Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1



Abstract
:1. Introduction
2. Results
2.1. COL7A1 Exon 10
2.2. COL7A1 Exon 73
2.3. Splicing Order Analysis
- Intron 72 versus intron 73. Primers that extend from exon 72 (primer 1) to intron 73 (primer 9) generate the spliced product primarily, with a hint of unspliced product, indicating that intron 72 is usually removed before intron 73. The unspliced product predominates over the spliced counterpart when amplified using primers extending from intron 72 (primer 2) to exon 74 (primer 10), indicating that intron 73 is usually removed after intron 72.
- Intron 73 versus intron 74. Primers that extend from exon 73 (primer 3) to intron 74 (primer 11) yield mainly the unspliced product, with traces of the spliced product, indicating intron 74 is usually removed before intron 73. Primers that extend from intron 73 (primer 4) to exon 75 (primer 12) again yield mainly the unspliced product, with a hint of the spliced product, indicating intron 73 is usually removed before intron 74. These results indicate no preference to the order of removal of introns 73 and 74.
- Intron 74 versus intron 75. Primers that extend from exon 74 (primer 5) to intron 75 (primer 13) yield the unspliced product, indicating intron 75 is usually removed before intron 74. Primers that extend from intron 74 (primer 6) to exon 76 (primer 14) again yield the unspliced product and the spliced product, indicating intron 75 is removed before intron 74.
- Intron 75 versus intron 76. Primers that extend from exon 75 (primer 7) to intron 76 (primer 15) yield both the unspliced and spliced transcripts, indicating intron 75 is removed before intron 76. Primers that extend from intron 75 (primer 8) to exon 76 (primer 16) yield solely the unspliced product, suggesting intron 75 is removed before intron 76.
- Intron 72 versus intron 74. Primers that extend from exon 72 (primer 1) to intron 74 (primer 11) yield mainly the unspliced products and transcripts missing intron 72 are a minor product. Primers that extend from intron 72 (primer 2) to exon 75 (primer 12) yield the unspliced product, and possibly transcripts either missing intron 74 or missing intron 73 + 74. Since a similar-sized product is also present in the genomic DNA amplification for this primer set, we cannot determine if intron 73 or 74 splicing occurred. The results from this primer set are, therefore, inconclusive but are suggestive of no preference to the order of removal of these introns.
- Intron 73 versus intron 75. Primers that extend from exon 73 (primer 3) to intron 75 (primer 13) yield mainly the unspliced product. Transcripts missing intron 73 or 74 individually, and transcripts missing introns 73 + 74, are minor products. Primers that extend from intron 73 (primer 4) to exon 76 (primer 14) yield mainly the unspliced product, transcripts missing intron 75, and transcripts missing both introns 74 + 75. Transcripts missing intron 74 is a minor product. Since the level of intron 75 spliced product is comparable to that of the unspliced molecule, intron 75 tends to be removed before intron 73.
- Intron 74 versus intron 76. Primers that extend from exon 74 (primer 5) to intron 76 (primer 15) yield mainly transcripts missing intron 75, and transcripts missing introns 74 + 75, with the unspliced product present as a minor product. Primers that extend from intron 74 (primer 6) to exon 77 (primer 16) yield primarily the unspliced product and the product missing intron 75. However, no products missing intron 76 are observed. This splicing pattern indicates intron 75, and a combination of introns 74 + 75 are spliced before intron 76.
3. Discussion
4. Materials and Methods
4.1. AO Design and Synthesis
4.2. Cell Propagation and Transfection
4.3. Molecular Analyses
4.4. In Silico Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DEB | Dystrophic epidermolysis bullosa |
| DDEB | Dominant DEB |
| RDEB | Recessive DEB |
| AO | Antisense oligomer |
| PMO | Phosphorodiamidate morpholino oligomer |
| 2′-OMe | 2′-O-methyl modified bases on a phosphorothioate backbone |
| MD | Muscular dystrophy |
| ESE | Exonic splicing enhancer |
References
- Dang, N.; Murrell, D.F. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp. Dermatol. 2008, 17, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Bruckner-Tuderman, L. Dystrophic epidermolysis bullosa: Pathogenesis and clinical features. Dermatol. Clin. 2010, 28, 107–114. [Google Scholar] [CrossRef] [PubMed]
- McClements, M.E.; MacLaren, R.E. Adeno-associated virus (AAV) dual vector strategies for gene therapy encoding large transgenes. Yale J. Biol. Med. 2017, 90, 611–623. [Google Scholar]
- Christiano, A. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics 1994, 21, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Bornert, O.; Kuhl, T.; Bremer, J.; van den Akker, P.C.; Pasmooij, A.M.; Nystrom, A. Analysis of the functional consequences of targeted exon deletion in COL7A1 reveals prospects for dystrophic epidermolysis bullosa therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1302–1311. [Google Scholar] [CrossRef] [Green Version]
- Bruckner-Tuderman, L. Immunohistochemical and mutation analyses demonstrate that procollagen VII is processed to collagen VII through removal of the NC-2 domain. J. Cell Biol. 1995, 131, 551–559. [Google Scholar] [CrossRef]
- Bruckner-Tuderman, L. Hereditary skin diseases of anchoring fibrils. J. Dermatol. Sci. 1999, 20, 122–133. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R.; Morris, N.P.; Burgeson, R.E. Type VII collagen is a major structural component of anchoring fibrils. J. Cell Biol. 1986, 103, 1577–1586. [Google Scholar] [CrossRef] [Green Version]
- Mecklenbeck, S.; Hammami-Hauasli, N.; Höpfner, B.; Schumann, H.; Kramer, A.; Küster, W.; Bruckner-Tuderman, L. Clustering of COL7A1 mutations in exon 73: Implications for mutation analysis in dystrophic epidermolysis bullosa. J. Investig. Dermatol. 1999, 112, 398–400. [Google Scholar] [CrossRef]
- Varki, R.; Sadowski, S.; Uitto, J.; Pfendner, E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype–genotype correlations in the dystrophic subtypes. J. Med. Genet. 2007, 44, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Decha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [Green Version]
- Kessler, O.; Jiang, Y.; Chasin, L.A. Order of intron removal during splicing of endogenous adenine phosphoribosyltransferase and dihydrofolate reductase pre-mRNA. Mol. Cell Biol. 1993, 13, 6211–6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Piovesan, A.; Caracausi, M.; Antonaros, F.; Pelleri, M.C.; Vitale, L. GeneBase 1.1: A tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database 2016, 2016, baw153. [Google Scholar] [CrossRef]
- Gazzoli, I.; Pulyakhina, I.; Verwey, N.E.; Ariyurek, Y.; Laros, J.F.J.; T Hoen, P.A.C.; Aartsma-Rus, A. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 2016, 13, 290–305. [Google Scholar] [CrossRef] [Green Version]
- Shimada, M.; Sasaki-Haraguchi, N.; Mayeda, A. Identification and validation of evolutionarily conserved unusually short pre-mRNA introns in the human genome. Int. J. Mol. Sci. 2015, 16, 10376–10388. [Google Scholar] [CrossRef] [Green Version]
- Tennyson, C.N.; Klamut, H.J.; Worton, R.G. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat. Genet. 1995, 9, 184–190. [Google Scholar] [CrossRef]
- Jonkers, I.; Kwak, H.; Lis, J.T. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Frey, U.H.; Bachmann, H.S.; Peters, J.; Siffert, W. PCR-amplification of GC-rich regions: ‘slowdown PCR’. Nat. Protoc. 2008, 3, 1312–1317. [Google Scholar] [CrossRef]
- McDowell, D.G.; Burns, N.A.; Parkes, H.C. Localised sequence regions possessing high melting temperatures prevent the amplification of a DNA mimic in competitive PCR. Nucleic Acids Res. 1998, 26, 3340–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Med. Biochem. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Swertz, M.A.; Dijkstra, M.; Adamusiak, T.; Van Der Velde, J.K.; Kanterakis, A.; Roos, E.T.; Lops, J.; Thorisson, G.A.; Arends, D.; Byelas, G.; et al. The MOLGENIS toolkit: Rapid prototyping of biosoftware at the push of a button. BMC Bioinform. 2010, 11, S12. [Google Scholar] [CrossRef] [Green Version]
- Van Den Akker, P.C.; Jonkman, M.F.; Rengaw, T.; Bruckner-Tuderman, L.; Has, C.; Bauer, J.W.; Klausegger, A.; Zambruno, G.; Castiglia, D.; Mellerio, J.E.; et al. The international dystrophic epidermolysis bullosa patient registry: An online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Human Mutat. 2011, 32, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Wertheim-Tysarowska, K.; Sobczyńska-Tomaszewska, A.; Kowalewski, C.; Skroński, M.; Święćkowski, G.; Kutkowska-Kaźmierczak, A.; Woźniak, K.; Bal, J. The COL7A1 mutation database. Hum. Mutat. 2012, 33, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Bornert, O.; Hogervorst, M.; Nauroy, P.; Bischof, J.; Swildens, J.; Athanasiou, I.; Tufa, S.F.; Keene, D.R.; Kiritsi, D.; Hainzl, S.; et al. QR-313, an antisense oligonucleotide, shows therapeutic efficacy for treatment ofdominant and recessive dystrophic epidermolysis bullosa: A preclinical study. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef]
- Flynn, L.L.; Ruohan, L.; Aung-Htut, M.T.; Pitout, I.L.; Cooper, J.; Hubbard, A.; Griffiths, L.; Bond, C.; Wilton, S.D.; Fox, A.H.; et al. Interaction of modified oligonucleotides with nuclear proteins, formation of novel nuclear structures and sequence-independent effects on RNA processing. bioRxiv 2018, 446773. [Google Scholar] [CrossRef]
- Adams, A.M.; Harding, P.L.; Iversen, P.L.; Coleman, C.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: Cocktails and chemistries. BMC Mol. Biol. 2007, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Aung-Htut, M.T.; McIntosh, C.S.; West, K.A.; Fletcher, S.; Wilton, S.D. In vitro validation of phosphorodiamidate morpholino oligomers. Molecules 2019, 24, 2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef]
- Bremer, J.; Bornert, O.; Nystrom, A.; Gostynski, A.; Jonkman, M.F.; Aartsma-Rus, A.; van den Akker, P.C.; Pasmooij, A.M. Antisense Oligonucleotide-Mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef]
- Shefer, K.; Sperling, J.; Sperling, R. The supraspliceosome—A multi-task machine for regulated pre-mRNA processing in the cell nucleus. Comput. Struct. Biotechnol. J. 2014, 11, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Bremer, J.; Van Der Heijden, E.H.; Eichhorn, D.S.; Meijer, R.; Lemmink, H.H.; Scheffer, H.; Sinke, R.J.; Jonkman, M.F.; Pasmooij, A.M.G.; Van Den Akker, P.C. Natural exon skipping sets the stage for exon skipping as therapy for dystrophic epidermolysis bullosa. Mol. Ther. Nucleic Acids 2019, 18, 465–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Hamada, T.; Ishii, N.; Fukuda, S.; Sakaguchi, S.; Nakano, H.; Tamai, K.; Sawamura, D.; Hashimoto, T. Exon 87 skipping of the COL7A1 gene in dominant dystrophic epidermolysis bullosa. J. Dermatol. 2011, 38, 489–492. [Google Scholar] [CrossRef]
- McGrath, J.A.; Ashton, G.H.; Mellerio, J.E.; Salas-Alanis, J.C.; Swensson, O.; McMillan, J.R.; Eady, R.A. Moderation of phenotypic severity in dystrophic and junctional forms of epidermolysis bullosa through in-frame skipping of exons containing non-sense or frameshift mutations. J. Investig. Dermatol. 1999, 113, 314–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Masunaga, T.; Ishiko, A. A novel de novo splice-site mutation in the COL7A1 gene in dominant dystrophic epidermolysis bullosa (DDEB): Specific exon skipping could be a prognostic factor for DDEB pruriginosa. Clin. Exp. Dermatol. 2009, 34, e934–e936. [Google Scholar] [CrossRef]
- Schwieger-Briel, A.; Weibel, L.; Chmel, N.; Leppert, J.; Kernland-Lang, K.; Gruninger, G.; Has, C. A COL7A1 variant leading to in-frame skipping of exon 15 attenuates disease severity in recessive dystrophic epidermolysis bullosa. Br. J. Dermatol. 2015, 173, 1308–1311. [Google Scholar] [CrossRef]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef]
- Aung-Htut, M.; McIntosh, C.; Ham, K.; Pitout, I.; Flynn, L.; Greer, K.; Fletcher, S.; Wilton, S. Systematic Approach to Developing Splice Modulating Antisense Oligonucleotides. Int. J. Mol. Sci. 2019, 20, 5030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilton, S.D.; Lim, L.; Dye, D.; Laing, N. Bandstab: A PCR-based alternative to cloning PCR products. Biotechniques 1997, 22, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef] [PubMed]




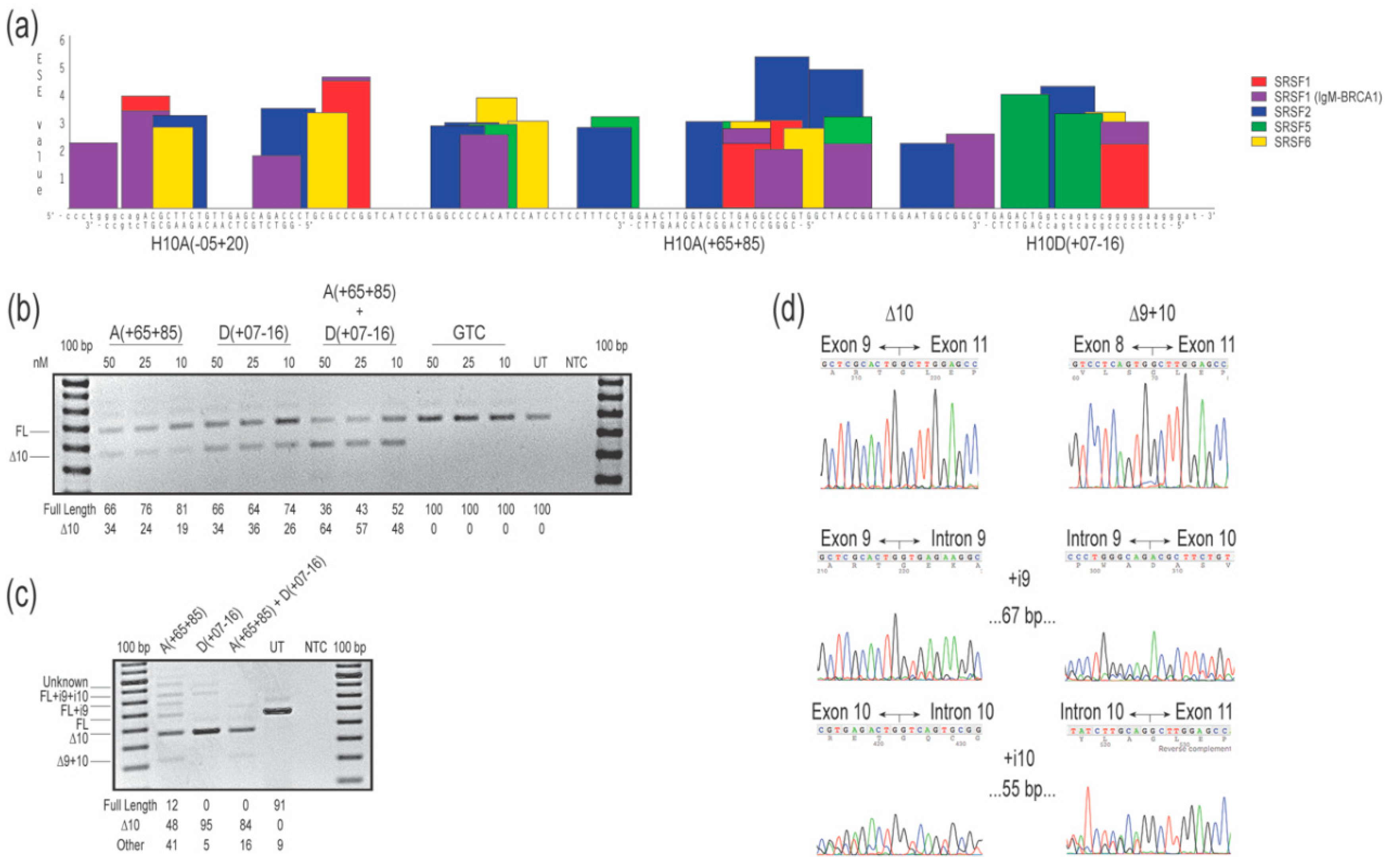{kind=link}
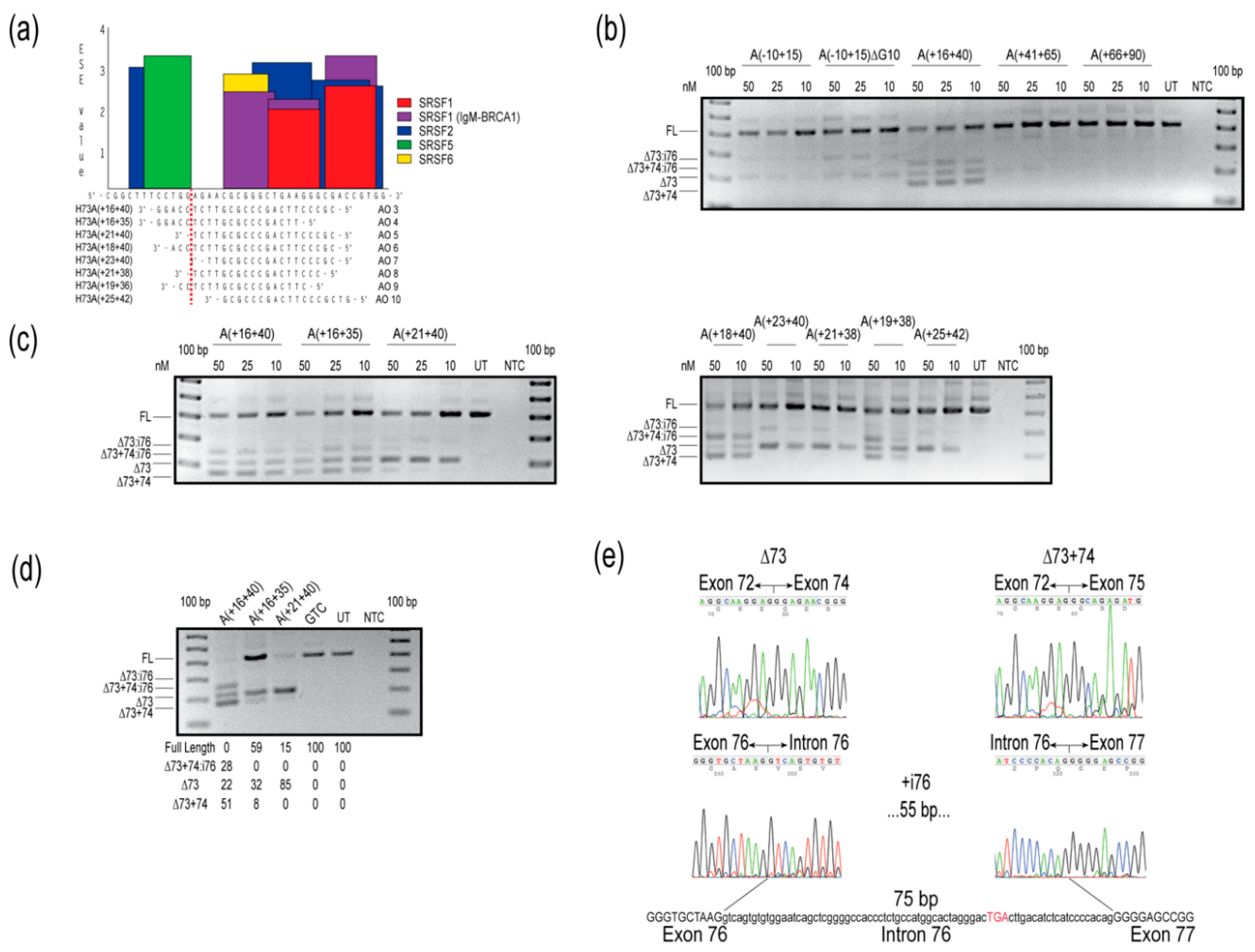{kind=link}
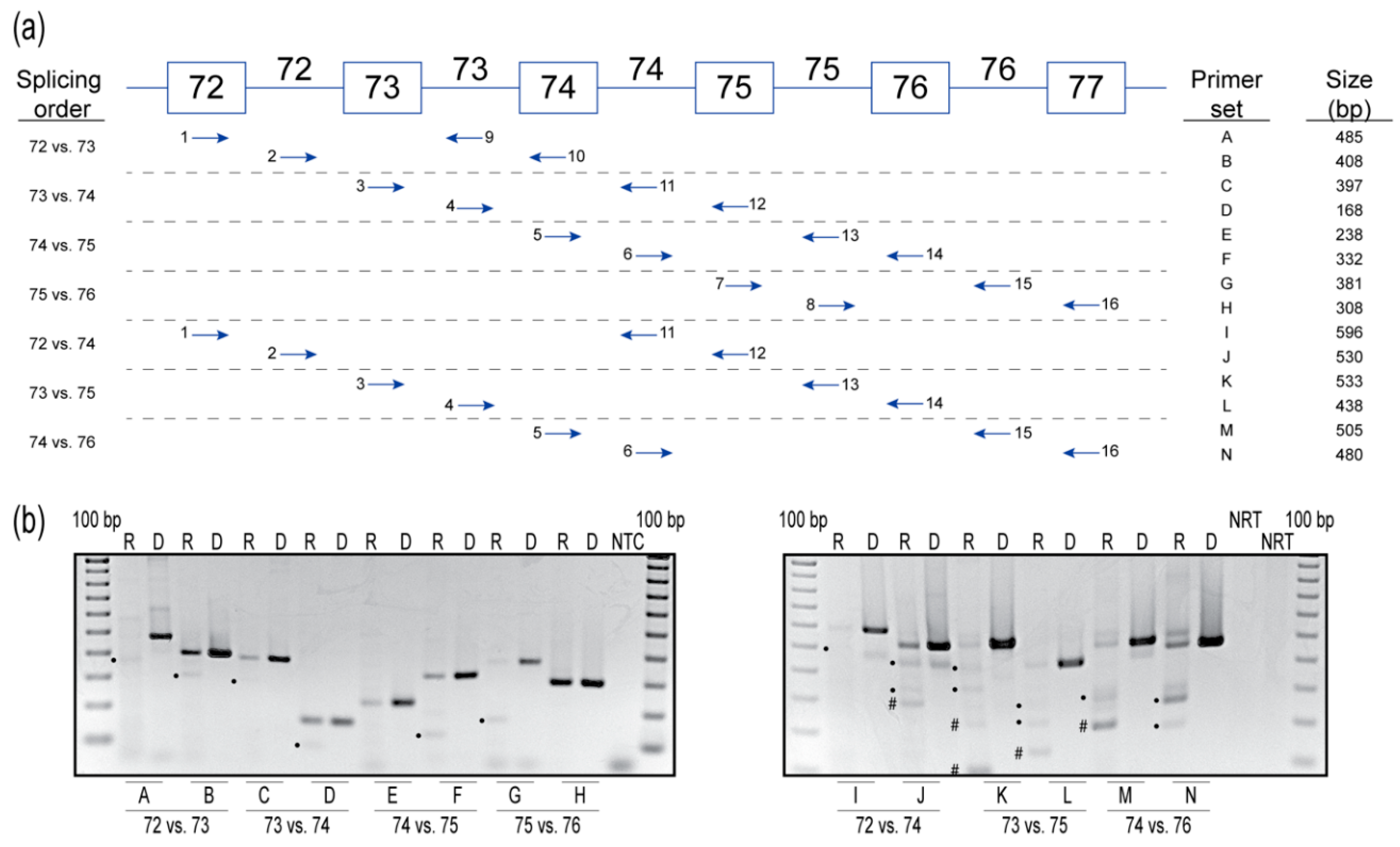{kind=link}
| Intron Pair | Primer Set | Intron Spliced | Spliced Product Expected Sizes (bp) | Order of Splicing |
|---|---|---|---|---|
| 72 vs. 73 | A | 72 | 399 | 72 generally removed before 73 |
| B | 73 1 | 312 | ||
| 73 vs. 74 | C | 73 1 | 301 | No preference 73 = 74 |
| D | 74 1 | 88 | ||
| 74 vs. 75 | E | None | 158 | 75 removed before 74 |
| F | 75 | 134 | ||
| 75 vs. 76 | G | 75 | 183 | 75 removed before 76 |
| H | None | 233 | ||
| 72 vs. 74 | I | 72 1 | 510; 500; 414 | Inconclusive |
| J | ? | 450; 434; 354 | ||
| 73 vs. 75 | K | 73 1 or 74 1, 73+74 1 | 458; 442; 362 | 75 removed before 73 |
| L | 74, 75, 74+75 | 413; 295; 215 | ||
| 74 vs. 76 | M | 74, 75, 74+75 | 429; 311; 231 | 74 and 75 removed before 76 |
| N | 75, 75+76 1 | 405; 282; 207 |
| AO Nomenclature and Annealing Coordinates | Sequence (5′–3′) |
|---|---|
| H10A(-05+20) | GGUCUGCUCAACAGAAGCGUCUGCC |
| H10A(+65+85) * | CGGGCCUCAGGCACCAAGUUC |
| H10D(+07-16) * | CUUCCCCCGCACUGACCAGUCUC |
| H73A(-10+15) | AAAGCCGAUGGGGCCCUGCAGGAGU |
| H73A(-03+17) | GGAAAGCCGAUGGGGCCCUG |
| H73A(+01+20) | CCAGGAAAGCCGAUGGGGCC |
| H73A(+04+23) | UCUCCAGGAAAGCCGAUGGG |
| H73A(+07+26) | CGUUCUCCAGGAAAGCCGAU |
| H73A(+10+29) | CCGCGUUCUCCAGGAAAGCC |
| H73A(+13+32) | AGCCCGCGUUCUCCAGGAAA |
| H73A(+16+35) * | UUCAGCCCGCGUUCUCCAGG |
| H73A(+16+40) * | CGCCCUUCAGCCCGCGUUCUCCAGG |
| H73A(+18+40) | CGCCCUUCAGCCCGCGUUCUCCA |
| H73A(+19+36) | CUUCAGCCCGCGUUCUCC |
| H73A(+21+38) | CCCUUCAGCCCGCGUUCU |
| H73A(+21+40) * | CGCCCUUCAGCCCGCGUUCU |
| H73A(+23+40) | CGCCCUUCAGCCCGCGUU |
| H73A(+25+40) | CGCCCUUCAGCCCGCG |
| H73A(+25+42) | GUCGCCCUUCAGCCCGCG |
| H73A(+27+40) | CGCCCUUCAGCCCG |
| H73A(+41+65) | CCCUGAGGGCCAGGGUCUCCACGGU |
| H73A(+66+90) | CUCCCCAAGGGCCAGACCAGGUGGC |
| H73A(+91+115) | GGCCGGAAGGCCCGGGGGGGCCCCU |
| H73A(+116+140) | CCAGGCUUUCCAGGCUCCCCGGCAA |
| H73A(+141+165) | AGCCCUGCCUGGGAGCCCGGGAAUA |
| H73A(+166+190) | GCCUUCCUGCCUCUCCCACACCCCC |
| H73D(+11-14) | GCCCCCAGCCUCACCCUCUCUCCUG |
| Standard control morpholino * | CCUCUUACCUCAGUUACAAUUUAUA |
| Primer Orientation | Sequence (5′–3′) | Length (bp) | PCR System | Cycling Conditions |
|---|---|---|---|---|
| Exon 8F | AACTGACCATCCAGAATACC | 698 | SSIII One-Step | 55 °C (30 min) and 94 °C (2 min); 30 cycles of 94 °C (30 s), 60 °C (1 min) and 68 °C (2 min) |
| Exon 13R | GTCATCCAAGTCGAATGCT | |||
| Exon 72F | AGATCGTGGAGACCTGGGATG | 521 | SSIV TaKaRa GC II | 94 °C (1 min); 35 cycles of 94 °C (30 s), 60 °C (1 min) and 72 °C (2 min) |
| Exon 77R | CCTGTCTCCTTTGGGACCTTG |
| Primer Number | Orientation | Region | Sequence (5′–3′) |
|---|---|---|---|
| 1 | Forward | Exon 72 | AGATCGTGGAGACCTGGGATG |
| 2 | Forward | Intron 72 | TGAGCAGAAGTGGCTCAGTG |
| 3 | Forward | Exon 73 | ATCGGCTTTCCTGGAGAACG |
| 4 | Forward | Intron 73 | ACCCGCTATTTGCATTTCAG |
| 5 | Forward | Exon 74 | AACGGGGAGAGAAAGGAGAA |
| 6 | Forward | Intron 74 | GCTGCCACCCCATTTTCTTG |
| 7 | Forward | Exon 75 | CCTCCTGGACTCCCTGGAAC |
| 8 | Forward | Intron 75 | GTGTGTGCCATAACCCTGGA |
| 9 | Reverse | Intron 73 | ATGCAAATAGCGGGTGAGGG |
| 10 | Reverse | Exon 74 | CGTTCTCCTTTCTCTCCCCG |
| 11 | Reverse | Intron 74 | CAAGAAAATGGGGTGGCAGC |
| 12 | Reverse | Exon 75 | GTTCCAGGGAGTCCAGGAG |
| 13 | Reverse | Intron 75 | ACCAAGCTAAGGGTGGCTTC |
| 14 | Reverse | Exon 76 | TGTTCTCCAGAGAGTCCAGG |
| 15 | Reverse | Intron 76 | GATGTCAAGTCAGTCCCTAGTGC |
| 16 | Reverse | Exon 77 | CCTGTCTCCTTTGGGACCTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ham, K.A.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D. Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1. Int. J. Mol. Sci. 2020, 21, 7705. https://doi.org/10.3390/ijms21207705
Ham KA, Aung-Htut MT, Fletcher S, Wilton SD. Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1. International Journal of Molecular Sciences. 2020; 21(20):7705. https://doi.org/10.3390/ijms21207705
Chicago/Turabian StyleHam, Kristin A., May Thandar Aung-Htut, Sue Fletcher, and Steve D. Wilton. 2020. "Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1" International Journal of Molecular Sciences 21, no. 20: 7705. https://doi.org/10.3390/ijms21207705
APA StyleHam, K. A., Aung-Htut, M. T., Fletcher, S., & Wilton, S. D. (2020). Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1. International Journal of Molecular Sciences, 21(20), 7705. https://doi.org/10.3390/ijms21207705