Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
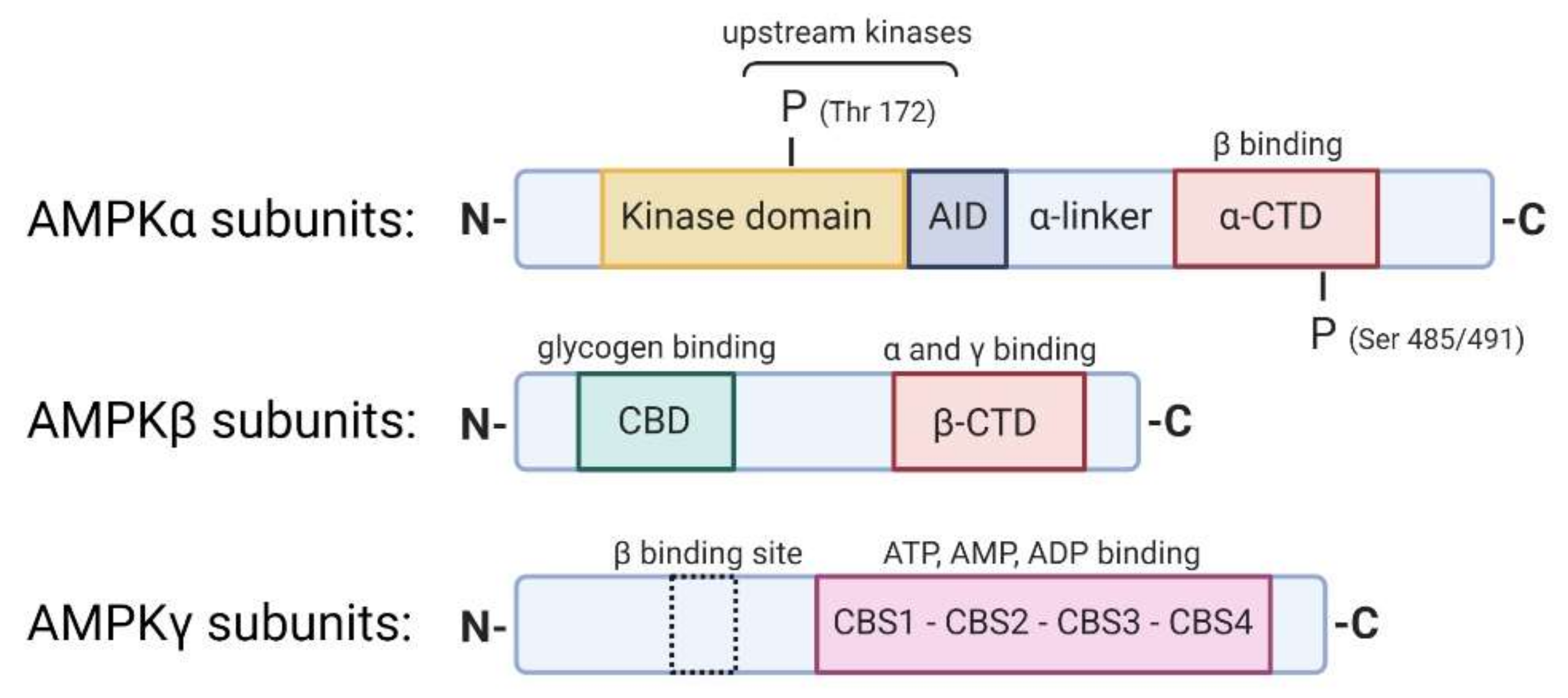
2. AMP-Activated Protein Kinase (AMPK): Structure, Renal Expression, and Function
2.1. An Allosteric Mechanism Activates AMPK
2.2. AMPK Exhibits a Dual Function in Cell Metabolism
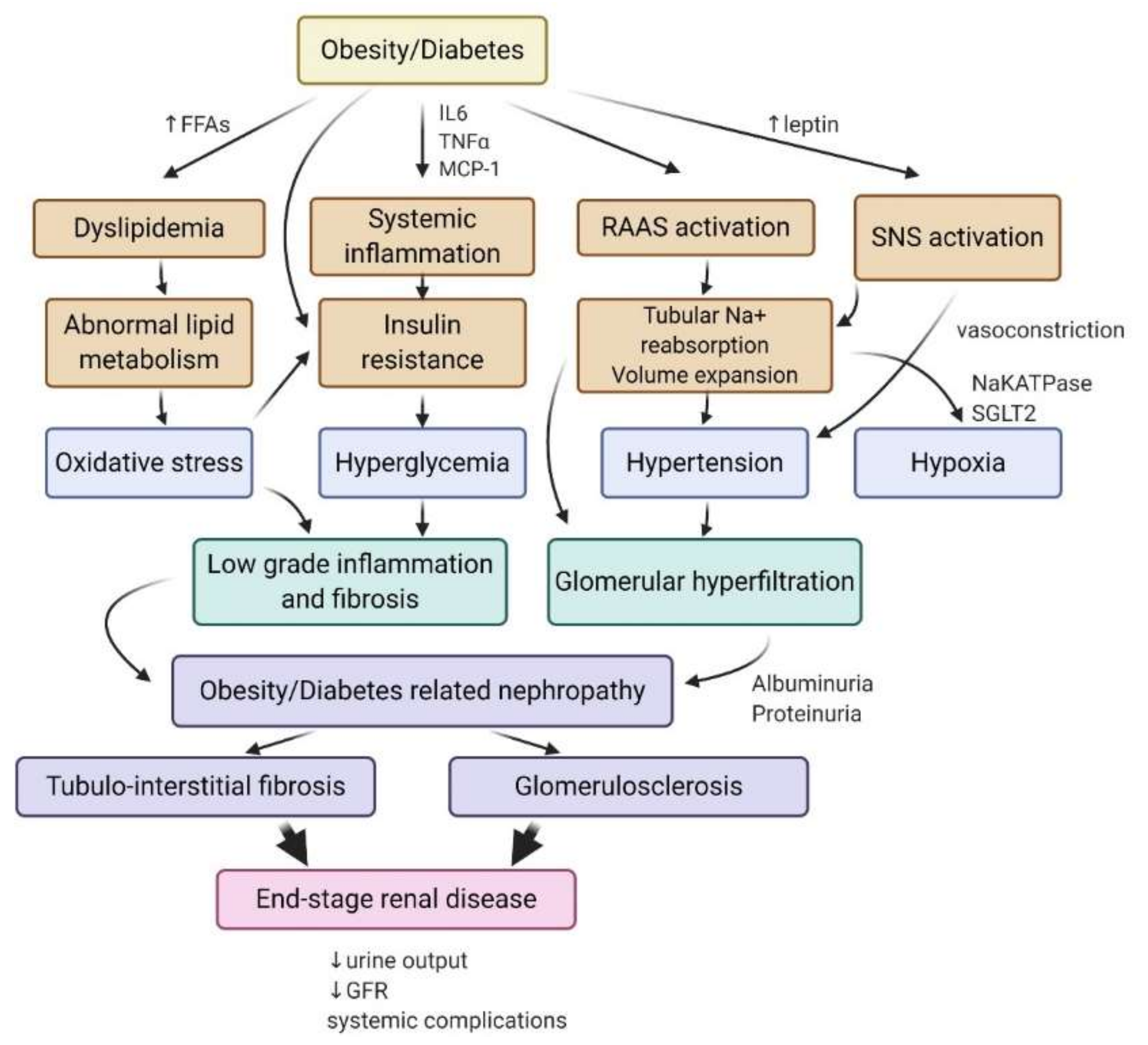
3. AMPK Activity in Obesity and Diabetes-Induced Chronic Kidney Disease (CKD)
4. AMPK in Renal Transport
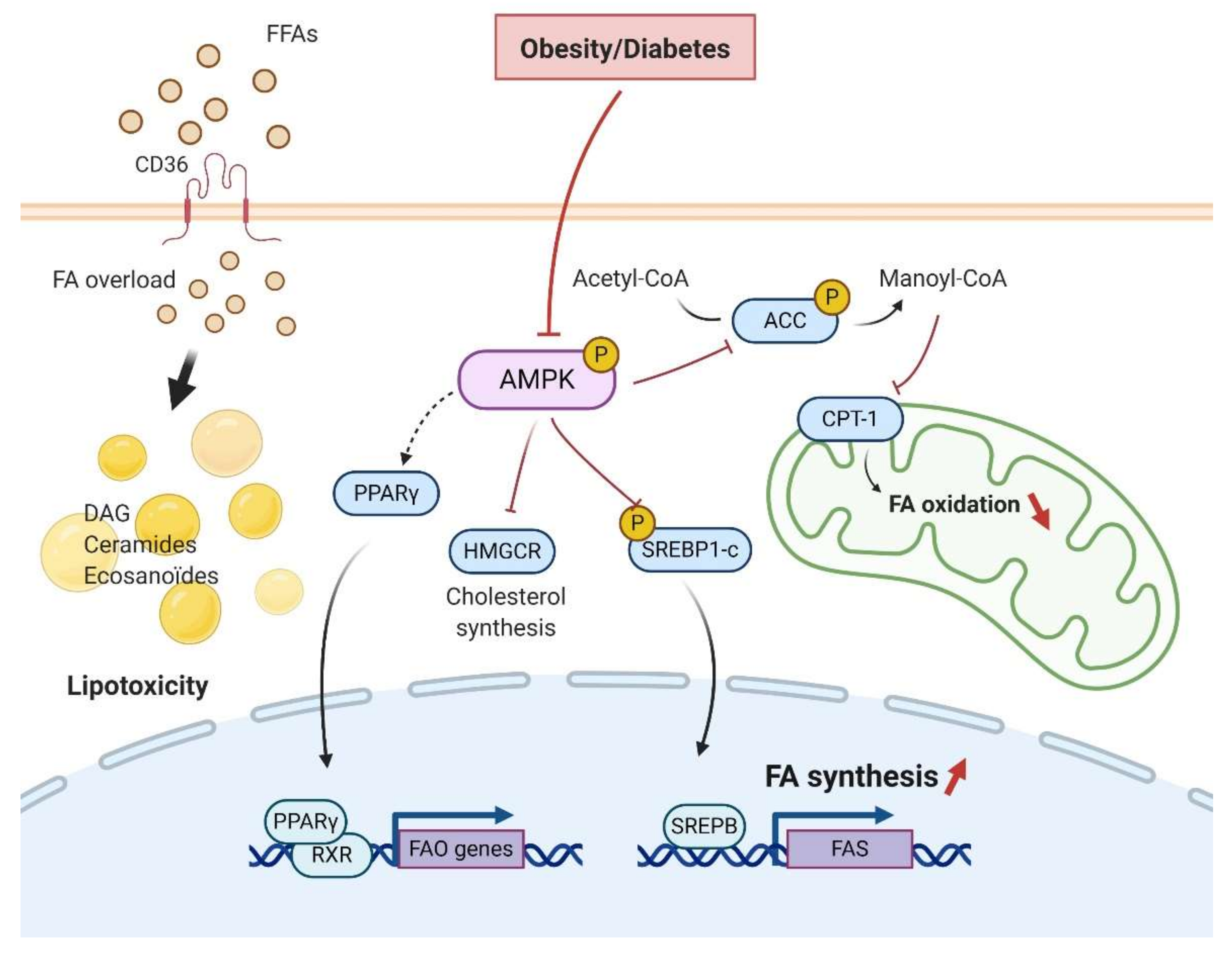
5. AMPK and Renal Lipid Metabolism
6. AMPK and Renal Glucose Metabolism
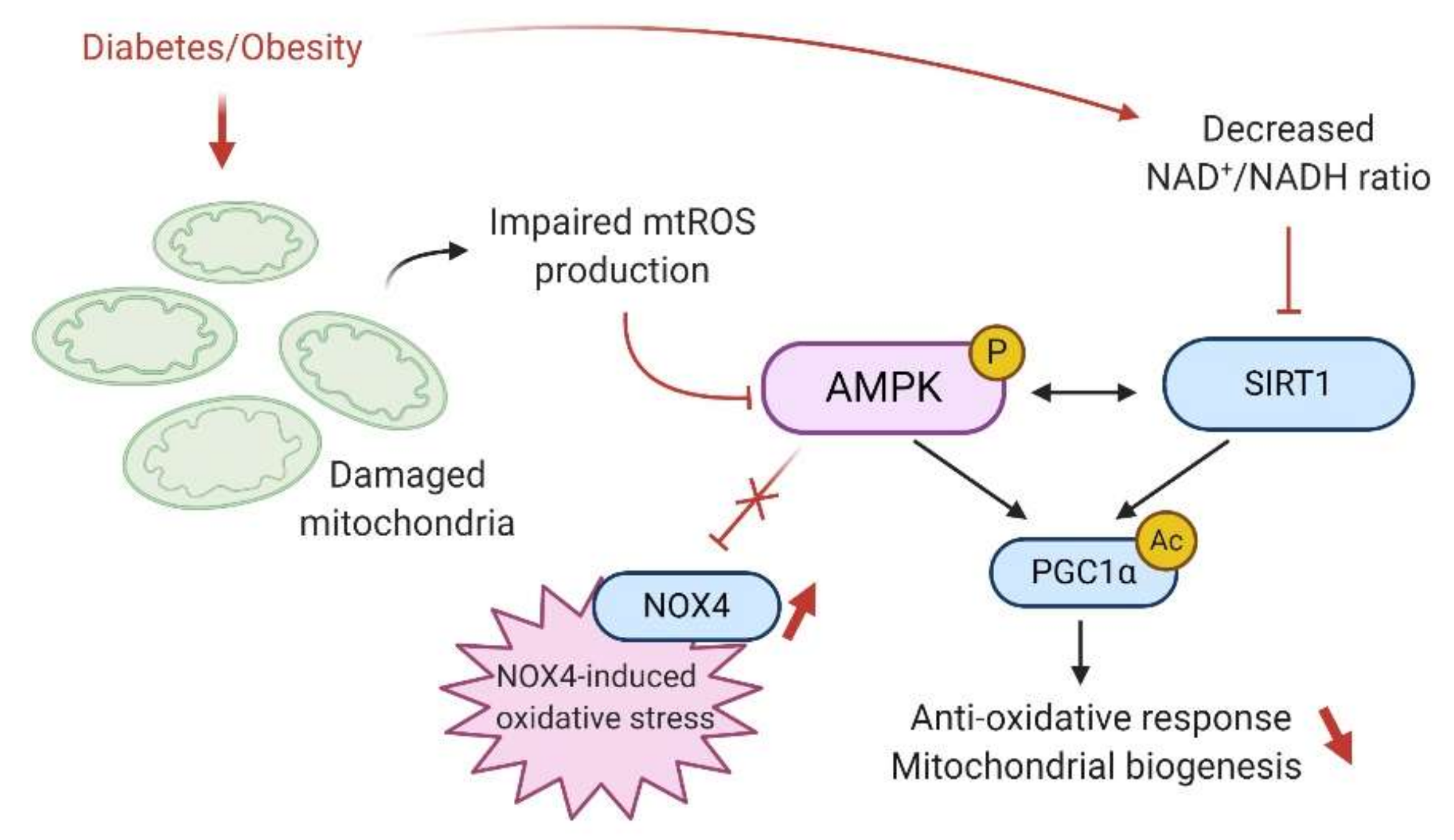
7. AMPK and Renal Mitochondrial Function and Dysfunction
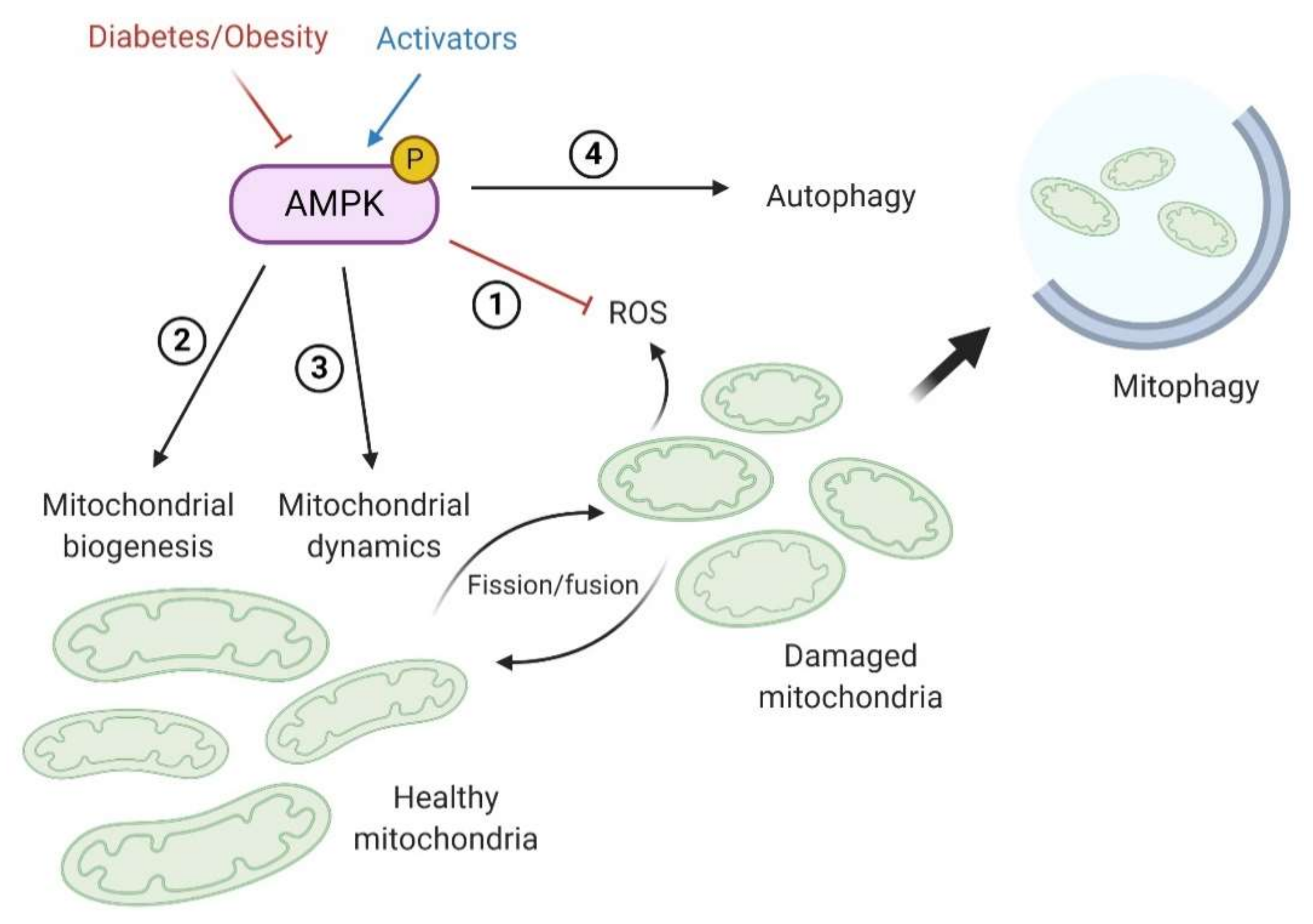
7.1. Mitochondrial Biogenesis and Dynamics
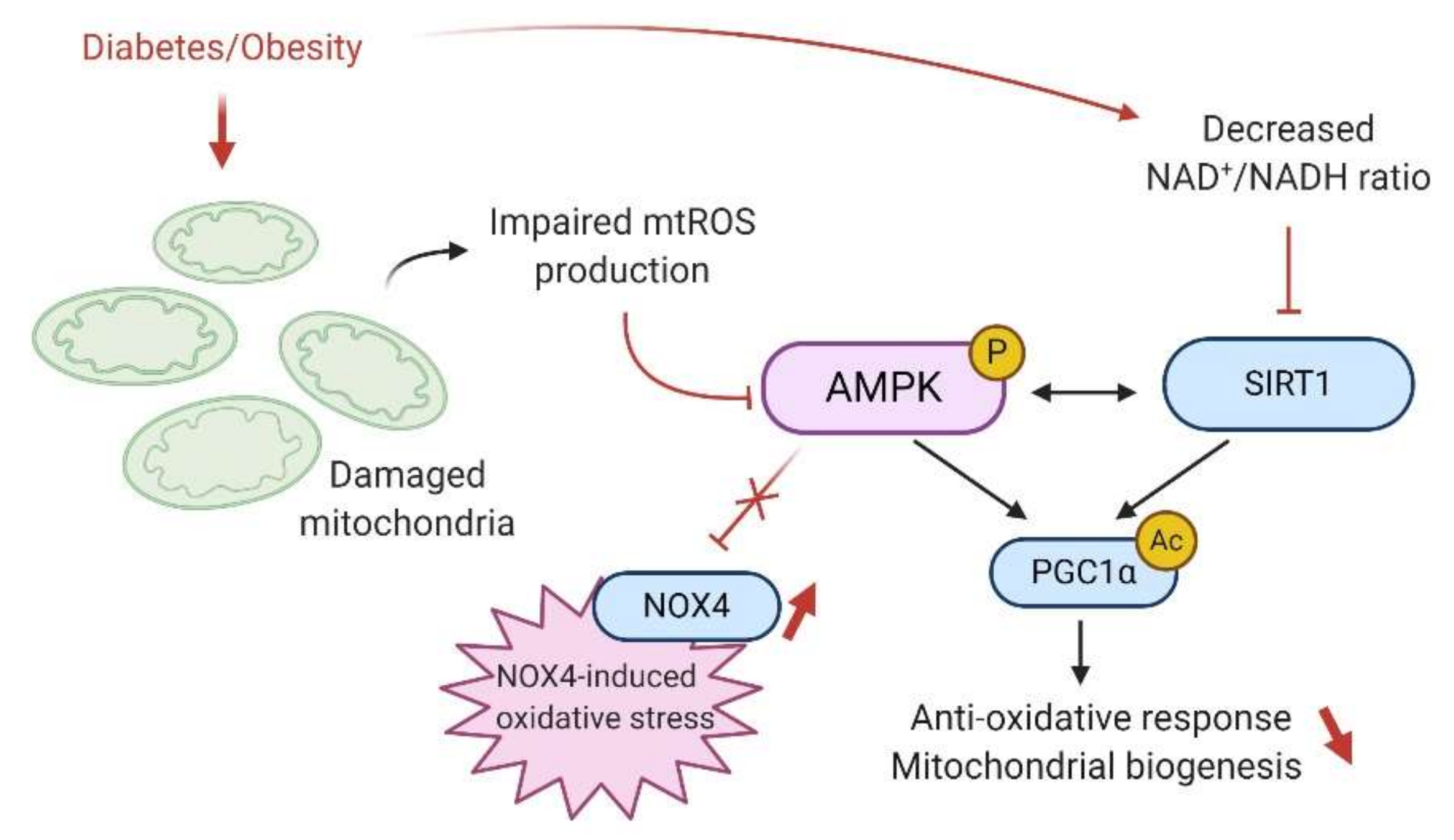
7.2. AMPK and Oxidative Stress
8. AMPK and the Regulation of Renal Autophagy and Mitophagy
9. AMPK and Sirtuins
10. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, V.; Vilme, H.; Maciejewski, M.L.; Boulware, L.E. The Economic Burden of Chronic Kidney Disease and End-Stage Renal Disease. Semin. Nephrol. 2016, 36, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Cornier, M.-A.; Dabelea, D.; Hernandez, T.L.; Lindstrom, R.C.; Steig, A.J.; Stob, N.R.; Van Pelt, R.E.; Wang, H.; Eckel, R.H. The metabolic syndrome. Endocr. Rev. 2008, 29, 777–822. [Google Scholar] [CrossRef] [PubMed]
- Nashar, K.; Egan, B.M. Relationship between chronic kidney disease and metabolic syndrome: Current perspectives. Diabetes Metab. Syndr. Obes. 2014, 7, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Raikou, V.D.; Gavriil, S. Metabolic Syndrome and Chronic Renal Disease. Diseases 2018, 6, 12. [Google Scholar] [CrossRef]
- Thomas, G.; Sehgal, A.R.; Kashyap, S.R.; Srinivas, T.R.; Kirwan, J.P.; Navaneethan, S.D. Metabolic syndrome and kidney disease: A systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 2364–2373. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, C.; Borrelli, S.; Minutolo, R.; Chiodini, P.; De Nicola, L.; Conte, G. A systematic review and meta-analysis suggests obesity predicts onset of chronic kidney disease in the general population. Kidney Int. 2017, 91, 1224–1235. [Google Scholar] [CrossRef]
- Whaley-Connell, A.; Sowers, J.R. Obesity and kidney disease: From population to basic science and the search for new therapeutic targets. Kidney Int. 2017, 92, 313–323. [Google Scholar] [CrossRef]
- Ejerblad, E. Obesity and Risk for Chronic Renal Failure. J. Am. Soc. Nephrol. 2006, 17, 1695–1702. [Google Scholar] [CrossRef]
- Bhupathiraju, S.N.; Hu, F.B. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ. Res. 2016, 118, 1723–1735. [Google Scholar] [CrossRef]
- Maric-Bilkan, C. Obesity and diabetic kidney disease. Med. Clin. N. Am. 2013, 97, 59–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, A.V.; Okada, S.; Sharma, K. Obesity related kidney disease. Curr. Diabetes Rev. 2011, 7, 41–49. [Google Scholar] [CrossRef]
- Goumenos, D.S.; Kawar, B.; El Nahas, M.; Conti, S.; Wagner, B.; Spyropoulos, C.; Vlachojannis, J.G.; Benigni, A.; Kalfarentzos, F. Early histological changes in the kidney of people with morbid obesity. Nephrol. Dial. Transplant. 2009, 24, 3732–3738. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Choi, M.-S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declèves, A.-E.; Sharma, K. Obesity and kidney disease: Differential effects of obesity on adipose tissue and kidney inflammation and fibrosis. Curr. Opin. Nephrol. Hypertens. 2015, 24, 28–36. [Google Scholar] [CrossRef] [Green Version]
- DeMarco, V.G.; Aroor, A.R.; Sowers, J.R. The pathophysiology of hypertension in patients with obesity. Nat. Rev. Endocrinol. 2014, 10, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Jorge, R.-F.; Rodrigo, D.-A.; Maria Ximena, C.-B.; Victor, L.-M.; Emilio, A.-F.; Nehomar, P.-G.; Jose, C.; José, C.; Manuel, C.; Amable, D.; et al. SGLT2 Inhibitors and nephroprotection in diabetic kidney disease: From mechanisms of action to the latest evidence in the literature. J. Clin. Nephrol. 2020, 4, 044–055. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [Green Version]
- D’Agati, V.D.; Chagnac, A.; de Vries, A.P.J.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef]
- Kaysen, G.A. Dyslipidemia in chronic kidney disease: Causes and consequences. Kidney Int. 2006, 70, S55–S58. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.G.; Bomback, A.S.; Radhakrishnan, J.; Herlitz, L.C.; Stokes, M.B.; Markowitz, G.S.; D’Agati, V.D. The modern spectrum of renal biopsy findings in patients with diabetes. Clin. J. Am. Soc. Nephrol. 2013, 8, 1718–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Han, Y.; Gao, P.; Yang, M.; Xiao, L.; Xiong, X.; Zhao, H.; Tang, C.; Chen, G.; Zhu, X.; et al. Disulfide-bond A oxidoreductase-like protein protects against ectopic fat deposition and lipid-related kidney damage in diabetic nephropathy. Kidney Int. 2019, 95, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Declèves, A.-E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMK-activated kinase in high fat diet–induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, A.P.J.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Barlovic, D.P.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Zhao, J.; Rui, H.-L.; Yang, M.; Sun, L.-J.; Dong, H.-R.; Cheng, H. CD36-Mediated Lipid Accumulation and Activation of NLRP3 Inflammasome Lead to Podocyte Injury in Obesity-Related Glomerulopathy. Mediat. Inflamm. 2019, 2019, 3172647. [Google Scholar]
- Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Kimura, T. High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J. Am. Soc. Nephrol. 2017, 28, 1534–1551. [Google Scholar] [CrossRef]
- Li, L.; Wang, C.; Yang, H.; Liu, S.; Lu, Y.; Fu, P.; Liu, J. Metabolomics reveal mitochondrial and fatty acid metabolism disorders that contribute to the development of DKD in T2DM patients. Mol. Biosyst. 2017, 13, 2392–2400. [Google Scholar] [CrossRef]
- Dugan, L.L.; You, Y.-H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.-E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.R.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Kim, Y.; Choi, B.S.; Kim, Y.-S.; Kim, H.W.; Lim, K.-M.; Kim, M.J.; et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism 2018, 85, 348–360. [Google Scholar] [CrossRef]
- Lee, M.-J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Mahimainathan, L.; Musi, N.; Foretz, M.; Viollet, B.; Weinberg, J.M.; Choudhury, G.G.; et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Ren. Physiol. 2007, 292, F617–F627. [Google Scholar] [CrossRef]
- Eid, A.A.; Ford, B.M.; Block, K.; Kasinath, B.S.; Gorin, Y.; Ghosh-Choudhury, G.; Barnes, J.L.; Abboud, H.E. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 2010, 285, 37503–37512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.H.; Malaga-Dieguez, L.; Chinga, F.; Kang, H.M.; Tao, J.; Reidy, K.; Susztak, K. Deletion of Lkb1 in Renal Tubular Epithelial Cells Leads to CKD by Altering Metabolism. J. Am. Soc. Nephrol. 2016, 27, 439–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasanvand, A.; Amini-Khoei, H.; Jahanabadi, S.; Hadian, M.R.; Abdollahi, A.; Tavangar, S.M.; Mehr, S.E.; Dehpour, A.R. Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through activation of AMPK signaling pathway. J. Nephropathol. 2018, 7, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Muise, E.S.; Haimbach, R.; Sebhat, I.K.; Zhu, Y.; Liu, F.; Souza, S.C.; Kan, Y.; Pinto, S.; Kelley, D.E.; et al. PAN-AMPK Activation Improves Renal Function in a Rat Model of Progressive Diabetic Nephropathy. J. Pharmacol. Exp. Ther. 2019, 371, 45–55. [Google Scholar] [CrossRef]
- Habib, S.L.; Yadav, A.; Kidane, D.; Weiss, R.H.; Liang, S. Novel protective mechanism of reducing renal cell damage in diabetes: Activation AMPK by AICAR increased NRF2/OGG1 proteins and reduced oxidative DNA damage. Cell Cycle 2016, 15, 3048–3059. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Xu, Z.-X.; Ding, Z.; Lu, Y.; Yu, Q.; Werle, K.D.; Zhou, G.; Park, Y.-Y.; Peng, G.; Gambello, M.J.; et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun. 2015, 6, 7926. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121. [Google Scholar] [CrossRef] [Green Version]
- Seo-Mayer, P.W.; Thulin, G.; Zhang, L.; Alves, D.S.; Ardito, T.; Kashgarian, M.; Caplan, M.J. Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am. J. Physiol. Ren. Physiol. 2011, 301, F1346–F1357. [Google Scholar] [CrossRef] [Green Version]
- Salatto, C.T.; Miller, R.A.; Cameron, K.O.; Cokorinos, E.; Reyes, A.; Ward, J.; Calabrese, M.F.; Kurumbail, R.G.; Rajamohan, F.; Kalgutkar, A.S.; et al. Selective Activation of AMPK beta1-Containing Isoforms Improves Kidney Function in a Rat Model of Diabetic Nephropathy. J. Pharmacol. Exp. Ther. 2017, 361, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Hallows, K.R.; Mount, P.F.; Pastor-Soler, N.M.; Power, D.A. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am. J. Physiol. Ren. Physiol. 2010, 298, F1067–F1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, D. Is TAK1 a Direct Upstream Kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef] [Green Version]
- Pulinilkunnil, T.; He, H.; Kong, D.; Asakura, K.; Peroni, O.D.; Lee, A.; Kahn, B.B. Adrenergic regulation of AMP-activated protein kinase in brown adipose tissue in vivo. J. Biol. Chem. 2011, 286, 8798–8809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.L.; Barré, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, K.A.; Valentine, R.J.; Sudit, B.S.; Allen, K.; Dagon, Y.; Kahn, B.B.; Ruderman, N.B.; Saha, A.K. PKD1 Inhibits AMPKα2 through Phosphorylation of Serine 491 and Impairs Insulin Signaling in Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 5664–5675. [Google Scholar] [CrossRef] [Green Version]
- Lazo-Fernandez, Y.; Baile, G.; Meade, P.; Torcal, P.; Martinez, L.; Ibanez, C.; Bernal, M.L.; Viollet, B.; Gimenez, I. Kidney-specific genetic deletion of both AMPK alpha-subunits causes salt and water wasting. Am. J. Physiol. Ren. Physiol. 2017, 312, F352–F365. [Google Scholar] [CrossRef]
- Kohlstedt, K.; Trouvain, C.; Boettger, T.; Shi, L.; Fisslthaler, B.; Fleming, I. AMP-activated protein kinase regulates endothelial cell angiotensin-converting enzyme expression via p53 and the post-transcriptional regulation of microRNA-143/145. Circ. Res. 2013, 112, 1150–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.-L.; Hsu, C.-N. AMP-Activated Protein Kinase as a Reprogramming Strategy for Hypertension and Kidney Disease of Developmental Origin. Int. J. Mol. Sci. 2018, 19, 1744. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Mudgett, J.; Bou-About, G.; Champy, M.-F.; Jacobs, H.; Monassier, L.; Pavlovic, G.; Sorg, T.; Herault, Y.; Petit-Demouliere, B.; et al. Physiological Expression of AMPKgamma2RG Mutation Causes Wolff-Parkinson-White Syndrome and Induces Kidney Injury in Mice. J. Biol. Chem. 2016, 291, 23428–23439. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B.; Horman, S.; Leclerc, J.; Lantier, L.; Foretz, M.; Billaud, M.; Giri, S.; Andreelli, F. AMPK inhibition in health and disease. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 276–295. [Google Scholar] [CrossRef] [Green Version]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchankova, G.; Nelson, L.E.; Gerhart-Hines, Z.; Kelly, M.; Gauthier, M.-S.; Saha, A.K.; Ido, Y.; Puigserver, P.; Ruderman, N.B. Concurrent regulation of AMP-activated protein kinase and SIRT1 in mammalian cells. Biochem. Biophys. Res. Commun. 2009, 378, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, H.; Sasaki, E.; Nomura, N.; Mori, T.; Minamishima, Y.A.; Yoshizaki, Y.; Takahashi, N.; Furusho, T.; Arai, Y.; Mandai, S.; et al. Failure to sense energy depletion may be a novel therapeutic target in chronic kidney disease. Kidney Int. 2019, 95, 123–137. [Google Scholar] [CrossRef]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Valentine, R.J.; Coughlan, K.A.; Ruderman, N.B.; Saha, A.K. Insulin inhibits AMPK activity and phosphorylates AMPK Ser485/491 through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch. Biochem. Biophys. 2014, 562, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Niu, M.; Xiang, L.; Liu, Y.; Zhao, Y.; Yuan, J.; Dai, X.; Chen, H. Adiponectin induced AMP-activated protein kinase impairment mediates insulin resistance in Bama mini-pig fed high-fat and high-sucrose diet. Asian-Australas. J. Anim. Sci. 2017, 30, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, C.; Durand, A.; Vidal, H.; Rieusset, J. Changes in adiponectin, its receptors and AMPK activity in tissues of diet-induced diabetic mice. Diabetes Metab. 2008, 34, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Sun, Y.; Gao, E.; Wei, X.; Lau, W.B.; Zheng, Q.; Wang, Y.; Yuan, Y.; Wang, X.; Tao, L.; et al. Reduced cardioprotective action of adiponectin in high-fat diet-induced type II diabetic mice and its underlying mechanisms. Antioxid. Redox Signal. 2011, 15, 1779–1788. [Google Scholar] [CrossRef] [Green Version]
- Lyons, C.L.; Roche, H.M. Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation. Int. J. Mol. Sci. 2018, 19, 3092. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.W.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Görgün, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajani, R.; Pastor-soler, N.M.; Hallows, K.R. Role of AMP-activated protein kinase in kidney tubular transport, metabolism, and disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 375–383. [Google Scholar] [CrossRef]
- Glosse, P.; Föller, M. AMP-Activated Protein Kinase (AMPK)-Dependent Regulation of Renal Transport. Int. J. Mol. Sci. 2018, 19, 3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spires, D.; Manis, A.D.; Staruschenko, A. Ion channels and transporters in diabetic kidney disease. Curr. Top. Membr. 2019, 83, 353–396. [Google Scholar] [PubMed]
- Liamis, G.; Liberopoulos, E.; Barkas, F.; Elisaf, M. Diabetes mellitus and electrolyte disorders. World J. Clin. Cases 2014, 2, 488–496. [Google Scholar] [CrossRef]
- Benziane, B.; Björnholm, M.; Pirkmajer, S.; Austin, R.L.; Kotova, O.; Viollet, B.; Zierath, J.R.; Chibalin, A.V. Activation of AMP-activated protein kinase stimulates Na+,K+-ATPase activity in skeletal muscle cells. J. Biol. Chem. 2012, 287, 23451–23463. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Zhu, S.; Guan, H.; Zheng, Y.; Li, F.; Zhang, X.; Guo, H.; Wang, X.; Ye, Z. AMPK alleviates high uric acid-induced Na+-K+-ATPase signaling impairment and cell injury in renal tubules. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Bhalla, V.; Oyster, N.M.; Fitch, A.C.; Wijngaarden, M.A.; Neumann, D.; Schlattner, U.; Pearce, D.; Hallows, K.R. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 2006, 281, 26159–26169. [Google Scholar] [CrossRef] [Green Version]
- Carattino, M.D.; Edinger, R.S.; Grieser, H.J.; Wise, R.; Neumann, D.; Schlattner, U.; Johnson, J.P.; Kleyman, T.R.; Hallows, K.R. Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 2005, 280, 17608–17616. [Google Scholar] [CrossRef] [Green Version]
- Fraser, S.A.; Gimenez, I.; Cook, N.; Jennings, I.; Katerelos, M.; Katsis, F.; Levidiotis, V.; Kemp, B.E.; Power, D.A. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem. J. 2007, 405, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, S.A.; Choy, S.-W.; Pastor-Soler, N.M.; Li, H.; Davies, M.R.P.; Cook, N.; Katerelos, M.; Mount, P.F.; Gleich, K.; McRae, J.L.; et al. AMPK couples plasma renin to cellular metabolism by phosphorylation of ACC1. Am. J. Physiol. Ren. Physiol. 2013, 305, F679–F690. [Google Scholar] [CrossRef] [Green Version]
- Dërmaku-Sopjani, M.; Almilaji, A.; Pakladok, T.; Munoz, C.; Hosseinzadeh, Z.; Blecua, M.; Sopjani, M.; Lang, F. Down-regulation of the Na+-coupled phosphate transporter NaPi-IIa by AMP-activated protein kinase. Kidney Blood Press. Res. 2013, 37, 547–556. [Google Scholar] [CrossRef] [PubMed]
- King, J.D.J.; Fitch, A.C.; Lee, J.K.; McCane, J.E.; Mak, D.-O.D.; Foskett, J.K.; Hallows, K.R. AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am. J. Physiol. Cell Physiol. 2009, 297, C94–C101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deji, N.; Kume, S.; Araki, S.; Soumura, M.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Koya, D.; Haneda, M.; et al. Structural and functional changes in the kidneys of high-fat diet-induced obese mice. Am. J. Physiol. Ren. Physiol. 2009, 296, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Kume, S.; Uzu, T.; Araki, S.-I.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Kubota, N.; Terauchi, Y.; Kadowaki, T.; et al. Role of Altered Renal Lipid Metabolism in the Development of Renal Injury Induced by a High-Fat Diet. J. Am. Soc. Nephrol. 2007, 18, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Proctor, G.; Jiang, T.; Iwahashi, M.; Wang, Z.; Li, J.; Levi, M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 2006, 55, 2502–2509. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Halaihel, N.; Zhang, W.; Rogers, H.; Levi, M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J. Biol. Chem. 2002, 277, 18919–18927. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Liebman, S.E.; Lucia, M.S.; Li, J.; Levi, M. Role of altered renal lipid metabolism and the sterol regulatory element binding proteins in the pathogenesis of age-related renal disease. Kidney Int. 2005, 68, 2608–2620. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced Obesity in C57BL/6J Mice Causes Increased Renal Lipid Accumulation and Glomerulosclerosis via a Sterol Regulatory Element-binding Protein-1c-dependent Pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y.; et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat. Commun. 2019, 10, 623. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.-J.; Kwon, S.-W.; Jung, B.-H.; Oh, S.-H.; Lee, B.-H. Role of the AMPK/SREBP-1 pathway in the development of orotic acid-induced fatty liver. J. Lipid Res. 2011, 52, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.-J.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorotea, D.; Koya, D.; Ha, H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front. Pharmacol. 2020, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-C.; Wu, M.-S.; Lin, Y.-F.; Chen, C.-R.; Chen, C.-Y.; Chen, C.-J.; Shen, C.-C.; Chen, K.-C.; Peng, C.-C. Nifedipine Modulates Renal Lipogenesis via the AMPK-SREBP Transcriptional Pathway. Int. J. Mol. Sci. 2019, 20, 1570. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Wang, W.; Miner, J.; Fromm, M. Cross regulation of sirtuin 1, AMPK, and PPARγ in conjugated linoleic acid treated adipocytes. PLoS ONE 2012, 7, e48874. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Kim, S.G. AMPK-Dependent Metabolic Regulation by PPAR Agonists. PPAR Res. 2010, 2010, 549101. [Google Scholar] [CrossRef]
- Sozio, M.S.; Lu, C.; Zeng, Y.; Liangpunsakul, S.; Crabb, D.W. Activated AMPK inhibits PPAR-{alpha} and PPAR-{gamma} transcriptional activity in hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G739–G747. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Seo, A.; Park, D.; Tao, J.; Sharma, K.; et al. Defective fatty acid oxidation in renal tubular epithelial cells plays a key role in kidney fibrosis development HHS Public Access. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Sas, K.M.; Nair, V.; Byun, J.; Kayampilly, P.; Zhang, H.; Saha, J.; Brosius, F.C., 3rd; Kretzler, M.; Pennathur, S. Targeted Lipidomic and Transcriptomic Analysis Identifies Dysregulated Renal Ceramide Metabolism in a Mouse Model of Diabetic Kidney Disease. J. Proteom. Bioinform. 2015. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Bandet, C.L.; Tan-Chen, S.; Bourron, O.; Le Stunff, H.; Hajduch, E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int. J. Mol. Sci. 2019, 20, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heathcote, H.R.; Mancini, S.J.; Strembitska, A.; Jamal, K.; Reihill, J.A.; Palmer, T.M.; Gould, G.W.; Salt, I.P. Protein kinase C phosphorylates AMP-activated protein kinase α1 Ser487. Biochem. J. 2016, 473, 4681–4697. [Google Scholar] [CrossRef] [Green Version]
- Declèves, A.-E.; Mathew, A.V.; Armando, A.M.; Han, X.; Dennis, E.A.; Quehenberger, O.; Sharma, K. AMP-activated protein kinase activation ameliorates eicosanoid dysregulation in high-fat-induced kidney disease in mice. J. Lipid Res. 2019, 60, 937–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogacka, D.; Piwkowska, A.; Audzeyenka, I.; Angielski, S.; Jankowski, M. Involvement of the AMPK-PTEN pathway in insulin resistance induced by high glucose in cultured rat podocytes. Int. J. Biochem. Cell Biol. 2014, 51, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Jauregui, A.N.; Merscher-Gomez, S.; Maiguel, D.; Muresan, C.; Mitrofanova, A.; Diez-Sampedro, A.; Szust, J.; Yoo, T.-H.; Villarreal, R.; et al. Podocyte-specific GLUT4-deficient mice have fewer and larger podocytes and are protected from diabetic nephropathy. Diabetes 2014, 63, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Özel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551–1566.e5. [Google Scholar] [CrossRef] [Green Version]
- Rachubik, P.; Szrejder, M.; Rogacka, D.; Audzeyenka, I.; Rychłowski, M.; Angielski, S.; Piwkowska, A. The TRPC6-AMPK Pathway is Involved in Insulin-Dependent Cytoskeleton Reorganization and Glucose Uptake in Cultured Rat Podocytes. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 51, 393–410. [Google Scholar] [CrossRef]
- Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2019, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chang, J.-H.; Buckley, A.F.; Spurney, R.F. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int. 2019, 95, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef]
- Rogacka, D.; Piwkowska, A.; Audzeyenka, I.; Angielski, S.; Jankowski, M. SIRT1-AMPK crosstalk is involved in high glucose-dependent impairment of insulin responsiveness in primary rat podocytes. Exp. Cell Res. 2016, 349, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Shati, A.A. Salidroside ameliorates diabetic nephropathy in rats by activating renal AMPK/SIRT1 signaling pathway. J. Food Biochem. 2020, 44, e13158. [Google Scholar] [CrossRef]
- Vallon, V. Molecular determinants of renal glucose reabsorption. Focus on “Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2”. Am. J. Physiol. Cell Physiol. 2011, 300, C6–C8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M. SGLT2 Inhibitors Produce Cardiorenal Benefits by Promoting Adaptive Cellular Reprogramming to Induce a State of Fasting Mimicry: A Paradigm Shift in Understanding Their Mechanism of Action. Diabetes Care 2020, 43, 508–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilla-Madrigal, R.; Barrenetxe, J.; Moreno-Aliaga, M.J.; Lostao, M.P. EPA blocks TNF-α-induced inhibition of sugar uptake in Caco-2 cells via GPR120 and AMPK. J. Cell. Physiol. 2018, 233, 2426–2433. [Google Scholar] [CrossRef]
- Di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.-P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, Y.; Liu, X.; Dagda, R.K.; Zhang, Y. How AMPK and PKA Interplay to Regulate Mitochondrial Function and Survival in Models of Ischemia and Diabetes. Oxid. Med. Cell. Longev. 2017, 2017, 4353510. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Root-McCaig, J.; Castellani, L.; Kemp, B.E.; Steinberg, G.R.; Wright, D.C. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 2014, 22, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullon, P.; Marin-Aguilar, F.; Roman-Malo, L. AMPK/Mitochondria in Metabolic Diseases. Exp. Suppl. 2016, 107, 129–152. [Google Scholar] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [PubMed] [Green Version]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Dong, Z. Mitochondrial dysfunction in obesity-related kidney disease: A novel therapeutic target. Kidney Int. 2016, 90, 930–933. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S. V Protection of mitochondria prevents high fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.Y.; Kang, J.M.; Jun, H.H.; Kim, D.-J.; Park, S.H.; Sung, M.J.; Heo, J.H.; Yang, D.H.; Lee, S.H.; Lee, S.-Y. Chloroquine and amodiaquine enhance AMPK phosphorylation and improve mitochondrial fragmentation in diabetic tubulopathy. Sci. Rep. 2018, 8, 8774. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.J.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-Y.; Kang, J.M.; Kim, D.-J.; Park, S.H.; Jeong, H.Y.; Lee, Y.H.; Kim, Y.G.; Yang, D.H.; Lee, S.H. PGC1α Activators Mitigate Diabetic Tubulopathy by Improving Mitochondrial Dynamics and Quality Control. J. Diabetes Res. 2017, 2017, 6483572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Banal, C.; Chow, B.S.M.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius-Perez, S.; Torres-Cuevas, I.; Millan, I.; Ortega, A.L.; Perez, S. PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, C.; Ehrenshaft, M.; Cleland, E.; Stadler, K. High-fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E1047–E1058. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K. Mitochondrial Hormesis and Diabetic Complications. Diabetes 2015, 64, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, F.G.; Virág, L.; Jagtap, P.; Szabó, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH oxidases, reactive oxygen species, and the kidney: Friend and foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, F.-R.; Wang, J.-N.; Gao, L.; Jiang, L.; Li, H.-D.; Ma, Q.; Liu, X.-Q.; Wei, B.; Zhou, L.; et al. Nox4 in renal diseases: An update. Free Radic. Biol. Med. 2018, 124, 466–472. [Google Scholar] [CrossRef]
- Muñoz, M.; López-Oliva, M.E.; Rodríguez, C.; Martínez, M.P.; Sáenz-Medina, J.; Sánchez, A.; Climent, B.; Benedito, S.; García-Sacristán, A.; Rivera, L.; et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020, 28, 101330. [Google Scholar] [CrossRef]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.J.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010, 299, F1348–F1358. [Google Scholar] [CrossRef]
- Jiang, F.; Lim, H.K.; Morris, M.J.; Prior, L.; Velkoska, E.; Wu, X.; Dusting, G.J. Systemic upregulation of NADPH oxidase in diet-induced obesity in rats. Redox Rep. 2011, 16, 223–229. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Xiong, J.; Nie, L.; Yu, Y.; Guan, X.; Xu, X.; Xiao, T.; Yang, K.; Liu, L.; Zhang, D.; et al. Resveratrol inhibits renal interstitial fibrosis in diabetic nephropathy by regulating AMPK/NOX4/ROS pathway. J. Mol. Med. 2016, 94, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, A.; Peixoto, E.B.M.I.; Silva, K.C.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Increase in AMPK brought about by cocoa is renoprotective in experimental diabetes mellitus by reducing NOX4/TGFβ-1 signaling. J. Nutr. Biochem. 2014, 25, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, A.; Peixoto, E.B.M.I.; Silva, K.C.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Inactivation of AMPK mediates high phosphate-induced extracellular matrix accumulation via NOX4/TGFß-1 signaling in human mesangial cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 34, 1260–1272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, C.-M.; Li, J.; Meng, Z.-J.; Wei, S.-N.; Li, J.; Bucala, R.; Li, Y.-L.; Chen, L. Berberine protects against palmitate-induced endothelial dysfunction: Involvements of upregulation of AMPK and eNOS and downregulation of NOX4. Mediat. Inflamm. 2013, 2013, 260464. [Google Scholar] [CrossRef]
- Kim, H.-R.; Kim, S.-Y. Perilla frutescens Sprout Extract Protect Renal Mesangial Cell Dysfunction against High Glucose by Modulating AMPK and NADPH Oxidase Signaling. Nutrients 2019, 11, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Lee, D.Y.; Mariappan, M.M.; Feliers, D.; Ghosh-Choudhury, G.; Abboud, H.E.; Gorin, Y.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced NADPH oxidase 4 expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells. J. Biol. Chem. 2017, 292, 5665–5675. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xia, T.; Li, R.; Tse, G.; Liu, T.; Li, G. Renal-Protective Effects of the Peroxisome Proliferator-Activated Receptor-γ Agonist Pioglitazone in ob/ob Mice. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 1582–1589. [Google Scholar] [CrossRef]
- Wang, Y.; An, W.; Zhang, F.; Niu, M.; Liu, Y.; Shi, R. Nebivolol ameliorated kidney damage in Zucker diabetic fatty rats by regulation of oxidative stress/NO pathway: Comparison with captopril. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1135–1148. [Google Scholar] [CrossRef]
- Eid, A.A.; Lee, D.-Y.; Roman, L.J.; Khazim, K.; Gorin, Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol. Cell. Biol. 2013, 33, 3439–3460. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Ma, Y.; Chen, Z.; Hu, J.; Chen, C.; Fan, Y.; Liang, W.; Ding, G. Sestrin-2 regulates podocyte mitochondrial dysfunction and apoptosis under high-glucose conditions via AMPK. Int. J. Mol. Med. 2020, 45, 1361–1372. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A.; Shah, S.V. Autophagy Function and Regulation in Kidney Disease. Biomolecules 2020, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.-E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzen, A.M.; Madsen, A.B.; Kleinert, M.; Treebak, J.T.; Lundsgaard, A.-M.; Jensen, T.E.; Richter, E.A.; Wojtaszewski, J.; Kiens, B.; Frøsig, C. Regulation of autophagy in human skeletal muscle: Effects of exercise, exercise training and insulin stimulation. J. Physiol. 2016, 594, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhao, X.; Hu, Y.; Sun, H.; Gong, G.; Huang, X.; Chen, X.; Xia, M.; Sun, C.; Huang, Q.; et al. Autophagy regulates the degeneration of the auditory cortex through the AMPK-mTOR-ULK1 signaling pathway. Int. J. Mol. Med. 2018, 41, 2086–2098. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Hwang, J.S.; Lai, T.H.; Zada, S.; Nguyen, H.Q.; Pham, T.M.; Yun, M.; Kim, D.R. Co-Expression Network Analysis of AMPK and Autophagy Gene Products during Adipocyte Differentiation. Int. J. Mol. Sci. 2018, 19, 1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Green, A.S.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D.; Tamburini, J. LKB1/AMPK/mTOR signaling pathway in hematological malignancies: From metabolism to cancer cell biology. Cell Cycle 2011, 10, 2115–2120. [Google Scholar] [CrossRef]
- Bork, T.; Liang, W.; Yamahara, K.; Lee, P.; Tian, Z.; Liu, S.; Schell, C.; Thedieck, K.; Hartleben, B.; Patel, K.; et al. Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy 2019, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Gan, Y.; Huang, W.F.; Wu, H.-L.; Zhang, X.-Q.; Zheng, H.J.; Liu, H.-F. Lysosome restoration to activate podocyte autophagy: A new therapeutic strategy for diabetic kidney disease. Cell Death Dis. 2019, 10, 806. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, S.; Hosojima, M.; Kaneko, R.; Aoki, H.; Nakano, D.; Sasagawa, T.; Kabasawa, H.; Kaseda, R.; Yasukawa, R.; Ishikawa, T.; et al. Megalin-Mediated Tubuloglomerular Alterations in High-Fat Diet-Induced Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1996–2008. [Google Scholar] [CrossRef] [Green Version]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, N.X.Y.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.W.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020, 2, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Min, Q.; Ouyang, C.; Lee, J.; He, C.; Zou, M.-H.; Xie, Z. AMPK activation prevents excess nutrient-induced hepatic lipid accumulation by inhibiting mTORC1 signaling and endoplasmic reticulum stress response. Biochim. Biophys. Acta 2014, 1842, 1844–1854. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A. Molecular Interactions Between Reactive Oxygen Species and Autophagy in Kidney Disease. Int. J. Mol. Sci. 2019, 20, 3791. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.; Kim, K.; Uddin, M.J.; Lee, G.; Hwang, I.; Kang, H.; Kim, H.; Lee, J.H.; Ha, H. Delayed treatment with fenofibrate protects against high-fat diet-induced kidney injury in mice: The possible role of AMPK autophagy. Am. J. Physiol. Ren. Physiol. 2017, 312, F323–F334. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Liu, S.; Ma, Q.; Xiao, D.; Chen, L. Berberine enhances the AMPK activation and autophagy and mitigates high glucose-induced apoptosis of mouse podocytes. Eur. J. Pharmacol. 2017, 794, 106–114. [Google Scholar] [CrossRef]
- Wang, X.; Gao, L.; Lin, H.; Song, J.; Wang, J.; Yin, Y.; Zhao, J.; Xu, X.; Li, Z.; Li, L. Mangiferin prevents diabetic nephropathy progression and protects podocyte function via autophagy in diabetic rat glomeruli. Eur. J. Pharmacol. 2018, 824, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, H.W.; Kim, M.Y.; Kim, T.W.; Kim, E.N.; Kim, Y.; Chung, S.; Kim, Y.S.; Choi, B.S.; Kim, Y.-S.; et al. Cinacalcet-mediated activation of the CaMKKbeta-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis. 2018, 9, 270. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1–11. [Google Scholar] [CrossRef]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Bharathi, S.S.; Zhang, Y.; Mohsen, A.-W.; Uppala, R.; Balasubramani, M.; Schreiber, E.; Uechi, G.; Beck, M.E.; Rardin, M.J.; Vockley, J.; et al. Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J. Biol. Chem. 2013, 288, 33837–33847. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Zeng, H.; Chen, J.-X. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J. Cell. Physiol. 2019, 234, 2252–2265. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Vaka, V.R.; He, X.; Booz, G.W.; Chen, J.X. High-fat diet induces cardiac remodelling and dysfunction: Assessment of the role played by SIRT3 loss. J. Cell. Mol. Med. 2015, 19, 1847–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.I.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic. Biol. Med. 2011, 51, 1258–1267. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Li, X.; Liu, Z.; Han, Y.; Xu, X.; Li, X.; Tang, Y.; Liu, Y.; Yu, T.; et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J. Cell. Physiol. 2019, 234, 23495–23506. [Google Scholar] [CrossRef]
- Guigas, B.; Viollet, B. Targeting AMPK: From Ancient Drugs to New Small-Molecule Activators. Exp. Suppl. 2016, 107, 327–350. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juszczak, F.; Caron, N.; Mathew, A.V.; Declèves, A.-E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7994. https://doi.org/10.3390/ijms21217994
Juszczak F, Caron N, Mathew AV, Declèves A-E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. International Journal of Molecular Sciences. 2020; 21(21):7994. https://doi.org/10.3390/ijms21217994
Chicago/Turabian StyleJuszczak, Florian, Nathalie Caron, Anna V. Mathew, and Anne-Emilie Declèves. 2020. "Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease" International Journal of Molecular Sciences 21, no. 21: 7994. https://doi.org/10.3390/ijms21217994
APA StyleJuszczak, F., Caron, N., Mathew, A. V., & Declèves, A.-E. (2020). Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. International Journal of Molecular Sciences, 21(21), 7994. https://doi.org/10.3390/ijms21217994