Wnt Pathway: An Integral Hub for Developmental and Oncogenic Signaling Networks

Abstract
1. Introduction
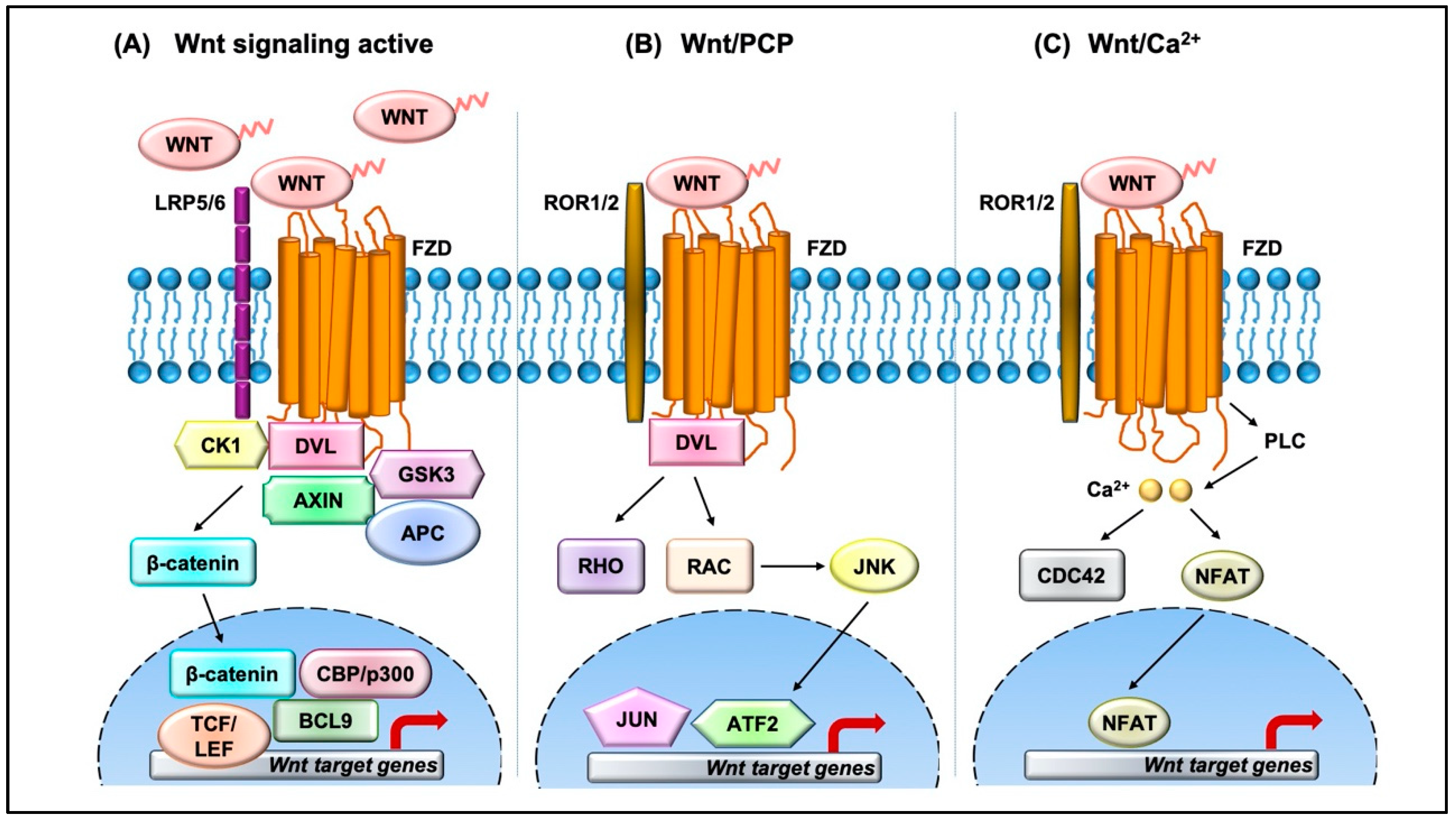
2. Overview of the Wnt Signaling Pathway
3. Wnt Signaling in Human Diseases
4. Wnt Signaling in Cancer
4.1. Hematological Malignancies
4.2. Breast Cancer
4.3. Colorectal Cancer
5. Insights into the Interplay between Wnt Signaling, Antitumor Immunity, and Stemness
5.1. Wnt Signaling and Antitumor Immunity
5.2. Wnt Signaling in Cancer Stem Cell Biology
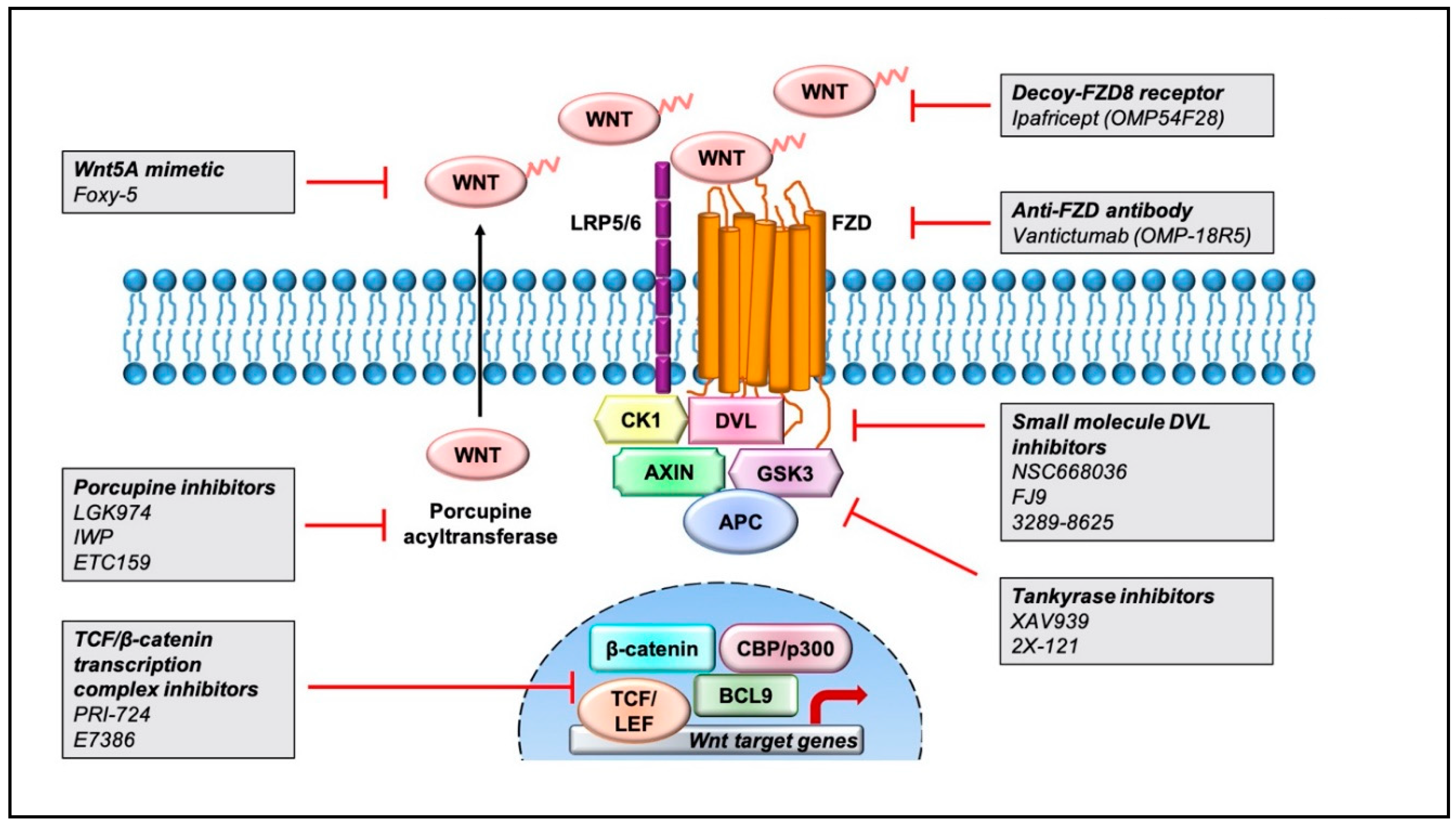
6. Wnt Pathway Inhibitors and Efforts in Clinical Trials
6.1. Inhibitors of Wnt-Receptors Complexes
6.2. Regulators of β-Catenin Destruction Complex
6.3. Inhibitors of TCF/β-Catenin Transcription Complex
7. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Vogelstein, B.; Kinzler, K.W. Association of the APC tumor suppressor protein with catenins. Science 1993, 262, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Willert, K. Wnt signaling: Is the party in the nucleus? Genes Dev. 2006, 20, 1394–1404. [Google Scholar] [CrossRef]
- Castro-Piedras, I.; Sharma, M.; Bakker, M.D.; Molehin, D.; Martinez, E.; Vartak, D.; Pruitt, W.M.; Deitrick, J.; Almodovar, S.; Pruitt, K. DVL1 and DVL3 differentially localize to CYP19A1 promoters and regulate aromatase mRNA in breast cancer cells. Oncotarget 2018, 9, 35639–35654. [Google Scholar] [CrossRef]
- Sharma, M.; Molehin, D.; Castro-Piedras, I.; Martinez, E.G.; Pruitt, K. Acetylation of conserved DVL-1 lysines regulates its nuclear translocation and binding to gene promoters in triple-negative breast cancer. Sci. Rep. 2019, 9, 16257. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, N.H.; Cho, E.S.; Yang, J.H.; Cha, Y.H.; Kang, H.E.; Yun, J.S.; Cho, S.B.; Lee, S.-H.; Paclikova, P.; et al. Dishevelled has a YAP nuclear export function in a tumor suppressor context-dependent manner. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, T.; Ninomiya-Tsuji, J.; Matsumoto, K. Regulation of lymphoid enhancer factor 1/T-cell factor by mitogen-activated protein kinase-related Nemo-like kinase-dependent phosphorylation in Wnt/beta-catenin signaling. Mol. Cell. Biol. 2003, 23, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef]
- Wallingford, J.B.; Habas, R. The developmental biology of Dishevelled: An enigmatic protein governingcell fate and cell polarity. Development 2005, 132, 4421–4436. [Google Scholar] [CrossRef] [PubMed]
- Lijam, N.; Paylor, R.; McDonald, M.P.; Crawley, J.N.; Deng, C.-X.; Herrup, K.; Stevens, K.; Maccaferri, G.; McBain, C.J.; Sussman, D.J.; et al. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell 1997, 90, 895–905. [Google Scholar] [CrossRef]
- Van Gijn, M.; Daemen, M.J.; Smits, J.F.; Blankesteijn, W.M. The wnt-frizzled cascade in cardiovascular disease. Cardiovasc. Res. 2002, 55, 16–24. [Google Scholar] [CrossRef]
- Hamblet, N.S.; Lijam, N.; Ruiz-Lozano, P.; Wang, J.; Yang, Y.; Luo, Z.; Mei, L.; Chien, K.R.; Sussman, D.J.; Wynshaw-Boris, A. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development 2002, 129, 5827–5838. [Google Scholar] [CrossRef]
- Pizzuti, A.; Novelli, G.; Mari, A.; Ratti, A.; Colosimo, A.; Amati, F.; Penso, D.; Sangiuolo, F.; Calabrese, G.; Palka, G.; et al. Human homologue sequences to the Drosophila dishevelled segment-polarity gene are deleted in the DiGeorge syndrome. Am. J. Hum. Genet. 1996, 58, 722–729. [Google Scholar]
- Chen, N.; Mi, J.; Wu, M.; Wang, W.; Gao, H. Expression of dishevelled gene in Hirschsprung’s disease. Int. J. Clin. Exp. Pathol. 2013, 6, 1791–1798. [Google Scholar]
- Person, A.D.; Beiraghi, S.; Sieben, C.M.; Hermanson, S.; Neumann, A.N.; Robu, M.E.; Schleiffarth, J.R., Jr.; Van Bokhoven, H.; Hoogeboom, J.M.; Mazzeu, J.F.; et al. WNT5Amutations in patients with autosomal dominant Robinow syndrome. Dev. Dyn. 2010, 239, 327–337. [Google Scholar] [CrossRef]
- White, J.; Mazzeu, J.F.; Hoischen, A.; Jhangiani, S.N.; Gambin, T.; Alcino, M.C.; Penney, S.; Saraiva, J.M.; Hove, H.; Skovby, F.; et al. DVL1 Frameshift Mutations Clustering in the Penultimate Exon Cause Autosomal-Dominant Robinow Syndrome. Am. J. Hum. Genet. 2015, 96, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Bunn, K.J.; Daniel, P.; Roesken, H.S.; O’Neill, A.C.; Cameron-Christie, S.R.; Morgan, T.; Brunner, H.G.; Lai, A.; Kunst, H.P.M.; Markie, D.M.; et al. Mutations in DVL1 Cause an Osteosclerotic Form of Robinow Syndrome. Am. J. Hum. Genet. 2015, 96, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Mansour, T.; Lucot, K.L.; Konopelski, S.E.; Dickinson, P.J.; Sturges, B.K.; Vernau, K.L.; Choi, S.; Stern, J.A.; Thomasy, S.; Döring, S.; et al. Whole genome variant association across 100 dogs identifies a frame shift mutation in DISHEVELLED 2 which contributes to Robinow-like syndrome in Bulldogs and related screw tail dog breeds. PLoS Genet. 2018, 14, e1007850. [Google Scholar] [CrossRef] [PubMed]
- White, J.J.; Mazzeu, J.F.; Hoischen, A.; Bayram, Y.; Withers, M.; Gezdirici, A.; Kimonis, V.; Steehouwer, M.; Jhangiani, S.N.; Muzny, D.M.; et al. DVL3 Alleles Resulting in a −1 Frameshift of the Last Exon Mediate Autosomal-Dominant Robinow Syndrome. Am. J. Hum. Genet. 2016, 98, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, N.-Q.; Yan, F.; Jin, H.; Zhou, S.-Y.; Shi, J.-S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef]
- Forrest, M.P.; Hill, M.J.; Quantock, A.J.; Martin-Rendon, E.; Blake, D.J. The emerging roles of TCF4 in disease and development. Trends Mol. Med. 2014, 20, 322–331. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 1–10. [Google Scholar] [CrossRef]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res. 2016, 118, 352–366. [Google Scholar] [CrossRef]
- Pai, S.-Y.; Kim, C.; Williams, D.A. Rac GTPases in Human Diseases. Dis. Markers 2010, 29, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Contini, A.; Ferri, N.; Bucci, R.; Lupo, M.G.; Erba, E.; Gelmi, M.L.; Pellegrino, S. Peptide modulators of Rac1/Tiam1 protein-protein interaction: An alternative approach for cardiovascular diseases. Pept. Sci. 2018, 110, e23089. [Google Scholar] [CrossRef] [PubMed]
- Izumi, D.; Toden, S.; Ureta, E.; Ishimoto, T.; Baba, H.; Goel, A. TIAM1 promotes chemoresistance and tumor invasiveness in colorectal cancer. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Contini, A.; Bernini, S.K.; Corsini, A. Role of Small GTPase Protein Rac1 in Cardiovascular Diseases: Development of new selective pharmacological inhibitors. J. Cardiovasc. Pharmacol. 2013, 62, 425–435. [Google Scholar] [CrossRef]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Miyaoka, T.; Seno, H.; Ishino, H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 1999, 38, 1–6. [Google Scholar] [CrossRef]
- Niemann, S.; Zhao, C.; Pascu, F.; Stahl, U.; Aulepp, U.; Niswander, L.; Weber, J.L.; Müller, U. Homozygous WNT3 Mutation Causes Tetra-Amelia in a Large Consanguineous Family. Am. J. Hum. Genet. 2004, 74, 558–563. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; Konrad, D.; Navratil, F.; Schoenle, E.J. A WNT4 Mutation Associated with Müllerian-Duct Regression and Virilization in a 46, XX Woman. N. Engl. J. Med. 2004, 351, 792–798. [Google Scholar] [CrossRef]
- Jordan, B.K.; Mohammed, M.; Ching, S.T.; Délot, E.; Chen, X.-N.; Dewing, P.; Swain, A.; Rao, P.N.; Elejalde, B.R.; Vilain, E. Up-Regulation of WNT-4 Signaling and Dosage-Sensitive Sex Reversal in Humans. Am. J. Hum. Genet. 2001, 68, 1102–1109. [Google Scholar] [CrossRef]
- Perantoni, A.O. Renal development: Perspectives on a Wnt-dependent process. Semin. Cell Dev. Biol. 2003, 14, 201–208. [Google Scholar] [CrossRef]
- Park, J.S.; Valerius, M.T.; McMahon, A.P. Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 2007, 134, 2533–2539. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, A.; Tsukada, S.; Sekine, A.; Tsunoda, T.; Takahashi, A.; Kashiwagi, A.; Tanaka, Y.; Babazono, T.; Matsuda, M.; Kaku, K.; et al. Association of the Gene Encoding Wingless-Type Mammary Tumor Virus Integration-Site Family Member 5B (WNT5B) with Type 2 Diabetes. Am. J. Hum. Genet. 2004, 75, 832–843. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, M.; Zhang, Y.; Zhou, W.; Zhu, T.; Ruan, Q.; Chen, H.; Fang, J.; Zhou, F.; Sun, J.; et al. WNT5B governs the phenotype of basal-like breast cancer by activating WNT signaling. Cell Commun. Signal. 2019, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.U.; Santos, S.; Musso, C.M.; Ezquina, S.A.; Opitz, J.M.; Kok, F.; Otto, P.; Mingroni-Netto, R.C. Santos syndrome is caused by mutation in the WNT7A gene. J. Hum. Genet. 2017, 62, 1073–1078. [Google Scholar] [CrossRef]
- Woods, C.G.; Stricker, S.; Seemann, P.; Stern, R.; Cox, J.; Sherridan, E.; Roberts, E.; Springell, K.; Scott, S.; Karbani, G.; et al. Mutations in WNT7A Cause a Range of Limb Malformations, Including Fuhrmann Syndrome and Al-Awadi/Raas-Rothschild/Schinzel Phocomelia Syndrome. Am. J. Hum. Genet. 2006, 79, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Horrell, J.; Snitow, M.; Cui, J.; Gochnauer, H.; Syrett, C.M.; Kallish, S.; Seykora, J.T.; Liu, F.; Gaillard, D.; et al. WNT10A mutation causes ectodermal dysplasia by impairing progenitor cell proliferation and KLF4-mediated differentiation. Nat. Commun. 2017, 8, 15397. [Google Scholar] [CrossRef]
- Adaimy, L.; Chouery, E.; Mégarbané, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; De Mazancourt, P.; Megarbane, A. Mutation in WNT10A Is Associated with an Autosomal Recessive Ectodermal Dysplasia: The Odonto-onycho-dermal Dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Christodoulides, C.; Scarda, A.; Granzotto, M.; Milan, G.; Nora, E.D.; Keogh, J.; De Pergola, G.; Stirling, H.; Pannacciulli, N.; Sethi, J.K.; et al. WNT10B mutations in human obesity. Diabetologia 2006, 49, 678–684. [Google Scholar] [CrossRef]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef]
- Türkmen, S.; Spielmann, M.; Güneş, N.; Knaus, A.; Flöttmann, R.; Mundlos, S.; Tüysüz, B. A Novel de novo FZD2 Mutation in a Patient with Autosomal Dominant Omodysplasia. Mol. Syndr. 2017, 8, 318–324. [Google Scholar] [CrossRef]
- Robitaille, J.; MacDonald, M.L.; Kaykas, A.; Sheldahl, L.C.; Zeisler, J.; Dubé, M.-P.; Zhang, L.-H.; Singaraja, R.R.; Guernsey, D.L.; Zheng, B.; et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 2002, 32, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Hayashi, H.; Oshima, K.; Tahira, T.; Hayashi, K. Frizzled 4 gene (FZD4) mutations in patients with familial exudative vitreoretinopathy with variable expressivity. Br. J. Ophthalmol. 2003, 87, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High Bone Density Due to a Mutation in LDL-Receptor–Related Protein 5. N. Engl. J. Med. 2002, 346, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Little, R.D.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.-C.; Eustace, B.; Lappe, M.M.; Spitzer, L.; Zweier, S.; Braunschweiger, K.; et al. A Mutation in the LDL Receptor–Related Protein 5 Gene Results in the Autosomal Dominant High–Bone-Mass Trait. Am. J. Hum. Genet. 2002, 70, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL Receptor-Related Protein 5 (LRP5) Affects Bone Accrual and Eye Development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef]
- Korvala, J.; Jüppner, H.; Mäkitie, O.; Sochett, E.; Schnabel, D.; Mora, S.; Bartels, C.F.; Warman, M.L.; Deraska, D.; Cole, W.G.; et al. Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med Genet. 2012, 13, 26. [Google Scholar] [CrossRef]
- Pefkianaki, M.; Hasanreisoglu, M.; Suchy, S.F.; Shields, C.L. Familial Exudative Vitreoretinopathy With a NovelLRP5Mutation. J. Pediatr. Ophthalmol. Strabismus 2016, 53, e39–e42. [Google Scholar] [CrossRef]
- Joiner, D.M.; Ke, J.; Zhong, Z.; Xu, H.E.; Williams, B.O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. 2013, 24, 31–39. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Papassotiropoulos, A.; Biechele, T.; De-Vrieze, F.W.; Avila, M.E.; Major, M.B.; Myers, A.; Sáez, K.; Henríquez, J.P.; Zhao, A.; et al. Common genetic variation within the Low-Density Lipoprotein Receptor-Related Protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 9434–9439. [Google Scholar] [CrossRef]
- Mani, A.; Radhakrishnan, J.; Wang, H.; Mani, M.-A.; Nelson-Williams, C.; Carew, K.S.; Mane, S.; Najmabadi, H.; Wu, D.; Lifton, R.P. LRP6 Mutation in a Family with Early Coronary Disease and Metabolic Risk Factors. Science 2007, 315, 1278–1282. [Google Scholar] [CrossRef]
- Velasco, J.; Zarrabeitia, M.T.; Prieto, J.R.; Pérez-Castrillón, J.L.; Perez-Aguilar, M.D.; Perez-Nuñez, M.I.; Sanudo, C.; Hernandez-Elena, J.; Calvo, I.; Ortiz, F.; et al. Wnt pathway genes in osteoporosis and osteoarthritis: Differential expression and genetic association study. Osteoporos. Int. 2009, 21, 109–118. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oates, N.A.; Van Vliet, J.; Duffy, D.L.; Kroes, H.Y.; Martin, N.G.; Boomsma, D.I.; Campbell, M.; Coulthard, M.; Whitelaw, E.; Chong, S. Increased DNA Methylation at the AXIN1 Gene in a Monozygotic Twin from a Pair Discordant for a Caudal Duplication Anomaly. Am. J. Hum. Genet. 2006, 79, 155–162. [Google Scholar] [CrossRef]
- Kroes, H.Y.; Takahashi, M.; Zijlstra, R.; Baert, J.; Kooi, K.; Hofstra, R.M.W.; Van Essen, A.J. Two cases of the caudal duplication anomaly including a discordant monozygotic twin. Am. J. Med Genet. 2002, 112, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, S.M.; Fearon, E.R. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 2014, 355, 1–8. [Google Scholar] [CrossRef]
- Picco, G.; Petti, C.; Centonze, A.; Torchiaro, E.; Crisafulli, G.; Novara, L.; Acquaviva, A.; Bardelli, A.; Medico, E. Loss of AXIN1 drives acquired resistance to WNT pathway blockade in colorectal cancer cells carrying RSPO3 fusions. EMBO Mol. Med. 2017, 9, 293–303. [Google Scholar] [CrossRef]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Dahmen, R.; Koch, A.; Denkhaus, D.; Tonn, J.C.; Sörensen, N.; Berthold, F.; Behrens, J.; Birchmeier, W.; Wiestler, O.D.; Pietsch, T. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res. 2001, 61, 7039–7043. [Google Scholar]
- Zhang, X.; Farrell, A.S.; Daniel, C.J.; Arnold, H.; Scanlan, C.; Laraway, B.; Janghorban, M.; Lum, L.; Chen, D.; Troxell, M.; et al. Mechanistic insight into Myc stabilization in breast cancer involving aberrant Axin1 expression. Proc. Natl. Acad. Sci. USA 2012, 109, 2790–2795. [Google Scholar] [CrossRef]
- Zhu, M.-J.; Ma, X.-Y.; Ding, P.-C.; Tang, H.-F.; Peng, R.; Lu, L.; Li, P.-Q.; Qiao, B.; Yang, X.-Y.; Zheng, Y.-F.; et al. Novel mutations of AXIN2 identified in a Chinese Congenital Heart Disease Cohort. J. Hum. Genet. 2019, 64, 427–435. [Google Scholar] [CrossRef]
- Lammi, L.; Arte, S.; Somer, M.; Järvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 Cause Familial Tooth Agenesis and Predispose to Colorectal Cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050. [Google Scholar] [CrossRef]
- Callahan, N.; Modesto, A.; Meira, R.; Seymen, F.; Patir, A.; Vieira, A. Axis inhibition protein 2 (AXIN2) polymorphisms and tooth agenesis. Arch. Oral Biol. 2009, 54, 45–49. [Google Scholar] [CrossRef]
- Hlouskova, A.; Bielik, P.; Bonczek, O.; Balcar, V.J.; Sery, O. Mutations in AXIN2 gene as a risk factor for tooth agenesis and cancer: A review. Neurol. Lett. 2017, 38, 131–137. [Google Scholar]
- Otero, L.; Lacunza, E.; Vasquez, V.; Arbelaez, V.; Cardier, F.; González, F. Variations in AXIN2 predict risk and prognosis of colorectal cancer. BDJ Open 2019, 5, 1–6. [Google Scholar] [CrossRef]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [PubMed]
- Egomez-Sintes, R.; Hernández, F.; Lucas, J.J.; Avila, J. GSK-3 mouse models to study neuronal apoptosis and neurodegeneration. Front. Mol. Neurosci. 2011, 4, 45. [Google Scholar]
- Kozlovsky, N.; Belmaker, R.H.; Agam, G. GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur. Neuropsychopharmacol. 2002, 12, 13–25. [Google Scholar] [CrossRef]
- Li, X.; Liu, M.; Cai, Z.; Wang, G.; Li, X. Regulation of glycogen synthase kinase-3 during bipolar mania treatment. Bipolar Disord. 2010, 12, 741–752. [Google Scholar] [CrossRef]
- Domoto, T.; Pyko, I.V.; Furuta, T.; Miyashita, K.; Uehara, M.; Shimasaki, T.; Nakada, M.; Minamoto, T. Glycogen synthase kinase-3beta is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; Van Wichen, D.; De Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/-colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-Catenin-Tcf Signaling in Colon Cancer by Mutations in beta-Catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Shen, Y. Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer’s disease. J Neurosci. 2009, 29, 6545–6557. [Google Scholar] [CrossRef] [PubMed]
- Tamberg, L.; Sepp, M.; Timmusk, T.; Palgi, M. Introducing Pitt-Hopkins syndrome-associated mutations of TCF4 to Drosophila daughterless. Biol. Open 2015, 4, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; van Es, J.H.; Huch, M.; Li, V.S.; José, A.; Hatzis, P.; Mokry, M.; Haegebarth, A.; van den Born, M.; Chambon, P.; et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 2012, 151, 1595–1607. [Google Scholar] [CrossRef]
- Saxena, M.; Dykes, S.S.; Malyarchuk, S.; Wang, A.E.; Cardelli, J.A.; Pruitt, K.M. The sirtuins promote Dishevelled-1 scaffolding of TIAM1, Rac activation and cell migration. Oncogene 2013, 34, 188–198. [Google Scholar] [CrossRef]
- Grainger, S.; Traver, D.; Willert, K. Wnt Signaling in Hematological Malignancies. Prog. Mol. Biol. Transl. Sci. 2018, 153, 321–341. [Google Scholar] [PubMed]
- Lento, W.; Congdon, K.; Voermans, C.; Kritzik, M.; Reya, T. Wnt Signaling in Normal and Malignant Hematopoiesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a008011. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Tschumper, R.C.; Wu, X.; Shanafelt, T.D.; Eckel-Passow, J.; Huddleston, P.M.; Slager, S.L.; Kay, N.E.; Jelinek, D.F. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood 2010, 116, 2975–2983. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Famili, F.; Perez, L.G.; Pike-Overzet, K. Aberrant Wnt Signaling in Leukemia. Cancers 2016, 8, 78. [Google Scholar] [CrossRef]
- Lu, D.; Zhao, Y.; Tawatao, R.; Cottam, H.B.; Sen, M.; Leoni, L.M.; Kipps, T.J.; Corr, M.; Carson, D.A. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 3118–3123. [Google Scholar] [CrossRef]
- Memarian, A.; Hojjat-Farsangi, M.; Asgarian-Omran, H.; Younesi, V.; Jeddi-Tehrani, M.; Sharifian, R.A.; Khoshnoodi, J.; Razavi, S.M.; Rabbani, H.; Shokri, F. Variation in WNT genes expression in different subtypes of chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, J.R.; Neuteboom, S.T.C.; Wancewicz, E.V.; Monia, B.P.; Downing, J.R.; Murre, C. Oncogenic homeodomain transcription factor E2A-Pbx1 activates a novel WNT gene in pre-B acute lymphoblastoid leukemia. Proc. Natl. Acad. Sci. USA 1999, 96, 11464–11469. [Google Scholar] [CrossRef] [PubMed]
- Pehlivan, M.; Çalışkan, C.; Yuce, Z.; Sercan, H.O. Secreted Wnt antagonists in leukemia: A road yet to be paved. Leuk. Res. 2018, 69, 24–30. [Google Scholar] [CrossRef]
- Liu, T.-H.; Raval, A.; Chen, S.-S.; Matkovic, J.J.; Byrd, J.C.; Plass, C. CpG Island Methylation and Expression of the Secreted Frizzled-Related Protein Gene Family in Chronic Lymphocytic Leukemia. Cancer Res. 2006, 66, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Castillejo, J.; Navarro, G.; Barrios, M.; Andreu, E.J.; Prósper, F.; Heiniger, A.; Torres, A. Transcriptional silencing of the Dickkopfs-3 (Dkk-3) gene by CpG hypermethylation in acute lymphoblastic leukaemia. Br. J. Cancer 2004, 91, 707–713. [Google Scholar] [CrossRef]
- Pehlivan, M.; Sercan, Z.; Sercan, H.O.; Sercan, H.O. SFRP1 promoter methylation is associated with persistent Philadelphia chromosome in chronic myeloid leukemia. Leuk. Res. 2009, 33, 1062–1067. [Google Scholar] [CrossRef]
- Cheng, C.K.; Li, L.; Cheng, S.H.; Ng, K.; Chan, N.P.H.; Ip, R.K.L.; Wong, R.S.M.; Shing, M.M.K.; Li, C.K.; Ng, M.H.L. Secreted-frizzled related protein 1 is a transcriptional repression target of the t(8;21) fusion protein in acute myeloid leukemia. Blood 2011, 118, 6638–6648. [Google Scholar] [CrossRef]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef]
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87. [Google Scholar] [CrossRef]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Mill, C.P.; Qian, Y.; Raina, K.; Rajapakshe, K.; Coarfa, C.; Soldi, R.; Bose, P.; et al. Targeting nuclear beta-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia 2019, 33, 1373–1386. [Google Scholar] [CrossRef]
- Bosch, F.; Jares, P.; Campo, E.; Lopez-Guillermo, A.; Piris, M.A.; Villamor, N. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: A highly specific marker of mantle cell lymphoma. Blood 1994, 84, 2726–2732. [Google Scholar] [CrossRef]
- Hegazy, S.A.; Alshareef, A.; Gelebart, P.; Anand, M.; Armanious, H.; Ingham, R.J.; Lai, R. Disheveled proteins promote cell growth and tumorigenicity in ALK-positive anaplastic large cell lymphoma. Cell. Signal. 2013, 25, 295–307. [Google Scholar] [CrossRef]
- Ge, X.; Lv, X.; Feng, L.; Liu, X.; Wang, X. High expression and nuclear localization of beta-catenin in diffuse large B-cell lymphoma. Mol. Med. Rep. 2012, 5, 1433–1437. [Google Scholar] [PubMed]
- Zhang, D.; O’Neil, M.F.; Cunningham, M.T.; Fan, F.; Olyaee, M.; Li, L. Abnormal Wnt signaling and stem cell activation in reactive lymphoid tissue and low-grade marginal zone lymphoma. Leuk. Lymphoma 2010, 51, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Groen, R.W.; Oud, M.E.; Schilder-Tol, E.J.; Overdijk, M.B.; ten Berge, D.; Nusse, R. Illegitimate WNT pathway activation by beta-catenin mutation or autocrine stimulation in T-cell malignancies. Cancer Res. 2008, 68, 6969–6977. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.; Peters, A.C.; Armanious, H.; Anand, M.; Gelebart, P.; Lai, R. Biological and clinical significance of GSK-3beta in mantle cell lymphoma—an immunohistochemical study. Int. J. Clin. Exp. Pathol. 2010, 3, 244–253. [Google Scholar] [PubMed]
- Koivula, S.; Valo, E.; Raunio, A.; Hautaniemi, S.; Leppa, S. Rituximab regulates signaling pathways and alters gene expression associated with cell death and survival in diffuse large B-cell lymphoma. Oncol. Rep. 2011, 25, 1183–1190. [Google Scholar]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef]
- Kimura, Y.; Arakawa, F.; Kiyasu, J.; Miyoshi, H.; Yoshida, M.; Ichikawa, A.; Niino, D.; Sugita, Y.; Okamura, T.; Doi, A.; et al. The Wnt signaling pathway and mitotic regulators in the initiation and evolution of mantle cell lymphoma: Gene expression analysis. Int. J. Oncol. 2013, 43, 457–468. [Google Scholar] [CrossRef]
- Derksen, P.W.B.; Tjin, E.; Meijer, H.P.; Klok, M.D.; Mac Gillavry, H.D.; Van Oers, M.H.J.; Lokhorst, H.M.; Bloem, A.C.; Clevers, H.; Nusse, R.; et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc. Natl. Acad. Sci.USA 2004, 101, 6122–6127. [Google Scholar] [CrossRef]
- Qiang, Y.-W.; Walsh, K.; Yao, L.; Kedei, N.; Blumberg, P.M.; Rubin, J.S.; Shaughnessy, J.; Rudikoff, S. Wnts induce migration and invasion of myeloma plasma cells. Blood 2005, 106, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Roelink, H.; Wagenaar, E.; Nusse, R. Amplification and proviral activation of several Wnt genes during progression and clonal variation of mouse mammary tumors. Oncogene 1992, 7, 487–492. [Google Scholar] [PubMed]
- Nusse, R.; Varmus, H.E. Wnt genes. Cell 1992, 69, 1073–1087. [Google Scholar] [CrossRef]
- Koval, A.; Katanaev, V.L. Dramatic dysbalancing of the Wnt pathway in breast cancers. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell. Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Xia, W.; Wang, J.C.; Kwong, K.Y.; Spohn, B.; Wen, Y.; Pestell, R.G.; Hung, M.-C. Beta-Catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci.USA 2000, 97, 4262–4266. [Google Scholar] [CrossRef]
- Jonsson, M.; Borg, A.; Nilbert, M.; Andersson, T. Involvement of adenomatous polyposis coli (APC)/beta-catenin signalling in human breast cancer. Eur. J. Cancer. 2000, 36, 242–248. [Google Scholar] [CrossRef]
- Dass, R.A.; Sarshad, A.A.; Carson, B.B.; Feenstra, J.M.; Kaur, A.; Obrdlik, A.; Parks, M.M.; Prakash, V.; Love, D.K.; Pietras, K.; et al. Wnt5a Signals through DVL1 to Repress Ribosomal DNA Transcription by RNA Polymerase I. PLoS Genet. 2016, 12, e1006217. [Google Scholar] [CrossRef]
- Holloway, K.R.; Calhoun, T.N.; Saxena, M.; Metoyer, C.F.; Kandler, E.F.; Rivera, C.A.; Pruitt, K. SIRT1 regulates Dishevelled proteins and promotes transient and constitutive Wnt signaling. Proc. Natl. Acad. Sci.USA 2010, 107, 9216–9221. [Google Scholar] [CrossRef]
- Simmons, G.E.; Pandey, S., Jr.; Nedeljkovic-Kurepa, A.; Saxena, M.; Wang, A.; Pruitt, K. Frizzled 7 expression is positively regulated by SIRT1 and beta-catenin in breast cancer cells. PLoS ONE 2014, 9, e98861. [Google Scholar] [CrossRef]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.-M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389–1406. [Google Scholar] [PubMed]
- Wend, P.; Holland, J.D.; Ziebold, U.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 2010, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt Pathway Activity in Breast Cancer Sub-Types and Stem-Like Cells. PloS ONE 2013, 8, e67811. [Google Scholar] [CrossRef]
- Van Schie, E.H.; van Amerongen, R. Aberrant WNT/CTNNB1 Signaling as a Therapeutic Target in Human Breast Cancer: Weighing the Evidence. Front. Cell. Dev. Biol. 2020, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic potential of targeting the Wnt/beta-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 2019, 110, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Polakis, P.; Hart, M.; Rubinfeld, B. Defects in the regulation of beta-catenin in colorectal cancer. Single Mol. Single Cell Seq. 1999, 470, 23–32. [Google Scholar]
- Polakis, P. The oncogenic activation of beta-catenin. Curr. Opin. Genet. Dev. 1999, 9, 15–21. [Google Scholar] [CrossRef]
- Yang, A.D.; Fan, F.; Camp, E.R.; Van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic Oxaliplatin Resistance Induces Epithelial-to-Mesenchymal Transition in Colorectal Cancer Cell Lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Novellasdemunt, L.; Antas, P.; Li, V.S.W. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am. J. Physiol. Physiol. 2015, 309, C511–C521. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.H.M.; Amin, N.; Flanagan, D.J.; Johanson, T.M.; Phesse, T.J.; Vincan, E. Wnt is necessary for mesenchymal to epithelial transition in colorectal cancer cells. Dev. Dyn. 2018, 247, 521–530. [Google Scholar] [CrossRef]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Spitzner, M.; Reineke, S.; Moller, J.; Auslander, N.; Kramer, F. Chemoradiotherapy Resistance in Colorectal Cancer Cells is Mediated by Wnt/beta-catenin Signaling. Mol. Cancer Res. 2017, 15, 1481–1490. [Google Scholar] [CrossRef]
- Xue, J.; Yu, X.; Xue, L.; Ge, X.; Zhao, W.; Peng, W. Intrinsic beta-catenin signaling suppresses CD8+ T-cell infiltration in colorectal cancer. Biomed. Pharmacother. 2019, 115, 108921. [Google Scholar] [CrossRef]
- Rahmani, F.; Avan, A.; Hashemy, S.I.; Hassanian, S.M. Role of Wnt/beta-catenin signaling regulatory microRNAs in the pathogenesis of colorectal cancer. J. Cell. Physiol. 2018, 233, 811–817. [Google Scholar] [CrossRef]
- Haseeb, M.; Pirzada, R.H.; Ain, Q.U.; Choi, S. Wnt Signaling in the Regulation of Immune Cell and Cancer Therapeutics. Cells 2019, 8, 1380. [Google Scholar] [CrossRef]
- Chae, W.-J.; Bothwell, A.L.M. Canonical and Non-Canonical Wnt Signaling in Immune Cells. Trends Immunol. 2018, 39, 830–847. [Google Scholar] [CrossRef]
- D’Amico, L.; Mahajan, S.; Capietto, A.H.; Yang, Z.; Zamani, A.; Ricci, B. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J. Exp. Med. 2016, 213, 827–840. [Google Scholar] [CrossRef]
- Qian, J.; Zheng, Y.; Zheng, C.; Wang, L.; Qin, H.; Hong, S. Active vaccination with Dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood 2012, 119, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kayama, H.; Shojima, K.; Matsumoto, S.; Koyama, H.; Minami, Y. The Wnt5a-Ror2 axis promotes the signaling circuit between interleukin-12 and interferon-gamma in colitis. Sci. Rep. 2015, 5, 10536. [Google Scholar] [CrossRef] [PubMed]
- Belinson, H.; Savage, A.K.; Fadrosh, D.; Kuo, Y.M.; Lin, D.; Valladares, R. Dual epithelial and immune cell function of Dvl1 regulates gut microbiota composition and intestinal homeostasis. JCI Insight 2016, 1, e85395. [Google Scholar] [CrossRef]
- Yang, D.; Li, S.; Duan, X.; Ren, J.; Liang, S.; Yakoumatos, L. TLR4 induced Wnt3a-Dvl3 restrains the intensity of inflammation and protects against endotoxin-driven organ failure through GSK3beta/beta-catenin signaling. Mol. Immunol. 2020, 118, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N. Immune suppression and resistance mediated by constitutive activation of Wnt/beta-catenin signaling in human melanoma cells. J Immunol. 2012, 189, 2110–2117. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Liang, X.; Cui, W.; Ober-Blobaum, J.L.; Vazzana, J.; Shrikant, P.A. Beta-Catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc. Natl. Acad. Sci. USA 2015, 112, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef]
- Taniguchi, H.; Moriya, C.; Igarashi, H.; Saitoh, A.; Yamamoto, H.; Adachi, Y.; Imai, K. Cancer stem cells in human gastrointestinal cancer. Cancer Sci. 2016, 107, 1556–1562. [Google Scholar] [CrossRef]
- Melo, F.D.S.E.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef]
- Yang, L.; Tang, H.; Kong, Y.; Xie, X.; Chen, J.; Song, C. LGR5 Promotes Breast Cancer Progression and Maintains Stem-Like Cells Through Activation of Wnt/beta-Catenin Signaling. Stem Cells 2015, 33, 2913–2924. [Google Scholar] [CrossRef]
- Wu, C.; Qiu, S.; Lu, L.; Zou, J.; Li, W.-F.; Wang, O.; Zhao, H.; Wang, H.; Tang, J.; Chen, L.; et al. RSPO2–LGR5 signaling has tumour-suppressive activity in colorectal cancer. Nat. Commun. 2014, 5, 3149. [Google Scholar] [CrossRef]
- Wang, X.; Jung, Y.-S.; Jun, S.; Lee, S.; Wang, W.; Schneider, A.; Oh, Y.S.; Lin, S.H.; Park, B.-J.; Chen, J.; et al. PAF-Wnt signaling-induced cell plasticity is required for maintenance of breast cancer cell stemness. Nat. Commun. 2016, 7, 10633. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, K.; Yamashita, Y.; Liu, W.; Hatanaka, H.; Kurashina, K.; Wada, T.; Takada, S.; Kaneda, R.; Choi, Y.L.; Fujiwara, S.-I.; et al. Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene 2006, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Suzuki, H.; Toyota, M.; Nojima, M.; Maruyama, R.; Sasaki, S.; Takagi, H.; Sogabe, Y.; Sasaki, Y.; Idogawa, M.; et al. Frequent epigenetic inactivation of DICKKOPF family genes in human gastrointestinal tumors. Carcinogenesis 2007, 28, 2459–2466. [Google Scholar] [CrossRef]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Oh, P.S.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFbetaR2) in colon cancer cells. Carcinogenesis 2012, 33, 68–76. [Google Scholar] [CrossRef]
- Yu, Y.; Sarkar, F.H.; Majumdar, A.P.N. Down-regulation of miR-21 Induces Differentiation of Chemoresistant Colon Cancer Cells and Enhances Susceptibility to Therapeutic Regimens. Transl. Oncol. 2013, 6, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Bitarte, N.; Bandres, E.; Boni, V.; Zarate, R.; Rodriguez, J.; Gonzalez-Huarriz, M.; Lopez, I.; Sola, J.J.; Alonso, M.M.; Fortes, P.; et al. MicroRNA-451 Is Involved in the Self-renewal, Tumorigenicity, and Chemoresistance of Colorectal Cancer Stem Cells. Stem Cells 2011, 29, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Isobe, T.; Hisamori, S.; Hogan, D.J.; Zabala, M.; Hendrickson, D.G.; Dalerba, P. MiR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife 2014, 3, e01977. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Runsheng, C.; Du, Y.; Zhu, P.; Huang, G.; Luo, J.; Yanying, W.; Ye, B.; Guanling, H.; Xia, P.; et al. The Long Noncoding RNA lncTCF7 Promotes Self-Renewal of Human Liver Cancer Stem Cells through Activation of Wnt Signaling. Cell Stem Cell 2015, 16, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.-J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.-J.; Bell, G.W.; Guo, W.; Rubin, J.S.; Richardson, A.L.; et al. Paracrine and Autocrine Signals Induce and Maintain Mesenchymal and Stem Cell States in the Breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Tai, D.; Wells, K.; Arcaroli, J.J.; Vanderbilt, C.; Aisner, D.L.; Messersmith, W.A.; Lieu, C.H. Targeting the WNT Signaling Pathway in Cancer Therapeutics. Oncologist 2015, 20, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Tran, F.H.; Zheng, J.J. Modulating the wnt signaling pathway with small molecules. Protein Sci. 2017, 26, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Ferri, M.; Liscio, P.; Carotti, A.; Asciutti, S.; Sardella, R.; Macchiarulo, A.; Camaioni, E. Targeting Wnt-driven cancers: Discovery of novel tankyrase inhibitors. Eur. J. Med. Chem. 2017, 142, 506–522. [Google Scholar] [CrossRef]
- Ho, S.Y.; Keller, T.H. The use of porcupine inhibitors to target Wnt-driven cancers. Bioorganic Med. Chem. Lett. 2015, 25, 5472–5476. [Google Scholar] [CrossRef] [PubMed]
- Du, F.-Y.; Zhou, Q.-F.; Sun, W.-J.; Chen, G.-L. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J. Stem Cells 2019, 11, 398–420. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Xu, Z.; Lee, A.Y.; Matsangou, M.; McCormick, F.; Jablons, D.M. A Monoclonal Antibody against Wnt-1 Induces Apoptosis in Human Cancer Cells. Neoplasia 2004, 6, 7–14. [Google Scholar] [CrossRef]
- Mikami, I.; You, L.; He, B.; Xu, Z.; Batra, S.; Lee, A.Y.; Mazieres, J.; Reguart, N.; Uematsu, K.; Koizumi, K.; et al. Efficacy of Wnt-1 monoclonal antibody in sarcoma cells. BMC Cancer 2005, 5, 1–7. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef]
- Mita, M.M.; Becerra, C.; Richards, D.A.; Mita, A.C.; Shagisultanova, E.; Osborne, C.R.C. Phase 1b study of WNT inhibitor vantictumab (VAN, human monoclonal antibody) with paclitaxel (P) in patients (pts) with 1st—to 3rd-line metastatic HER2-negative breast cancer (BC). J. Clin. Oncol. 2016, 34 (Suppl. S15), 2516. [Google Scholar] [CrossRef]
- Davies, S.; Cardin, D.B.; Shahda, S.; Lenz, H.-J.; Dotan, E.; O’Neil, B.H.; Kapoun, A.M.; Stagg, R.J.; Berlin, J.; Messersmith, W.A.; et al. A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab-paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Investig. New Drugs 2019, 38, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Rosen, L.S.; Chugh, R.; Goldman, J.W.; Xu, L.; Kapoun, A.; Brachmann, R.K.; Dupont, J.; Stagg, R.J.; Tolcher, A.W.; et al. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J. Clin. Oncol. 2013, 31 (Suppl. S15), 2540. [Google Scholar] [CrossRef]
- Moore, K.N.; Gunderson, C.C.; Sabbatini, P.; McMeekin, D.S.; Mantia-Smaldone, G.M.; Burger, R.A.; Morgan, M.; Kapoun, A.M.; Brachmann, R.K.; Stagg, R.; et al. A phase 1b dose escalation study of ipafricept (OMP 54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2019, 154, 294–301. [Google Scholar] [CrossRef]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.-Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef] [PubMed]
- Dotan, E.; Cardin, D.B.; Lenz, H.-J.; Messersmith, W.A.; O’Neil, B.; Cohen, S.J.; Denlinger, C.S.; Shahda, S.; Kapoun, A.M.; Brachmann, R.K.; et al. Phase Ib study of WNT inhibitor ipafricept (IPA) with nab-paclitaxel (Nab-P) and gemcitabine (G) in patients (pts) with previously untreated stage IV pancreatic cancer (mPC). J. Clin. Oncol. 2019, 37 (Suppl. S4), 369. [Google Scholar] [CrossRef]
- Säfholm, A.; Tuomela, J.; Rosenkvist, J.; Dejmek, J.; Härkönen, P.; Andersson, T. The Wnt-5a-Derived Hexapeptide Foxy-5 Inhibits Breast Cancer Metastasis In vivo by Targeting Cell Motility. Clin. Cancer Res. 2008, 14, 6556–6563. [Google Scholar] [CrossRef] [PubMed]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a Is Correlated with Aggressiveness of Gastric Cancer by Stimulating Cell Migration and Invasion. Cancer Res. 2006, 66, 10439–10448. [Google Scholar] [CrossRef]
- Soerensen, P.G.; Andersson, T.; Buhl, U.; Moelvadgaard, T.; Jensen, P.B.; Brünner, N.; Nielsen, D. Phase I dose-escalating study to evaluate the safety, tolerability, and pharmacokinetic and pharmacodynamic profiles of Foxy-5 in patients with metastatic breast, colorectal, or prostate cancer. J. Clin. Oncol. 2014, 32 (Suppl. S15), TPS1140. [Google Scholar] [CrossRef]
- Agarwal, P.; Zhang, B.; Ho, Y.; Cook, A.; Li, L.; Mikhail, F.M.; Wang, Y.; McLaughlin, M.E.; Bhatia, R. Enhanced targeting of CML stem and progenitor cells by inhibition of porcupine acyltransferase in combination with TKI. Blood 2017, 129, 1008–1020. [Google Scholar] [CrossRef]
- Koo, B.-K.; Van Es, J.H.; Born, M.V.D.; Clevers, H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43; Znrf3-mutant neoplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 7548–7550. [Google Scholar] [CrossRef]
- Chen, B.; Dodge, M.E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.-W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small molecule–mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Shetti, D.; Zhang, B.; Fan, C.; Mo, C.; Lee, B.H.; Wei, K. Low Dose of Paclitaxel Combined with XAV939 Attenuates Metastasis, Angiogenesis and Growth in Breast Cancer by Suppressing Wnt Signaling. Cells 2019, 8, 892. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, X.; Han, Y.; Lv, Y.; Lan, F.; Zhao, J. XAV939 inhibits the proliferation and migration of lung adenocarcinoma A549 cells through the WNT pathway. Oncol. Lett. 2018, 15, 8973–8982. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Shen, F.; Xiao, W.; Chen, J.; Pan, F. Wnt inhibitor XAV939 suppresses the viability of small cell lung cancer NCI-H446 cells and induces apoptosis. Oncol. Lett. 2017, 14, 6585–6591. [Google Scholar] [CrossRef]
- Wu, X.; Luo, F.; Li, J.; Zhong, X.; Liu, K. Tankyrase 1 inhibitior XAV939 increases chemosensitivity in colon cancer cell lines via inhibition of the Wnt signaling pathway. Int. J. Oncol. 2016, 48, 1333–1340. [Google Scholar] [CrossRef]
- Arques, O.; Chicote, I.; Puig, I.; Tenbaum, S.P.; Argiles, G.; Dienstmann, R. Tankyrase Inhibition Blocks Wnt/beta-Catenin Pathway and Reverts Resistance to PI3K and AKT Inhibitors in the Treatment of Colorectal Cancer. Clin. Cancer Res. 2016, 22, 644–656. [Google Scholar] [CrossRef]
- Shan, J.; Shi, D.L.; Wang, J.; Zheng, J. Identification of a specific inhibitor of the dishevelled PDZ domain. Biochemistry 2005, 44, 15495–15503. [Google Scholar] [CrossRef]
- Fujii, N.; You, L.; Xu, Z.; Uematsu, K.; Shan, J.; He, B.; Mikami, I.; Edmondson, L.R.; Neale, G.; Zheng, J.; et al. An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res. 2007, 67, 573–579. [Google Scholar] [CrossRef]
- Grandy, D.; Shan, J.; Zhang, X.; Rao, S.; Akunuru, S.; Li, H.; Zhang, Y.; Alpatov, I.; Zhang, X.A.; Lang, R.A.; et al. Discovery and Characterization of a Small Molecule Inhibitor of the PDZ Domain of Dishevelled. J. Biol. Chem. 2009, 284, 16256–16263. [Google Scholar] [CrossRef]
- Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.-J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R.R. Final results of a phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34 (Suppl. S15), e15721. [Google Scholar] [CrossRef]
- Yamada, K.; Hori, Y.; Yamaguchi, A.; Matsuki, M.; Tsukamoto, S.; Yokoi, A.; Semba, T.; Ozawa, Y.; Inoue, S.; Yamamoto, Y.; et al. Abstract 5177: E7386: First-in-class orally active CBP/beta-catenin modulator as an anticancer agent. Exp. Mol. Ther. 2017, 77 (Suppl. S13), 5177. [Google Scholar]
- Lepourcelet, M.; Chen, Y.N.; France, D.S.; Wang, H.; Crews, P.; Petersen, F. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell 2004, 5, 91–102. [Google Scholar] [CrossRef]
- Gonsalves, F.C.; Klein, K.; Carson, B.B.; Katz, S.; Ekas, L.A.; Evans, S.; Nagourney, R.; Cardozo, T.; Brown, A.M.C.; Dasgupta, R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 5954–5963. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Dysfunction | Associated Disease | References |
|---|---|---|---|
| Wnt 1 | Gain of function | Schizophrenia | [36] |
| Wnt 3 | Loss of function | Tetra-amelia | [37] |
| Wnt 4 | Gain and loss of function | Intersex phenotype (GOF), kidney development, Mullerian duct regression and virilization (LOF) | [38,39,40,41] |
| Wnt 5B | Gain of function | Type II diabetes, breast tumorigenesis | [42,43] |
| Wnt 7A | Loss of function | Fuhrmann syndrome, Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome, Santos syndrome | [44,45] |
| Wnt 10A | Loss of function | Hypohidrotic ectodermal dysplasia, odonto-onycho-dermal dysplasia | [46,47] |
| Wnt 10B | Loss of function | Obesity, reduced bone mass | [48,49] |
| Fzd 2 | Heterozygous mutations | Omodysplasia, cardiovascular disease | [16,50] |
| Fzd 4 | Loss of function | Familial exudative vitreoretinopathy (FEVR) | [51,52] |
| LRP5 | Gain and loss of function | High bone mass (GOF), osteoporosis-pseudoglioma syndrome (LOF), familial exudative vitreoretinopathy (FEVR) | [53,54,55,56,57] |
| LRP6 | Gain and loss of function | Alzheimer’s disease (LOF), coronary artery disease (LOF), osteoarthritis (GOF) | [58,59,60,61] |
| DVL1 | Gain and loss of function | Robinow syndrome (frameshift mutation), Schwartz–Jampel syndrome, Charcot–Marie–Tooth disease type 2A and DiGeorge syndrome, myocardial infarction, Hirschsprung’s disease (GOF), autism spectrum disorders | [18,19,21,22,25] |
| DVL2 | Loss of function | Robinow syndrome, defect in cardiac outflow tract formation | [17,23] |
| DVL3 | Gain and loss of function | Hirschsprung’s disease (GOF), autism spectrum disorders | [19,25] |
| Axin 1 | Loss of function | Caudal duplication anomalies, gastrointestinal cancers, colorectal cancer, hepatocellular carcinomas, sporadic medulloblastoma, breast cancer | [62,63,64,65,66,67,68] |
| Axin 2 | Loss of function | Congenital heart defects, familial tooth agenesis, predisposition to multiple cancers including hepatocellular carcinoma and colorectal, prostate, ovarian, and lung cancers | [69,70,71,72,73] |
| APC | Loss of function | Familial adenomatous polyposis (FAP), colon cancer | [3,74] |
| GSK3β | Altered activity | Alzheimer’s disease, diabetes, schizophrenia, bipolar disorder, and cancer | [26,75,76,77,78,79] |
| β-Catenin | Gain and loss of function | Cancer (GOF), Alzheimer’s disease (LOF) | [80,81,82] |
| TCF4 | Transcript variants | Pitt–Hopkins syndrome, schizophrenia, Fuchs’ endothelial corneal dystrophy, primary sclerosing cholangitis, type II diabetes | [27,83,84] |
| JNK | Altered activity | Obesity, type II diabetes, nonalcoholic fatty liver disease (NAFLD) | [28] |
| Rho/Rac | Altered activity | Alzheimer’s disease, cardiovascular disease, leukocyte adhesion deficiency (LAD) | [29,30,31] |
| TIAM1 | Altered activity | Cardiovascular disease and cancer | [32,33,85] |
| NFAT | Loss of function | Down’s syndrome | [35] |
| Agent | Target Component | Trial Phase and Identifier | Condition or Disease | Collaborator and Sponsor |
|---|---|---|---|---|
| WNT974 | Porcupine inhibitor | Phase 1 NCT01351103 | Pancreatic cancer, BRAF mutant colorectal cancer, melanoma, triple-negative breast cancer, head and neck squamous cell cancer, cervical squamous cell cancer, esophageal squamous cell cancer, and lung squamous cell cancer | Novartis Pharmaceuticals |
| WNT974 (in combination with LGX818 and cetuximab) | Porcupine inhibitor | Phase 1 and 2 NCT02278133 | Metastatic colorectal cancer with BRAFV600-mutant mCRC with RNF43 mutations or RSPO fusions. | Array BioPharma |
| Foxy-5 | Wnt-5A mimetic | Phase 1 and 2 NCT02020291 NCT02655952 NCT03883802 | Metastatic breast cancer, colorectal cancer, prostate cancer, colon cancer | WntResearch AB, SMS-Oncology BV, SAGA Diagnostics AB, Unilabs A/S, BioVica AB, Catalan Institute of Oncology |
| ETC-159 | Porcupine inhibitor | Phase 1 NCT02521844 | Solid tumors | EDDC (Experimental Drug Development Centre), A*STAR Research Entities, PPD |
| Ipafricept (OMP54F28) | Decoy Fzd8-receptor | Phase 1 NCT01608867 | Solid tumors | Bayer; OncoMed Pharmaceuticals, Inc. |
| Vantictumab (OMP-18R5) | Anti-FZD antibody | Phase 1 NCT01345201 NCT02005315 NCT01957007 NCT01973309 | Solid tumors, pancreatic cancer stage IV, pancreatic cancer, non-small-cell lung cancer, metastatic breast cancer | OncoMed Pharmaceuticals, Inc. |
| 2X-121 | Tankyrase inhibitors | Phase 2 NCT03562832 NCT03878849 | Metastatic breast cancer, advanced ovarian cancer | Oncology Venture, Smerud Medical Research International AS, Danish Breast Cancer Cooperative Group, Alcedis GmbH, Amarex Clinical Research |
| PRI-724 | Inhibitor of TCF/β-catenin transcription complex | Phase 1 and 2 NCT01606579 | Advanced myeloid malignancies | inVentiv Health Clinical; Prism Pharma Co., Ltd. |
| PRI-724 | Inhibitor of TCF/β-catenin transcription complex | Phase 1 NCT01302405 | Advanced solid tumors | inVentiv Health Clinical; Prism Pharma Co., Ltd. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; Pruitt, K. Wnt Pathway: An Integral Hub for Developmental and Oncogenic Signaling Networks. Int. J. Mol. Sci. 2020, 21, 8018. https://doi.org/10.3390/ijms21218018
Sharma M, Pruitt K. Wnt Pathway: An Integral Hub for Developmental and Oncogenic Signaling Networks. International Journal of Molecular Sciences. 2020; 21(21):8018. https://doi.org/10.3390/ijms21218018
Chicago/Turabian StyleSharma, Monica, and Kevin Pruitt. 2020. "Wnt Pathway: An Integral Hub for Developmental and Oncogenic Signaling Networks" International Journal of Molecular Sciences 21, no. 21: 8018. https://doi.org/10.3390/ijms21218018
APA StyleSharma, M., & Pruitt, K. (2020). Wnt Pathway: An Integral Hub for Developmental and Oncogenic Signaling Networks. International Journal of Molecular Sciences, 21(21), 8018. https://doi.org/10.3390/ijms21218018