Acids Res. 2019, 47, 5852–5866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galati, D.; Srinivasan, S.; Raza, H.; Prabu, S.K.; Hardy, M.; Chandran, K.; Lopez, M.; Kalyanaraman, B.; Avadhani, N.G. Role of nuclear-encoded subunit Vb I the assembly and stability of cytochrome c oxidase complex: Implications in mitochondrial dysfunction and ROS production. Biochem. J. 2009, 420, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalimonchuk, O.; Bird, A.; Winge, D.R. Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J. Biol. Chem. 2007, 282, 17442–17449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Serrano, M.; Camafeita, E.; Lopez, J.A.; Rubio, M.A.; Breton, I.; Garcia-Consuegra, I.; Garcia-Santos, E.; Lago, J.; Sanchez-Pernaute, A.; Torres, A.; et al. Differential proteomic and oxidative profiles unveil dysfunctional protein import to adipocyte mitochondria in obesity-associated aging and diabetes. Redox Biol. 2017, 11, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Rampello, N.G.; Stenger, M.; Westermann, B.; Osiewacz, H.D. Impact of F1Fo-ATP-synthase dimer assembly factors on mitochondrial function and organismic aging. Microb. Cell 2018, 5, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Tauchi, H.; Sato, T. Age changes in size and number of mitochondria of human hepatic cells. J. Gerontol. 1968, 23, 454–461. [Google Scholar] [CrossRef]
- Herbener, G.H. A morphometric study of age-dependent changes in mitochondrial population of mouse liver and heart. J. Gerontol. 1976, 31, 8–12. [Google Scholar] [CrossRef]
- El’darov, C.M.; Vays, V.B.; Vangeli, I.M.; Kolosova, N.G.; Bakeeva, L.E. Morphometric Examination of Mitochondrial Ultrastructure in Aging Cardiomyocytes. Biochem. (Mosc.) 2015, 80, 604–609. [Google Scholar] [CrossRef]
- Houmard, J.A.; Weidner, M.L.; Gavigan, K.E.; Tyndall, G.L.; Hickey, M.S.; Alshami, A. Fiber type and citrate synthase activity in the human gastrocnemius and vastus lateralis with aging. J. Appl. Physiol. 1998, 85, 1337–1341. [Google Scholar] [CrossRef]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.S.; Ongwijitwat, S.; Wong-Riley, M.T. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J. Biol. Chem. 2008, 283, 3120–3129. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, Z.; Lu, Y.; Du, R.; Katiyar, S.; Yang, J.; Fu, M.; Leader, J.E.; Quong, A.; Novikoff, P.M.; et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc. Natl. Acad. Sci. USA 2006, 103, 11567–11572. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Scarpulla, R.C. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol. Cell Biol. 2001, 21, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Leung, L.; Kwong, M.; Hou, S.; Lee, C.; Chan, J.Y. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 2003, 278, 48021–48029. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Buja, L.M.; Scarpulla, R.C.; McMillin, J.B. Electrical stimulation of neonatal cardiomyocytes results in the sequential activation of nuclear genes governing mitochondrial proliferation and differentiation. Proc. Natl. Acad. Sci. USA 1997, 94, 11399–11404. [Google Scholar] [CrossRef] [Green Version]
- Ojuka, E.O.; Jones, T.E.; Han, D.H.; Chen, M.; Holloszy, J.O. Raising Ca2 + in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J. 2003, 17, 675–681. [Google Scholar] [CrossRef]
- Suliman, H.B.; Carraway, M.S.; Tatro, L.G.; Piantadosi, C.A. A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 2007, 120, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Villena, J.A.; Kralli, A. ERRalpha: A metabolic function for the oldest orphan. Trends Endocrinol. Metab. 2008, 19, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid hormone receptor and ERRalpha coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 2018, 11, 536. [Google Scholar] [CrossRef] [Green Version]
- Perry, M.C.; Dufour, C.R.; Tam, I.S.; B’Chir, W.; Giguere, V. Estrogen-related receptor-alpha coordinates transcriptional programs essential for exercise tolerance and muscle fitness. Mol. Endocrinol. 2014, 28, 2060–2071. [Google Scholar] [CrossRef] [Green Version]
- Alaynick, W.A.; Kondo, R.P.; Xie, W.; He, W.; Dufour, C.R.; Downes, M.; Jonker, J.W.; Giles, W.; Naviaux, R.K.; Giguere, V.; et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007, 6, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Ennequin, G.; Boisseau, N.; Caillaud, K.; Chavanelle, V.; Gerbaix, M.; Metz, L.; Etienne, M.; Walrand, S.; Masgrau, A.; Guillet, C.; et al. Exercise training and return to a well-balanced diet activate the neuregulin 1/ErbB pathway in skeletal muscle of obese rats. J. Physiol. 2015, 593, 2665–2677. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.X.; Shi, X.C.; Chen, T.; Tan, Z.N.; Lin, Q.Q.; Du, S.J.; Tian, Z.J. Exercise training activates neuregulin 1/ErbB signaling and promotes cardiac repair in a rat myocardial infarction model. Life Sci. 2016, 149, 1–9. [Google Scholar] [CrossRef]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalfalah, F.; Sobek, S.; Bornholz, B.; Gotz-Rosch, C.; Tigges, J.; Fritsche, E.; Krutmann, J.; Kohrer, K.; Deenen, R.; Ohse, S.; et al. Inadequate mito-biogenesis in primary dermal fibroblasts from old humans is associated with impairment of PGC1A-independent stimulation. Exp. Gerontol. 2014, 56, 59–68. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP- activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Weiqiang, K.; Qing, Z.; Pengju, Z.; Yi, L. Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKalpha. Exp. Mol. Med. 2007, 39, 53543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barger, J.L.; Anderson, R.M.; Newton, M.A.; da Silva, C.; Vann, J.A.; Pugh, T.D.; Someya, S.; Prolla, T.A.; Weindruch, R. A conserved transcriptional signature of delayed aging and reduced disease vulnerability is partially mediated by SIRT3. PLoS ONE 2015, 10, e0120738. [Google Scholar] [CrossRef]
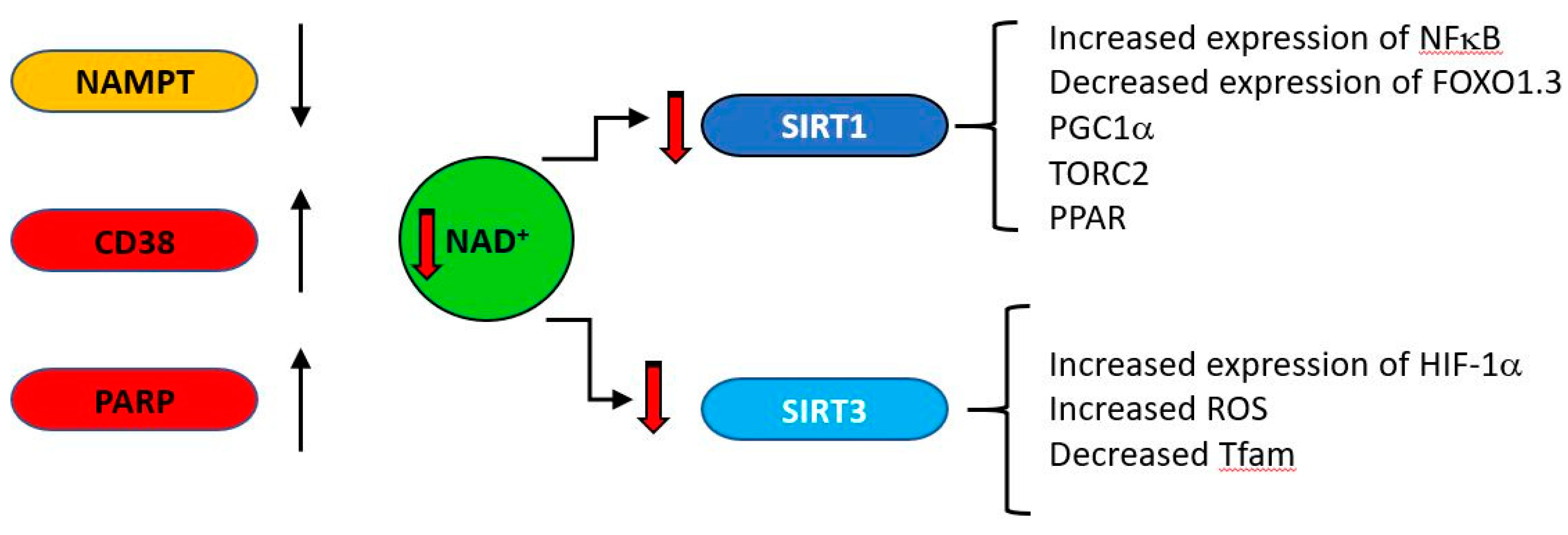
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Lee, H.J.; Kang, M.G.; Cha, H.Y.; Kim, Y.M.; Lim, Y.; Yang, S.J. Effects of Piceatannol and Resveratrol on Sirtuins and Hepatic Inflammation in High-Fat Diet-Fed Mice. J. Med. Food 2019, 22, 833–840. [Google Scholar] [CrossRef]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcazar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurina, Y.Y.; Witt, E. Role of coenzyme Q and superoxide in vitamin E cycling. Subcell. Biochem. 1998, 30, 491–507. [Google Scholar]
- Beyer, R.E.; Burnett, B.A.; Cartwright, K.J.; Edington, D.W.; Falzon, M.J.; Kreitman, K.R.; Kuhn, T.W.; Ramp, B.J.; Rhee, S.Y.; Rosenwasser, M.J.; et al. Tissue coenzyme Q (ubiquinone) and protein concentrations over the life span of the laboratory rat. Mech. Ageing Dev. 1985, 32, 267–281. [Google Scholar] [CrossRef] [Green Version]
- Kalen, A.; Appelkvist, E.L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef]
- Diaz-Castro, J.; Moreno-Fernandez, J.; Chirosa, I.; Chirosa, L.J.; Guisado, R.; Ochoa, J.J. Beneficial Effect of Ubiquinol on Hematological and Inflammatory Signaling during Exercise. Nutrients 2020, 12, 424. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; Gonzalez-Garcia, P.; Battino, M.; Lopez, L.C.; Varela-Lopez, A. The Paradox of Coenzyme Q10 in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Beriault, R.; Lemire, J.; Singh, R.; Chenier, D.R.; Hamel, R.D.; Appanna, V.D. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS ONE 2007, 2, e690. [Google Scholar] [CrossRef] [Green Version]
- Zdzisinska, B.; Zurek, A.; Kandefer-Szerszen, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Ther. Exp. 2017, 65, 21–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kauppinen, A.; Hiltunen, M.; Kaarniranta, K. Krebs cycle intermediates regulate DNA and histone methylation: Epigenetic impact on the aging process. Ageing Res. Rev. 2014, 16, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Delaval, E.; Perichon, M.; Friguet, B. Age-related impairment of mitochondrial matrix aconitase and ATP-stimulated protease in rat liver and heart. Eur. J. Biochem. 2004, 271, 4559–4564. [Google Scholar] [CrossRef] [PubMed]
- Yarian, C.S.; Toroser, D.; Sohal, R.S. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech. Ageing Dev. 2006, 127, 79–84. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef]
- Gilissen, J.; Jouret, F.; Pirotte, B.; Hanson, J. Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther. 2016, 159, 56–65. [Google Scholar] [CrossRef]
- Hu, J.; Li, T.; Du, X.; Wu, Q.; Le, Y.Z. G protein-coupled receptor 91 signaling in diabetic retinopathy and hypoxic retinal diseases. Vis. Res. 2017, 139, 59–64. [Google Scholar] [CrossRef]
- Knauf, F.; Mohebbi, N.; Teichert, C.; Herold, D.; Rogina, B.; Helfand, S.; Gollasch, M.; Luft, F.C.; Aronson, P.S. The life-extending gene Indy encodes an exchanger for Krebs-cycle intermediates. Biochem. J. 2006, 397, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.J.; Liu, J.C.; Inoue, K.; Zhuang, L.; Miyake, K.; Miyauchi, S.; Ganapathy, V. Relevance of NAC-2, an Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem. J. 2004, 379, 191–198. [Google Scholar] [CrossRef]
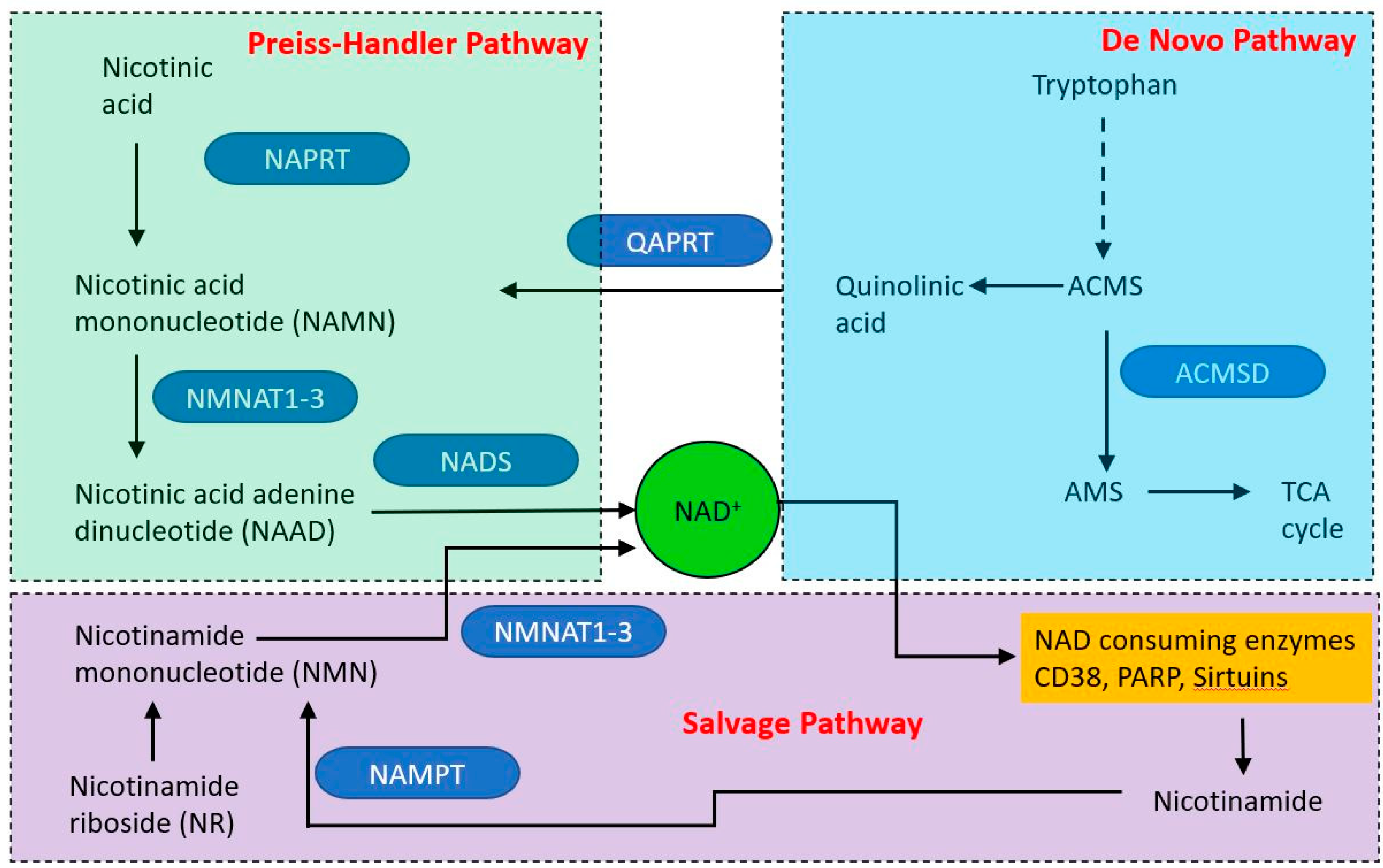
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; DʼAmico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Davila, A.; Liu, L.; Chellappa, K.; Redpath, P.; Nakamaru-Ogiso, E.; Paolella, L.M.; Zhang, Z.; Migaud, M.E.; Rabinowitz, J.D.; Baur, J.A. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife 2018, 7, e33246. [Google Scholar] [CrossRef]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Ethier, C.; Tardif, M.; Arul, L.; Poirier, G.G. PARP-1 modulation of mTOR signaling in response to a DNA alkylating agent. PLoS ONE 2012, 7, e47978. [Google Scholar] [CrossRef]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef] [Green Version]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Chen, S.D.; Jou, S.B.; Lin, T.K.; Chen, S.F.; Chen, N.C.; Hsu, C.Y. Sirtuin 1 Regulates Mitochondrial Biogenesis and Provides an Endogenous Neuroprotective Mechanism Against Seizure-Induced Neuronal Cell Death in the Hippocampus Following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 3588. [Google Scholar] [CrossRef] [Green Version]
- Torrens-Mas, M.; Hernandez-Lopez, R.; Pons, D.G.; Roca, P.; Oliver, J.; Sastre-Serra, J. Sirtuin 3 silencing impairs mitochondrial biogenesis and metabolism in colon cancer cells. Am. J. Physiol. Cell Physiol. 2019, 317, C398–C404. [Google Scholar] [CrossRef]
- Li, P.; Liu, Y.; Burns, N.; Zhao, K.S.; Song, R. SIRT1 is required for mitochondrial biogenesis reprogramming in hypoxic human pulmonary arteriolar smooth muscle cells. Int. J. Mol. Med. 2017, 39, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagne, J.P.; de Moura, M.B.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014, 8, 1183–1819. [Google Scholar] [CrossRef] [Green Version]
- Mota, R.A.; Sanchez-Bueno, F.; Saenz, L.; Hernandez-Espinosa, D.; Jimeno, J.; Tornel, P.L.; Martinez-Torrano, A.; Ramirez, P.; Parrilla, P.; Yelamos, J. Inhibition of poly(ADP-ribose) polymerase attenuates the severity of acute pancreatitis and associated lung injury. Lab. Investig. 2005, 85, 1250–1262. [Google Scholar] [CrossRef] [Green Version]
- Grube, K.; Burkle, A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc. Natl. Acad. Sci. USA 1992, 89, 11759–11763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muiras, M.L.; Muller, M.; Schachter, F.; Burkle, A. Increased poly(ADP-ribose) polymerase activity in lymphoblastoid cell lines from centenarians. J. Mol. Med. 1998, 76, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Vida, A.; Abdul-Rahman, O.; Miko, E.; Brunyanszki, A.; Bai, P. Poly(ADP-Ribose) Polymerases in Aging—Friend or Foe? Curr. Protein Pept. Sci. 2016, 17, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Boesten, D.M.; de Vos-Houben, J.M.; Timmermans, L.; den Hartog, G.J.; Bast, A.; Hageman, G.J. Accelerated aging during chronic oxidative stress: A role for PARP-1. Oxid. Med. Cell Longev. 2013, 2013, 680414. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.; Mielke, N.; Grimstead, J.W.; Jones, R.E.; Nguyen, T.; Mueller, M.; Baird, D.M.; Hendrickson, E.A. PARP1 is required for preserving telomeric integrity but is dispensable for A-NHEJ. Oncotarget 2018, 9, 34821–34837. [Google Scholar] [CrossRef] [Green Version]
- Gomez, M.; Wu, J.; Schreiber, V.; Dunlap, J.; Dantzer, F.; Wang, Y.; Liu, Y. PARP1 Is a TRF2-associated poly(ADP-ribose)polymerase and protects eroded telomeres. Mol. Biol. Cell 2006, 17, 1686–1696. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Herrmann, M.; Pusceddu, I.; Marz, W.; Herrmann, W. Telomere biology and age-related diseases. Clin. Chem. Lab. Med. 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Turner, K.J.; Vasu, V.; Griffin, D.K. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Takasawa, S.; Tohgo, A.; Noguchi, N.; Koguma, T.; Nata, K.; Sugimoto, T.; Yonekura, H.; Okamoto, H. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J. Biol. Chem. 1993, 268, 26052–26054. [Google Scholar] [PubMed]
- Lee, H.C. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amici, S.A.; Young, N.A.; Narvaez-Miranda, J.; Jablonski, K.A.; Arcos, J.; Rosas, L.; Papenfuss, T.L.; Torrelles, J.B.; Jarjour, W.N.; Guerau-De-Arellano, M. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front. Immunol. 2018, 9, 1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P.S. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, K.W.; Yu, B.P.; Chung, H.Y. The effect of age on cyclooxygenase-2 gene expression: NF-kappaB activation and IkappaBalpha degradation. Free. Radic. Biol. Med. 2000, 28, 683–692. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Liu, J.; Geletka, L.; Delaney, C.; DelProposto, J.; Desai, A.; Oatmen, K.; Martinez-Santibanez, G.; Julius, A.; Garg, S.; et al. Aging Is Associated with an Increase in T Cells and Inflammatory Macrophages in Visceral Adipose Tissue. J. Immunol. 2011, 187, 6208–6216. [Google Scholar] [CrossRef]
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 2012, 11, 867–875. [Google Scholar] [CrossRef]
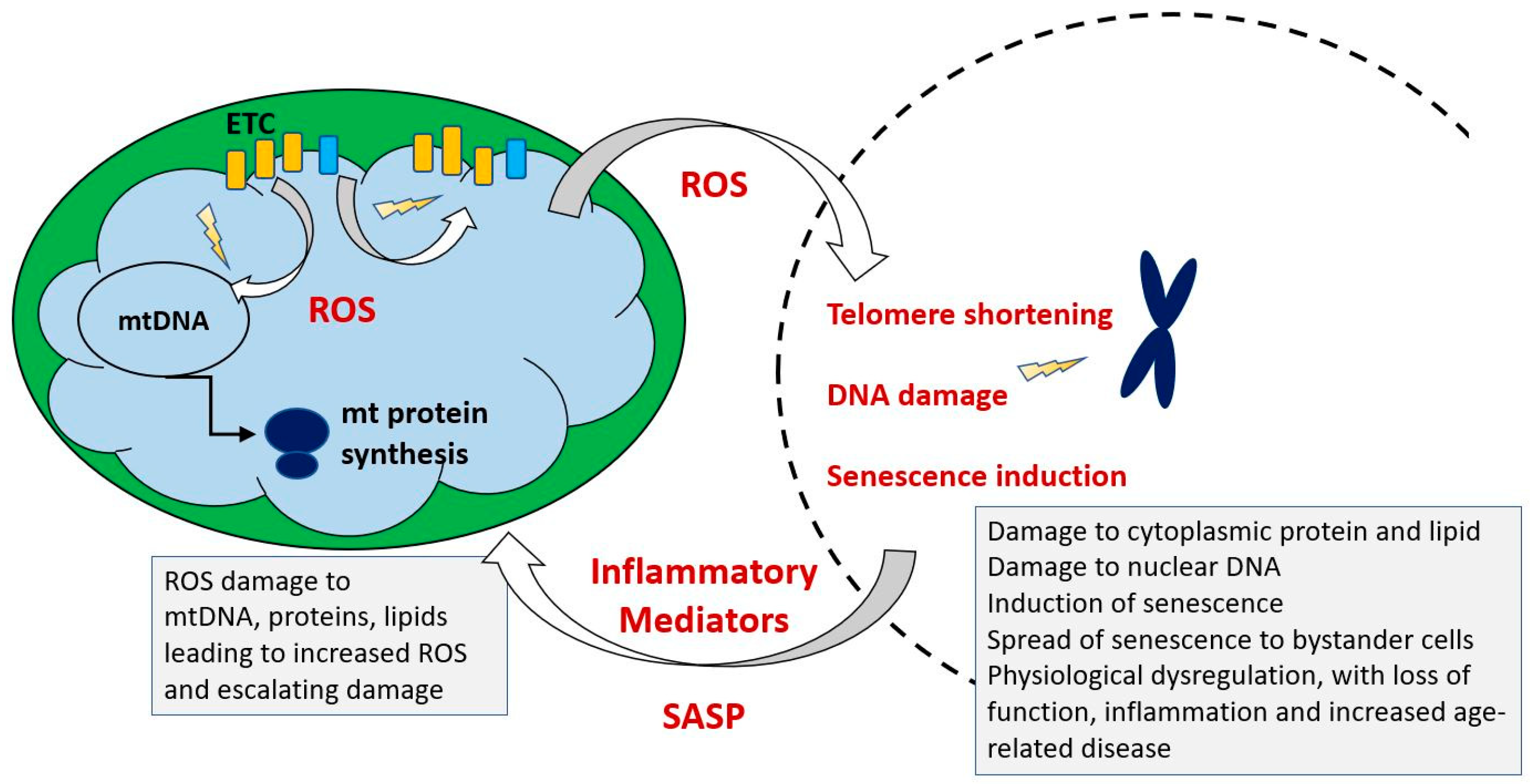
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E.; et al. Redefining Chronic Inflammation in Aging and Age-Related Diseases: Proposal of the Senoinflammation Concept. Aging Dis. 2019, 10, 367–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; De Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095.e10. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.; Langer, T.; Baker, M.J. Proteolytic control of mitochondrial function and morphogenesis. Biochim. Biophys. Acta 2013, 1833, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Quirós, P.M.; Langer, T.; López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Bertelsen, B.; Melchior, L.; Jensen, L.R.; Groth, C.; Glenthøj, B.; Rizzo, R.; Debes, N.M.M.M.; Skov, L.; Brøndum-Nielsen, K.; Paschou, P.; et al. Intragenic deletions affecting two alternative transcripts of the IMMP2L gene in patients with Tourette syndrome. Eur. J. Hum. Genet. 2014, 22, 1283–1289. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Lee, J.R.; Grimes, D.A.; Racacho, L.; Ye, D.; Yang, H.; Ross, O.A.; Farrer, M.J.; McQuibban, G.A.; Bulman, D.E. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, B.M.; Haynes, C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011, 36, 254–261. [Google Scholar] [CrossRef]
- Lionaki, E.; Tavernarakis, N. Oxidative stress and mitochondrial protein quality control in aging. J. Proteom. 2013, 92, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Español, Y.; Acín-Pérez, R.; Rodríguez, F.; Bárcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernández-García, M.S.; Fueyo, A.; et al. ATP-Dependent Lon Protease Controls Tumor Bioenergetics by Reprogramming Mitochondrial Activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef] [Green Version]
- Ngo, J.K.; Davies, K.J. Importance of the Lon protease in mitochondrial maintenance and the significance of declining Lon in aging. Ann. N. Y. Acad. Sci. 2007, 1119, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [Green Version]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [Green Version]
- Koltai, E.; Hart, N.; Taylor, A.W.; Goto, S.; Ngo, J.K.; Davies, K.E.; Radak, Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Integr. Comp. Physiol. 2012, 303, R127–R134. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Sík, A.; Passer, B.J.; Koonin, E.V.; Pellegrini, L. Self-regulated cleavage of the mitochondrial intramembrane-cleaving protease PARL yields Pbeta, a nuclear-targeted peptide. J. Biol. Chem. 2004, 279, 15323–15329. [Google Scholar] [CrossRef] [Green Version]
- Civitarese, A.E.; MacLean, P.S.; Carling, S.; Kerr-Bayles, L.; McMillan, R.P.; Pierce, A.; Becker, T.C.; Moro, C.; Finlayson, J.; Lefort, N.; et al. Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metab. 2010, 11, 412–426. [Google Scholar] [CrossRef] [Green Version]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, 6256. [Google Scholar] [CrossRef]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Kang, S.; Fernandes-Alnemri, T.; Alnemri, E.S. A novel role for the mitochondrial HTRA2/OMI protease in aging. Autophagy 2013, 9, 420–421. [Google Scholar] [CrossRef] [Green Version]
- George, S.K.; Jiao, Y.; Bishop, C.E.; Lu, B. Mitochondrial peptidase IMMP2L mutation causes early onset of age-associated disorders and impairs adult stem cell self-renewal. Aging Cell 2011, 10, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Zhai, L.; Qian, L.; Huang, D.; Ding, Y.; Xiang, H.; Liu, X.; Thompson, J.W.; Liu, J.; He, Y.H.; et al. Switching off IMMP2L signaling drives senescence via simultaneous metabolic alteration and blockage of cell death. Cell Res. 2018, 28, 625–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta 2016, 1, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.N.; McElhaney, R.N. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta 2009, 1788, 2069–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Guan, Z.; Murphy, A.N.; Wiley, S.E.; Perkins, G.A.; Worby, C.A.; Engel, J.L.; Heacock, P.; Nguyen, O.K.; Wang, J.H.; et al. Mitochondrial Phosphatase PTPMT1 Is Essential for Cardiolipin Biosynthesis. Cell Metab. 2011, 13, 690–700. [Google Scholar] [CrossRef] [Green Version]
- Claypool, S.M. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2059–2068. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, M. Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta 2009, 1788, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Musatov, A.; Sedlák, E. Role of cardiolipin in stability of integral membrane proteins. Biochimie 2017, 142, 102–111. [Google Scholar] [CrossRef]
- Lange, C.; Nett, J.H.; Trumpower, B.L.; Hunte, C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 2001, 20, 6591–6600. [Google Scholar] [CrossRef] [Green Version]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Stepanyants, N.; Macdonald, P.J.; Francy, C.A.; Mears, J.A.; Qi, X.; Ramachandran, R. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol. Biol. Cell 2015, 26, 3104–3116. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin CoordinateMitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nature 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Chu, C.T.; Tyurina, Y.Y.; Cheikhi, A.; Bayir, H. Cardiolipin asymmetry, oxidation and signaling. Chem. Phys. Lipids 2014, 179, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000, 466, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species generated by the mitochondrial respiratory chain affect the complex III activity via cardiolipin peroxidation in beef-heart submitochondrial particles. Mitochondrion 2001, 1, 151–159. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochrome c release. FEBS Lett. 2006, 580, 6311–6316. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signaling platform to launch apoptosis. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2022–2031. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.-L.; et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008, 183, 681–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, T.; Sen, N.; Jana, S.; Khan, F.H.; Chatterjee, U.; Chakrabarti, S. Depolarization and cardiolipin depletion in aged rat brain mitochondria: Relationship with oxidative stress and electron transport chain activity. Neurochem. Int. 2007, 50, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Vorbeck, M.L.; Martin, A.P.; Long, J.W., Jr.; Smith, J.M.; Orr, R.R., Jr. Aging-dependent modification of lipid composition and lipid structural order parameter of hepatic mitochondria. Arch. Biochem. Biophys. 1982, 217, 351–361. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: Role of cardiolipin. FEBS Lett. 1997, 406, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [Green Version]
- Sutovsky, P.; Moreno, R.D.; Ramalho-Santos, J.; Dominko, T.; Simerly, C.; Schatten, G. Ubiquitin tag for sperm mitochondria. Nature 1999, 402, 371–372. [Google Scholar] [CrossRef]
- Cann, R.L.; Stoneking, M.; Wilson, A.C. Mitochondrial DNA and human evolution. Nature 1987, 325, 31–36. [Google Scholar] [CrossRef]
- Ramos, A.; Planchat, M.; Melo, A.R.V.; Raposo, M.; Shamim, U.; Suroliya, V.; Srivastava, A.K.; Faruq, M.; Morino, H.; Ohsawa, R.; et al. Mitochondrial DNA haplogroups and age at onset of Machado–Joseph disease/spinocerebellar ataxia type 3: A study in patients from multiple populations. Eur. J. Neurol. 2019, 26, 506–512. [Google Scholar] [CrossRef]
- Veronese, N.; Stubbs, B.; Koyanagi, A.; Vaona, A.; Demurtas, J.; Schofield, P.; Maggi, S. Mitochondrial genetic haplogroups and cardiovascular diseases: Data from the Osteoarthritis Initiative. PLoS ONE 2019, 14, e0213656. [Google Scholar] [CrossRef]
- Chalkia, D.; Singh, L.N.; Leipzig, J.; Lvova, M.; Derbeneva, O.; Lakatos, A.; Hadley, D.; Hakonarson, H.; Wallace, D.C. Association Between Mitochondrial DNA Haplogroup Variation and Autism Spectrum Disorders. JAMA Psychiatry 2017, 74, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez Povedano, C.; Salgado, J.; Gil, C.; Robles, M.; Patino-Garcia, A.; Garcia-Foncillas, J. Analysis of BRCA1 and mtDNA haplotypes and mtDNA polymorphism in familial breast cancer. Mitochondrial DNA 2015, 26, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Vyshkina, T.; Sylvester, A.; Sadiq, S.; Bonilla, E.; Canter, J.A.; Perl, A.; Kalman, B. Association of common mitochondrial DNA variants with multiple sclerosis and systemic lupus erythematosus. Clin. Immunol. 2008, 129, 31–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Taylor, G.A.; Howell, N.; Andrews, R.M.; Morris, C.M.; Taylor, R.W.; McKeith, I.G.; Perry, R.H.; Edwardson, J.A.; Turnbull, D.M. Mitochondrial DNA haplogroups and susceptibility to AD and dementia with Lewy bodies. Neurology 2000, 55, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Kirou, K.A. What Causes Lupus Flares? Curr. Rheumatol. Rep. 2016, 18, 14. [Google Scholar] [CrossRef]
- DiMauro, S.; Garone, C. Historical perspective on mitochondrial medicine. Dev. Disabil. Res. Rev. 2010, 16, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; McCormack, R.; Curran, M.D.; Duguid, R.A.; Barnett, Y.A.; Rea, I.M.; Middleton, D. Mitochondrial DNA polymorphism: Its role in longevity of the Irish population. Exp. Gerontol. 2001, 36, 1161–1178. [Google Scholar] [CrossRef]
- Niemi, A.K.; Hervonen, A.; Hurme, M.; Karhunen, P.J.; Jylha, M.; Majamaa, K. Mitochondrial DNA polymorphisms associated with longevity in a Finnish population. Hum. Genet. 2003, 112, 29–33. [Google Scholar] [CrossRef]
- Shlush, L.I.; Atzmon, G.; Weisshof, R.; Behar, D.; Yudkovsky, G.; Barzilai, N.; Skorecki, K. Ashkenazi Jewish centenarians do not demonstrate enrichment in mitochondrial haplogroup J. PLoS ONE 2008, 3, e3425. [Google Scholar] [CrossRef] [Green Version]
- Dato, S.; Passarino, G.; Rose, G.; Altomare, K.; Bellizzi, D.; Mari, V.; Feraco, E.; Franceschi, C.; De Benedictis, G. Association of the mitochondrial DNA haplogroup J with longevity is population specific. Eur. J. Hum. Genet. 2004, 12, 1080–1082. [Google Scholar] [CrossRef] [Green Version]
- Tranah, G.J.; Santaniello, A.; Caillier, S.J.; D’Alfonso, S.; Boneschi, F.M.; Hauser, S.L.; Oksenberg, J.R. Mitochondrial DNA sequence variation in multiple sclerosis. Neurology 2015, 85, 325–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcuello, A.; Martinez-Redondo, D.; Dahmani, Y.; Casajus, J.A.; Ruiz-Pesini, E.; Montoya, J.; Lopez-Perez, M.J.; Diez-Sanchez, C. Human mitochondrial variants influence on oxygen consumption. Mitochondrion 2009, 9, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Raule, N.; Chomyn, A.; Attardi, G. Decreased reactive oxygen species production in cells with mitochondrial haplogroups associated with longevity. PLoS ONE 2012, 7, e46473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, J.R.; Schaerf, T.M.; Beekman, M. Selection against heteroplasmy explains the evolution of uniparental inheritance of mitochondria. PLoS Genet. 2015, 11, e1005112. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Bogenhagen, D.; Clayton, D.A. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell 1977, 11, 719–727. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What auses mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 259–275. [Google Scholar] [CrossRef]
- Reeve, A.K.; Krishnan, K.J.; Elson, J.L.; Morris, C.M.; Bender, A.; Lightowlers, R.N.; Turnbull, D.M. Nature of mitochondrial DNA deletions in substantia nigra neurons. Am. J. Hum. Genet. 2008, 82, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Liu, V.W.; Zhang, C.; Nagley, P. Mutations in mitochondrial DNA accumulate differentially in three different human tissues during ageing. Nucleic Acids Res. 1998, 26, 1268–1275. [Google Scholar] [CrossRef] [Green Version]
- McDonald, S.A.; Greaves, L.C.; Gutierrez–Gonzalez, L.; Rodriguez–Justo, M.; Deheragoda, M.; Leedham, S.J.; Taylor, R.W.; Lee, C.Y.; Preston, S.L.; Lovell, M.; et al. Mechanisms of Field Cancerization in the Human Stomach: The Expansion and Spread of Mutated Gastric Stem Cells. Gastroenterology 2008, 134, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Lee, Y.; Hayama, T.; Van Dyken, C.; Marti-Gutierrez, N.; Li, Y.; Ahmed, R.; Koski, A.; Kang, E.; Darby, H.; et al. Germline and somatic mtDNA mutations in mouse aging. PLoS ONE 2018, 13, e0201304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.; Wang, X.; Tippner-Hedges, R.; Ma, H.; Folmes, C.D.; Gutierrez, N.M.; Lee, Y.; Van Dyken, C.; Ahmed, R.; Li, Y.; et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell Stem Cell 2016, 18, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Wilson, I.J.; Yu-Wai-Man, P.; Coxhead, J.; Deehan, D.; Horvath, R.; Taylor, R.W.; Samuels, D.C.; Santibanez-Koref, M.; Chinnery, P.F. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 2013, 22, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [Green Version]
- Elson, J.L.; Samuels, D.C.; Turnbull, D.M.; Chinnery, P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 2001, 68, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Amunts, A.; Brown, A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu. Rev. Biochem. 2016, 85, 77–101. [Google Scholar] [CrossRef]
- Sylvester, J.E.; Fischel-Ghodsian, N.; Mougey, E.B.; OʼBrien, T.W. Mitochondrial ribosomal proteins: Candidate genes for mitochondrial disease. Genet. Med. 2004, 6, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Tiosano, D.; Guran, T.; Baris, H.N.; Bayram, Y.; Mory, A.; Shapiro-Kulnane, L.; A Hodges, C.; Akdemir, Z.C.; Turan, S.; et al. Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum. Mol. Genet. 2018, 27, 1913–1926. [Google Scholar] [CrossRef] [Green Version]
- Bugiardini, E.; Mitchell, A.L.; Rosa, I.D.; Horning-Do, H.-T.; Pitmann, A.M.; Poole, O.V.; Holton, J.L.; Shah, S.; Woodward, C.; Hargreaves, I.P.; et al. MRPS25 mutations impair mitochondrial translation and cause encephalomyopathy. Hum. Mol. Genet. 2019, 28, 2711–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borna, N.N.; Kishita, Y.; Kohda, M.; Lim, S.C.; Shimura, M.; Wu, Y.; Mogushi, K.; Yatsuka, Y.; Harashima, H.; Hisatomi, Y.; et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 2019, 20, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Akbergenov, R.; Duscha, S.; Fritz, A.K.; Juskeviciene, R.; Oishi, N.; Schmitt, K.; Shcherbakov, D.; Teo, Y.; Boukari, H.; Freihofer, P.; et al. Mutant MRPS 5 affects mitoribosomal accuracy and confers stress-related behavioral alterations. EMBO Rep. 2018, 19, e46193. [Google Scholar] [CrossRef]
- González-Serrano, L.E.; Chihade, J.W.; Sissler, M. When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J. Biol. Chem. 2019, 294, 5309–5320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Nagao, A.; Suzuki, T. Human mitochondrial tRNAs: Biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011, 45, 299–329. [Google Scholar] [CrossRef]
- Barić, I.; Fumić, K.; Petković Ramadža, D.; Sperl, W.; Zimmermann, F.A.; Muačević-Katanec, D.; Mitrović, Z.; Pažanin, L.; Cvitanović Šojat, L.; Kekez, T.; et al. Mitochondrial myopathy associated with a novel 5522G > A mutation in the mitochondrial tRNA(Trp) gene. Eur. J. Hum. Genet. 2013, 21, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, D.; Schubert, K.; Joshi, P.R.; Hardy, S.A.; Tuppen, H.A.L.; Baty, K.; Blakely, E.L.; Bamberg, C.; Zierz, S.; Deschauer, M.; et al. Pathogenic mitochondrial mt-tRNA(Ala) variants are uniquely associated with isolated myopathy. Eur. J. Hum. Genet. 2015, 23, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Ji, Y.; Guan, M.X. Mitochondrial tRNA mutations associated with deafness. Mitochondrion 2012, 12, 406–413. [Google Scholar] [CrossRef]
- Zheng, J.; Bai, X.; Xiao, Y.; Ji, Y.; Meng, F.; Aishanjiang, M.; Gao, Y.; Wang, H.; Fu, Y.; Guan, M.X. Mitochondrial tRNA mutations in 887 Chinese subjects with hearing loss. Mitochondrion 2020, 52, 163–172. [Google Scholar] [CrossRef]
- Djordjevic, D.; Brady, L.; Bai, R.; Tarnopolsky, M.A. Two novel mitochondrial tRNA mutations, A7495G (tRNASer(UCN)) and C5577T (tRNATrp), are associated with seizures and cardiac dysfunction. Mitochondrion 2016, 31, 40–44. [Google Scholar] [CrossRef]
- Saoura, M.; Powell, C.A.; Kopajtich, R.; Alahmad, A.; Al-Balool, H.H.; Albash, B.; Alfadhel, M.; Alston, C.L.; Bertini, E.; Bonnen, P.E.; et al. Mutations in ELAC2 associated with hypertrophic cardiomyopathy impair mitochondrial tRNA 3′-end processing. Hum. Mutat. 2019, 40, 1731–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatz, C.; D’Aco, K.; Smith, S.; Sondheimer, N. Mutation in the mitochondrial tRNA(Val) causes mitochondrial encephalopathy, lactic acidosis and stroke-like episodes. Mitochondrion 2011, 11, 615–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, K.; Kaufmann, P.; Naini, A.B.; Lu, J.; Parsons, T.C.; Wang, D.; Willey, J.Z.; Shanske, S.; Hirano, M.; Bonilla, E.; et al. A novel tRNAVal mitochondrial DNA mutation causing MELAS. J. Neurol. Sci. 2008, 270, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Ferrín, G.; Acín-Pérez, R.; Gallardo, M.E.; Viscomi, C.; Pérez-Martos, A.; Zeviani, M.; Fernandez-Silva, P.; Enriquez, J.A. Evolution Meets Disease: Penetrance and Functional Epistasis of Mitochondrial tRNA Mutations. PLoS Genet. 2011, 7, e1001379. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, T.; Suzuki, T.; Ishii, N.; Ohta, S.; Watanabe, K. Wobble modification defect in tRNA disturbs codon-anticodon interaction in a mitochondrial disease. EMBO J. 2001, 20, 4794–4802. [Google Scholar] [CrossRef]
- Richter, U.; Evans, M.E.; Clark, W.C.; Marttinen, P.; Shoubridge, E.A.; Suomalainen, A.; Wredenberg, A.; Wedell, A.; Pan, T.; Battersby, B.J. RNA modification landscape of the human mitochondrial tRNALys regulates protein synthesis. Nat. Commun. 2018, 9, 3966. [Google Scholar] [CrossRef]
- Elson, J.L.; Smith, P.M.; Greaves, L.C.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.; Taylor, R.W.; Vila-Sanjurjo, A. The presence of highly disruptive 16S rRNA mutations in clinical samples indicates a wider role for mutations of the mitochondrial ribosome in human disease. Mitochondrion 2015, 25, 17–27. [Google Scholar] [CrossRef]
- Smith, P.M.; Elson, J.L.; Greaves, L.C.; Wortmann, S.B.; Rodenburg, R.J.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Taylor, R.W.; Vila-Sanjurjo, A. The role of the mitochondrial ribosome in human disease: Searching for mutations in 12S mitochondrial rRNA with high disruptive potential. Hum. Mol. Genet. 2014, 23, 949–967. [Google Scholar] [CrossRef] [Green Version]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Öztas, S.; Qiu, W.-Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic–induced and non–syndromic deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef]
- Zhao, H.; Li, R.; Wang, Q.; Yan, Q.; Deng, J.-H.; Han, D.; Bai, Y.; Young, W.-Y.; Guan, M.-X. Maternally Inherited Aminoglycoside-Induced and Nonsyndromic Deafness Is Associated with the Novel C1494T Mutation in the Mitochondrial 12S rRNA Gene in a Large Chinese Family. Am. J. Hum. Genet. 2004, 74, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Coulbault, L.; Deslandes, B.; Herlicoviez, D.; Read, M.; Leporrier, N.; Schaeffer, S.; Mouadil, A.; Lombès, A.; Chapon, F.; Jauzac, P.; et al. A novel mutation 3090 G > A of the mitochondrial 16S ribosomal RNA associated with myopathy. Biochem. Biophys. Res. Commun. 2007, 362, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, Y.; Li, D.; He, X.; Li, S.; Wu, B.; Wang, W.; Gu, S.; Zhu, X.; Wang, X.; et al. The novel mitochondrial 16S rRNA 2336T > C mutation is associated with hypertrophic cardiomyopathy. J. Med. Genet. 2014, 51, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichart, G.; Mayer, J.; Tokay, T.; Lange, F.; Johne, C.; Baltrusch, S.; Tiedge, M.; Fuellen, G.; Ibrahim, S.; Köhling, R. Combination of mitochondrial tRNA and OXPHOS mutation reduces lifespan and physical condition in aged mice. bioRxiv 2017, 233593. [Google Scholar] [CrossRef] [Green Version]
- Filograna, R.; Koolmeister, C.; Upadhyay, M.; Pajak, A.; Clemente, P.; Wibom, R.; Simard, M.L.; Wredenberg, A.; Freyer, C.; Stewart, J.B.; et al. Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci. Adv. 2019, 5, eaav9824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, D.C.; Li, C.; Li, B.; Song, Z.; Torstenson, E.; Clay, H.B.; Rokas, A.; Thornton-Wells, T.A.; Moore, J.H.; Hughes, T.M.; et al. Recurrent Tissue-Specific mtDNA Mutations Are Common in Humans. PLoS Genet. 2013, 9, e1003929. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Cavelier, L.; Collins-Schramm, H.E.; Seldin, M.F.; McGrogan, M.; Savontaus, M.-L.; Cortopassi, G.A. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum. Mol. Genet. 2002, 11, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
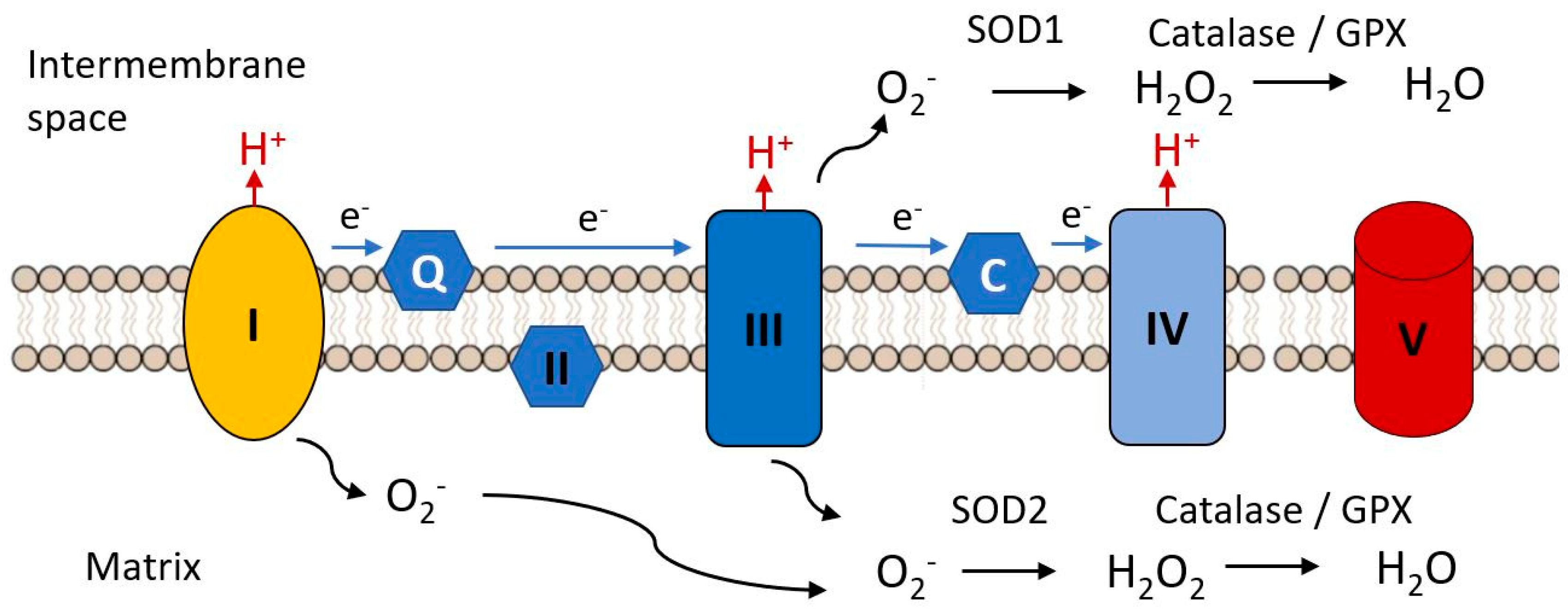
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Cand, F.; Verdetti, J. Superoxide dismutase, glutathione peroxidase, catalase, and lipid peroxidation in the major organs of the aging rats. Free Radic. Biol. Med. 1989, 7, 59–63. [Google Scholar] [CrossRef]
- Lewis, K.N.; Andziak, B.; Yang, T.; Buffenstein, R. The naked mole-rat response to oxidative stress: Just deal with it. Antioxid. Redox Signal. 2013, 19, 1388–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Li, J.; Hekimi, S. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics 2007, 177, 2063–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 5785–5790. [Google Scholar] [CrossRef] [Green Version]
- Doonan, R.; McElwee, J.J.; Matthijssens, F.; Walker, G.A.; Houthoofd, K.; Back, P.; Matscheski, A.; Vanfleteren, J.R.; Gems, D. Against the oxidative damage theory of aging: Superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008, 22, 3236–3241. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Barja, G. The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 2014, 127, 1–27. [Google Scholar]
- Barja, G. Towards a unified mechanistic theory of aging. Exp. Gerontol. 2019, 124, 110627. [Google Scholar] [CrossRef]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hamalainen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Gotz, A.; Forsstrom, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.; Pickrell, A.M.; Wang, X.; Bacman, S.R.; Yu, A.; Hida, A.; Dillon, L.M.; Morton, P.D.; Malek, T.R.; Williams, S.L.; et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ. 2017, 24, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.T.; Dou, Y.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef]
- Tapia, P.C. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med. Hypotheses 2006, 66, 832–843. [Google Scholar]
- Van Zant, G.; Liang, Y. The role of stem cells in aging. Exp. Hematol. 2003, 31, 659–672. [Google Scholar] [CrossRef]
- Anso, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.Q.; Suda, T. Reactive Oxygen Species and Mitochondrial Homeostasis as Regulators of Stem Cell Fate and Function. Antioxid. Redox Signal. 2018, 29, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Jacobs, J.J.; de Lange, T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr. Biol. 2004, 14, 2302–2308. [Google Scholar] [CrossRef] [Green Version]
- Prattichizzo, F.; Giuliani, A.; Recchioni, R.; Bonafè, M.; Marcheselli, F.; De Carolis, S.; Campanati, A.; Giuliodori, K.; Rippo, M.R.; Brugè, F.; et al. Anti-TNF-α treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget 2016, 7, 11945–11958. [Google Scholar] [CrossRef] [Green Version]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Brandenburg, S.; Demaria, M. Induction and Validation of Cellular Senescence in Primary Human Cells. J. Vis. Exp. 2018, 136, 55782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millerand, M.; Berenbaum, F.; Jacques, C. Danger signals and inflammaging in osteoarthritis. Clin. Exp. Rheumatol. 2019, 3, 48–56. [Google Scholar]
- Nie, L.; Zhang, P.; Wang, Q.; Zhou, X.; Wang, Q. lncRNA—Triggered Macrophage Inflammaging Deteriorates Age-Related Diseases. Mediat. Inflamm. 2019, 2019, 4260309. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Byun, H.O.; Cho, H.; Kim, B.K.; Yoon, G. Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induce senescence—Associated growth arrest. J. Biol. Chem. 2003, 278, 51577–51586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Huang, Q.; Huang, S.; Yang, X.; Zhu, T.; Wang, W.; Wang, H.; He, S.; Ji, L.; Wang, Y.; et al. Senescence Inducer Shikonin ROS-Dependently Suppressed Lung Cancer Progression. Front. Pharmacol. 2018, 9, 519. [Google Scholar] [CrossRef] [PubMed]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef]
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 2003, 5, R234–R240. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1162–1552. [Google Scholar] [CrossRef]
- Jylhava, J.; Nevalainen, T.; Marttila, S.; Jylha, M.; Hervonen, A.; Hurme, M. Characterization of the role of distinct plasma cell-free DNA species in age-associated inflammation and frailty. Aging Cell 2013, 12, 388–397. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Schwartz, M. NIX-ing mitochondria: From development to pathology. EMBO J. 2017, 36, 1650–1652. [Google Scholar] [CrossRef] [PubMed]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–195055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, M.; Lindsten, T.; Yang, C.Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef]
- Rojansky, R.; Cha, M.Y.; Chan, D.C. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 2016, 5, e17896. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Narendra, D.P.; Jin, S.M.; Tekle, E.; Banerjee, S.; Youle, R.J. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J. Cell Biol. 2013, 200, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin At Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; E Haigh, S.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Abeliovich, H.; Zarei, M.; Rigbolt, K.T.; Youle, R.J.; Dengjel, J. Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat. Commun. 2013, 4, 2789. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmstrom, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, A.M.; Staveley, B.E. Expression of Pink1 with alpha-synuclein in the dopaminergic neurons of Drosophila leads to increases in both lifespan and healthspan. Genet. Mol. Res. 2012, 11, 1497–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Liu, Y.; Tu, Z.; Xiao, C.; Yan, S.; Ma, X.; Guo, X.; Chen, X.; Yin, P.; Yang, Z.; et al. CRISPR/Cas9-mediated PINK1 deletion leads to neurodegeneration in rhesus monkeys. Cell Res. 2019, 29, 334–336. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Li, S.; Li, X.J. A CRISPR monkey model unravels a unique function of PINK1 in primate brains. Mol. Neurodegener. 2019, 14, 17. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Prat, L.; Munoz-Canoves, P.; Martinez-Vicente, M. Dysfunctional autophagy is a driver of muscle stem cell functional decline with aging. Autophagy 2016, 12, 612–613. [Google Scholar] [CrossRef] [Green Version]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Gioscia-Ryan, R.A.; Hearon, C.M., Jr.; Seals, D.R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 2013, 134, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef] [Green Version]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Felix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Jahn, H.J.; Li, J.; Ling, L.; Guo, H.; Zhu, X.; Preedy, V.; Lu, H.; Bohr, V.A.; et al. A research agenda for aging in China in the 21st century. Ageing Res. Rev. 2015, 24, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Hengst, J.A.; Dick, T.E.; Sharma, A.; Doi, K.; Hegde, S.; Tan, S.F.; Geffert, L.M.; Fox, T.E.; Sharma, A.K.; Desai, D.; et al. SKI-A Multitargeted Inhibitor of Sphingosine Kinase and Microtubule Dynamics Demonstrating Therapeutic Efficacy in Acute Myeloid Leukemia Models. Cancer Transl. Med. 2017, 3, 109–121. [Google Scholar]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chan, D.C. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Tondera, D.; Czauderna, F.; Paulick, K.; Schwarzer, R.; Kaufmann, J.; Santel, A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J. Cell Sci. 2005, 118, 3049–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Hausenloy, D.J. Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 2014, 171, 1890–1906. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.J.; Soh, M.S.; Chandhok, G.; Vijayaraghavan, T.; Teoh, J.S.; Crawford, S.; Cobham, A.E.; Yapa, N.M.B.; Mirth, C.K.; Neumann, B. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell Mol. Life Sci. 2019, 76, 1967–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhardt, D.; Müller, M.; Reichert, A.S.; Osiewacz, H.D. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci. Rep. 2015, 5, 7885. [Google Scholar] [CrossRef] [Green Version]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.J.; Vallee, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2017, 9, 6128–6143. [Google Scholar] [CrossRef] [Green Version]
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J. Mol. Cell Cardiol. 2019, 130, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Ebert, A.E.; Haileselassie, B.; Mochly-Rosen, D. Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington’s disease. J. Mol. Cell Cardiol. 2019, 127, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, J.; Wang, Q.; Zhao, S.; Yang, S.; Tian, L.; Meng, P.; Li, J.; Li, H. Hyperglycaemia Stress-Induced Renal Injury is Caused by Extensive Mitochondrial Fragmentation, Attenuated MKP1 Signalling, and Activated JNK-CaMKII-Fis1 Biological Axis. Cell. Physiol. Biochem. 2018, 51, 1778–1798. [Google Scholar] [CrossRef]
- Zhang, Z.; Sliter, D.A.; Bleck, C.K.E.; Ding, S. Fis1 deficiencies differentially affect mitochondrial quality in skeletal muscle. Mitochondrion 2019, 49, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Shi, M.; Li, M.; Cheng, L.; Yang, J.; Huang, X. Sirtuin 3 inhibition induces mitochondrial stress in tongue cancer by targeting mitochondrial fission and the JNK-Fis1 biological axis. Cell Stress Chaperones 2019, 24, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.-M.; Liou, Y.-C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, D.; Mottis, A.; Potenza, F.; Sorrentino, V.; Li, H.; Romani, M.; Lemos, V.; Schoonjans, K.; Zamboni, N.; Knott, G.; et al. The RNA-Binding Protein PUM2 Impairs Mitochondrial Dynamics and Mitophagy During Aging. Mol. Cell 2019, 73, 775–787.e10. [Google Scholar] [CrossRef] [Green Version]
- Navratil, M.; Terman, A.; Arriaga, E.A. Giant mitochondria do not fuse and exchange their contents with normal mitochondria. Exp. Cell Res. 2008, 314, 164–172. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W. 2nd, Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malena, A.; Loro, E.; Di Re, M.; Holt, I.J.; Vergani, L. Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum. Mol. Genet. 2009, 18, 3407–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, R.; Kakimoto, Y.; Furuta, S.; Itoh, K.; Sesaki, H.; Endo, T.; Tamura, Y. Maintenance of Cardiolipin and Crista Structure Requires Cooperative Functions of Mitochondrial Dynamics and Phospholipid Transport. Cell Rep. 2019, 26, 518–528.e6. [Google Scholar] [CrossRef] [Green Version]
- Partikian, A.; Olveczky, B.; Swaminathan, R.; Li, Y.; Verkman, A.S. Rapid diffusion of green fluorescent protein in the mitochondrial matrix. J. Cell Biol. 1998, 140, 821–829. [Google Scholar] [CrossRef]
- Legros, F.; Malka, F.; Frachon, P.; Lombes, A.; Rojo, M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 2004, 117, 2653–2662. [Google Scholar] [CrossRef] [Green Version]
- Mouli, P.K.; Twig, G.; Shirihai, O.S. Frequency and selectivity of mitochondrial fusion are key to its quality maintenance function. Biophys. J. 2009, 96, 3509–3518. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
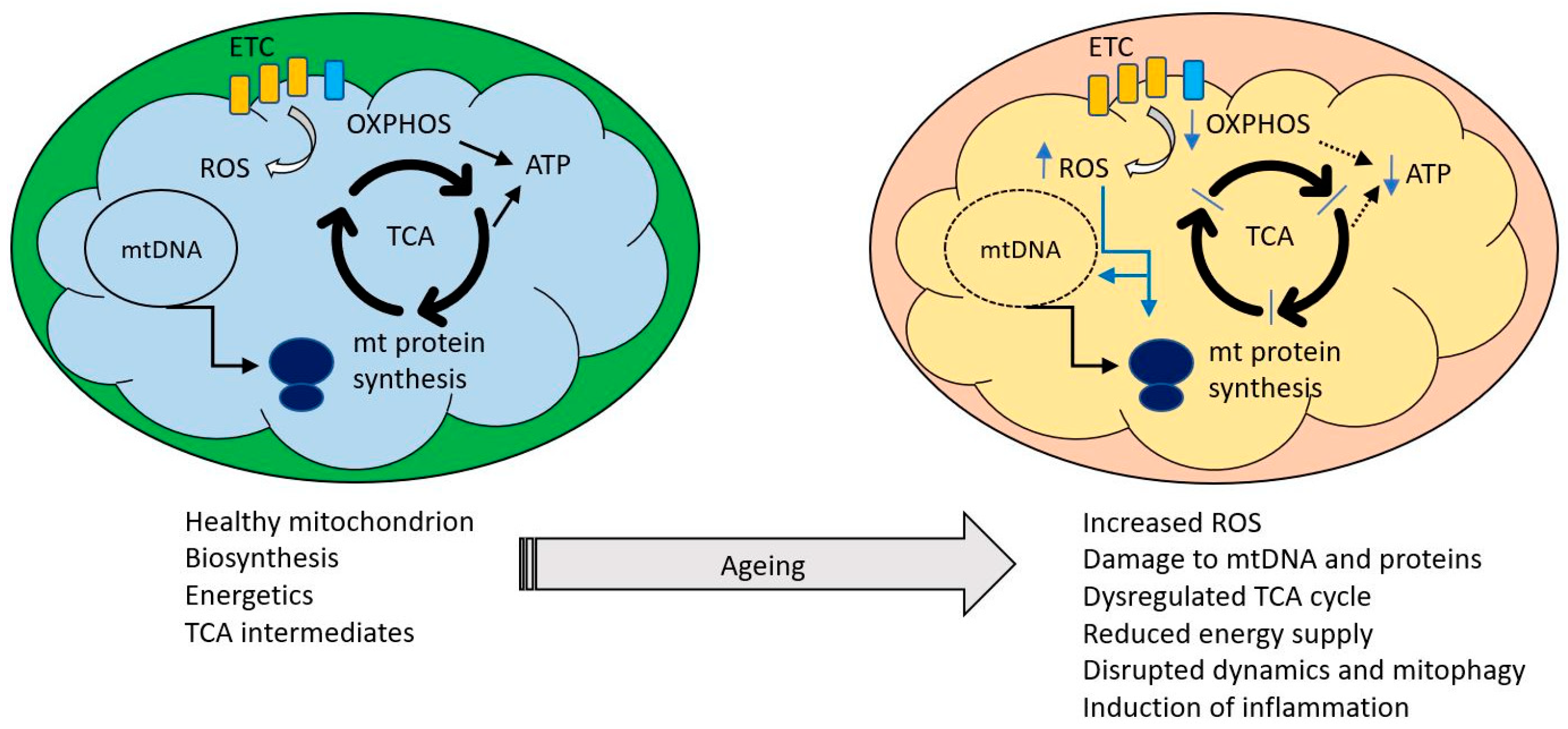

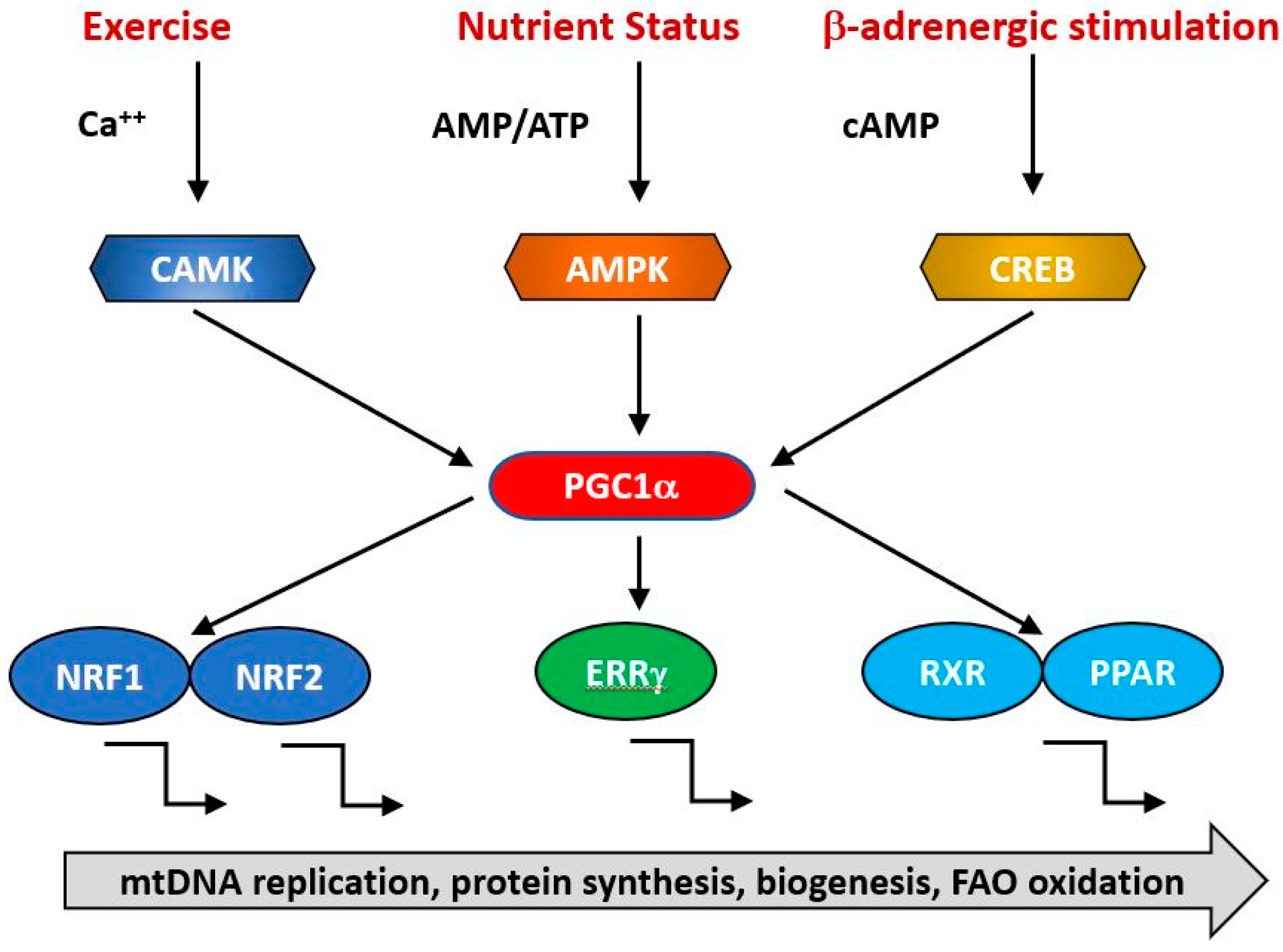


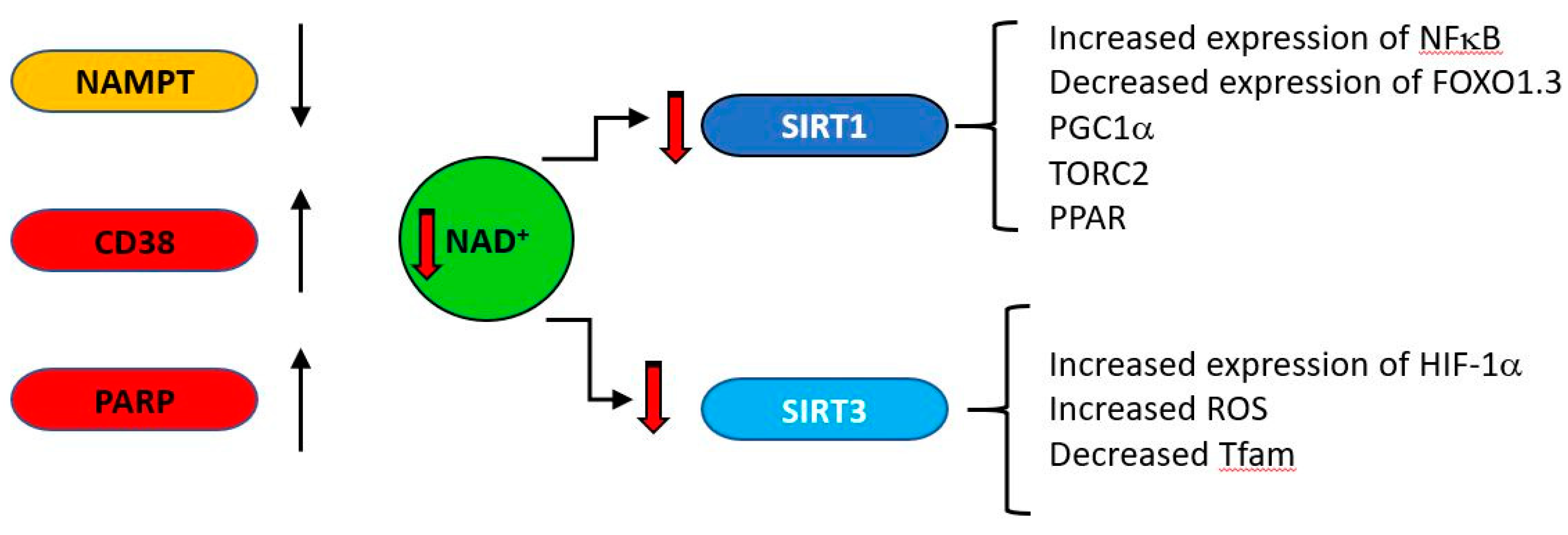




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webb, M.; Sideris, D.P. Intimate Relations—Mitochondria and Ageing. Int. J. Mol. Sci. 2020, 21, 7580. https://doi.org/10.3390/ijms21207580
Webb M, Sideris DP. Intimate Relations—Mitochondria and Ageing. International Journal of Molecular Sciences. 2020; 21(20):7580. https://doi.org/10.3390/ijms21207580
Chicago/Turabian StyleWebb, Michael, and Dionisia P. Sideris. 2020. "Intimate Relations—Mitochondria and Ageing" International Journal of Molecular Sciences 21, no. 20: 7580. https://doi.org/10.3390/ijms21207580
APA StyleWebb, M., & Sideris, D. P. (2020). Intimate Relations—Mitochondria and Ageing. International Journal of Molecular Sciences, 21(20), 7580. https://doi.org/10.3390/ijms21207580