Multiplicity of α-Synuclein Aggregated Species and Their Possible Roles in Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. α-Synuclein Aggregation and Synucleinopathies
3. α-Synuclein Aggregation Mechanisms and Pathways
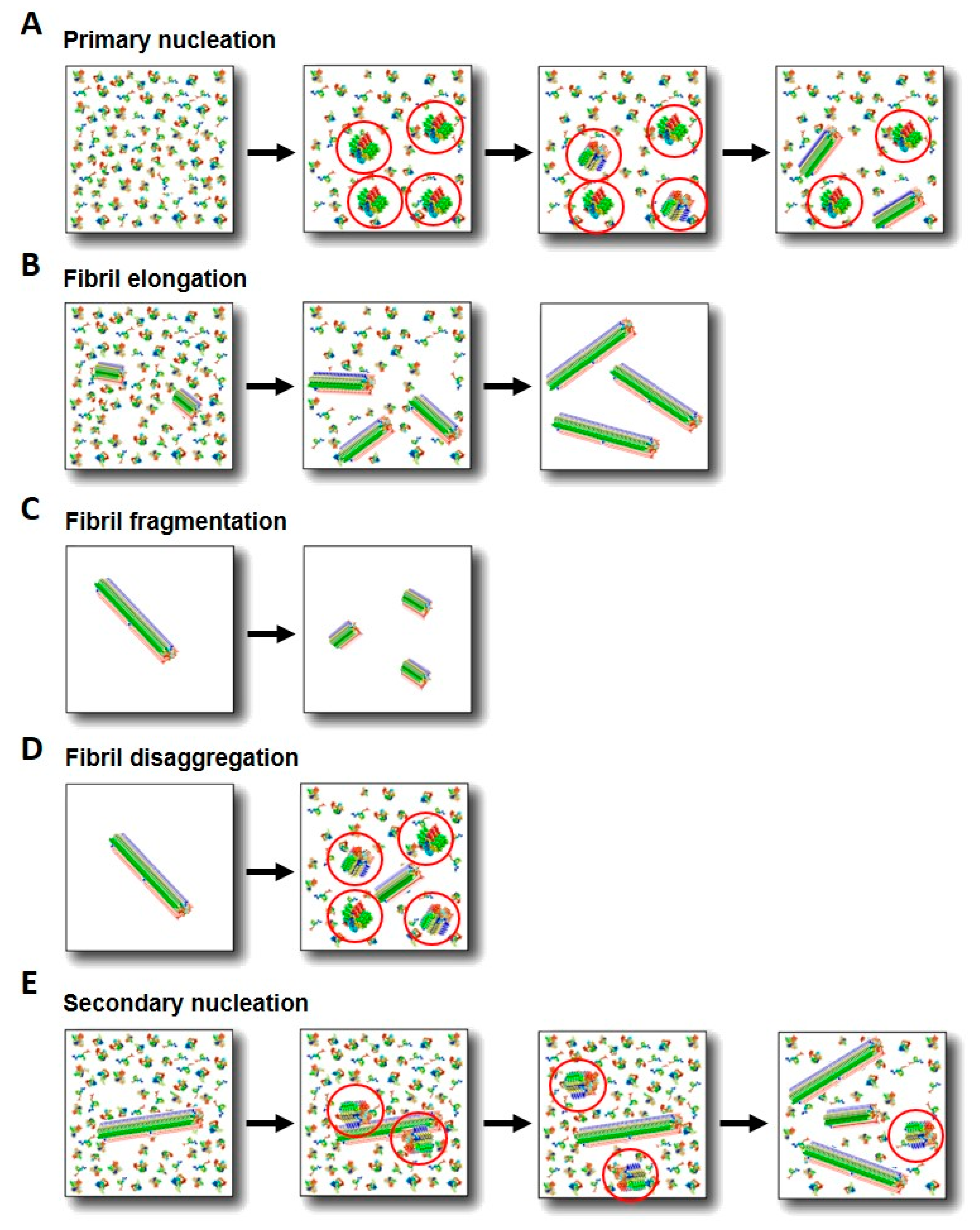
3.1. Primary Nucleation
3.2. Secondary Processes
3.3. Physicochemical Factors Affecting Amyloid Aggregation Process
4. Multiplicity of α-Synuclein Aggregated Species
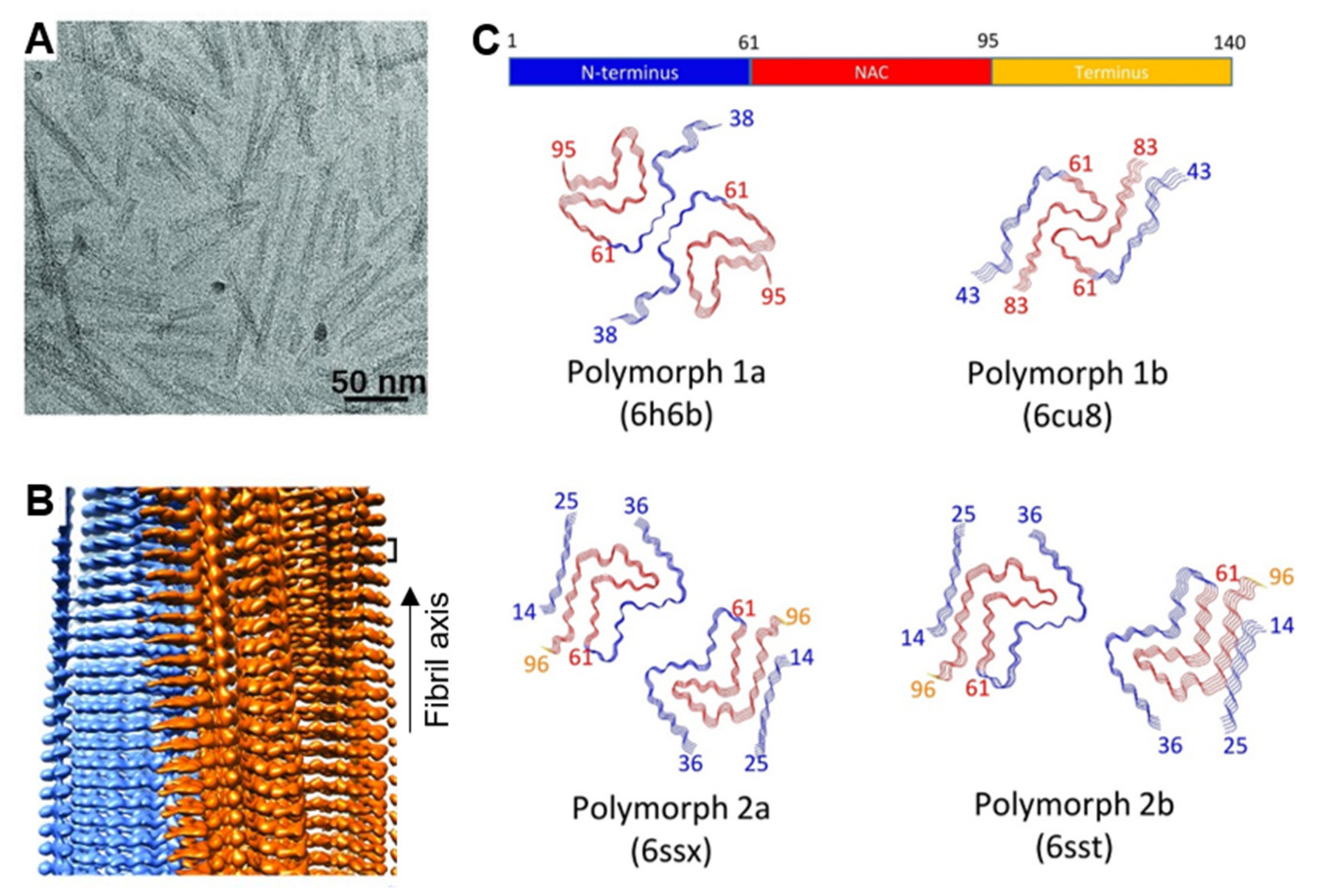
4.1. Fibrillar Polymorphs
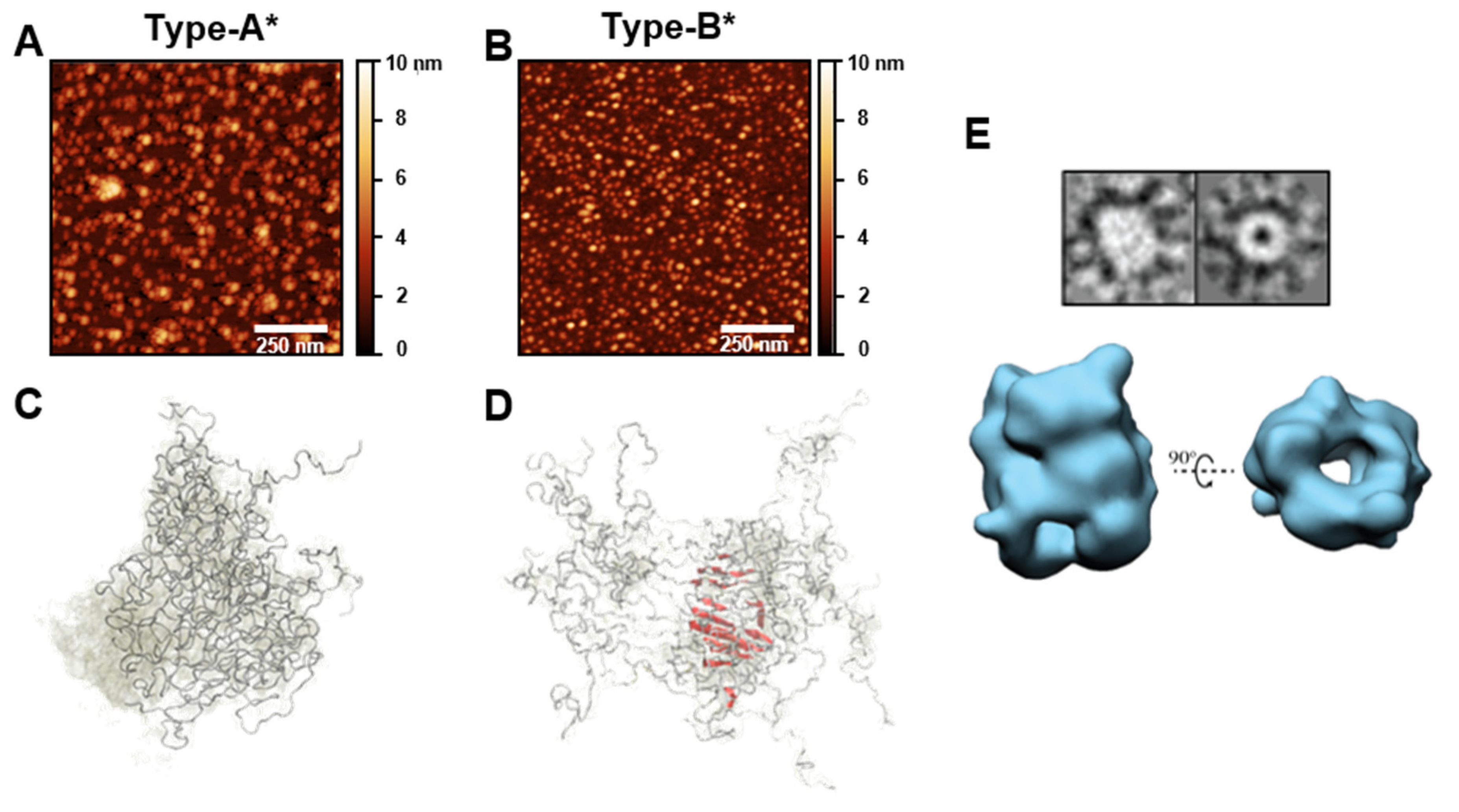
4.2. Oligomeric Polymorphs
5. Possible Roles of α-Synuclein Species in Cell Toxicity
6. Possible Roles of α-Synuclein Species in Cell-to-Cell Propagation
7. Post-Translational Modifications
8. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C.F. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.L.; Guijarro, J.I.; Orlova, E.; Zurdo, J.; Dobson, C.M.; Sunde, M.; Saibil, H.R. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 1999, 18, 815–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fändrich, M.; Dobson, C.M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002, 21, 5682–5690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.J.; White, D.A.; Abate, A.R.; Agresti, J.J.; Cohen, S.I.A.; Sperling, R.A.; De Genst, E.J.; Dobson, C.M.; Weitz, D.A. Observation of spatial propagation of amyloid assembly from single nuclei. Proc. Natl. Acad. Sci. USA 2011, 108, 14746–14751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peelaerts, W.; Bousset, L.; Van Der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van Den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers. Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein Aggregation in the Brain: The Molecular Basis for Alzheimer’s and Parkinson’s Diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Pickart, C.M. The ubiquitin-proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol. 2004, 14, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, J.T.; Chen, S.W.; Borg, C.B.; Ness, S.; Bahl, J.M.; Heegaard, N.H.H.; Dobson, C.M.; Hemmingsen, L.; Cremades, N.; Teilum, K. Amyloid-β and α-Synuclein Decrease the Level of Metal-Catalyzed Reactive Oxygen Species by Radical Scavenging and Redox Silencing. J. Am. Chem. Soc. 2016, 138, 3966–3969. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxidants Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [Green Version]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.C.; Kuperstein, I.; Wilkinson, H.; Maes, E.; Vanbrabant, M.; Jonckheere, W.; Van Gelder, P.; Hartmann, D.; D’Hooge, R.; De Strooper, B.; et al. Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008, 27, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; Cho, M.-K.; Riedel, D.; Fernandez, C.O.; Zweckstetter, M. Dissociation of Amyloid Fibrils of α-Synuclein in Supercooled Water. Angew. Chemie 2008, 120, 5124–5126. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.S.; Ansaloni, A.; Mezzenga, R.; Lashuel, H.A.; Dietler, G. Novel mechanistic insight into the molecular basis of amyloid polymorphism and secondary nucleation during amyloid formation. J. Mol. Biol. 2013, 425, 1765–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Brain region-dependent differential expression of alpha-synuclein. J. Comp. Neurol. 2016, 524, 1236–1258. [Google Scholar] [CrossRef]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, E.R.; Ramlall, T.F.; Borbat, P.P.; Freed, J.H.; Eliezer, D. Membrane-Bound α-Synuclein Forms an Extended Helix: Long-Distance Pulsed ESR Measurements Using Vesicles, Bicelles, and Rodlike Micelles. J. Am. Chem. Soc. 2008, 130, 12856–12857. [Google Scholar] [CrossRef] [Green Version]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar]
- Viennet, T.; Wördehoff, M.M.; Uluca, B.; Poojari, C.; Shaykhalishahi, H.; Willbold, D.; Strodel, B.; Heise, H.; Buell, A.K.; Hoyer, W.; et al. Structural insights from lipid-bilayer nanodiscs link α-Synuclein membrane-binding modes to amyloid fibril formation. Commun. Biol. 2018, 1, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, A.R.; Lacy, M.M.; Ducas, V.C.; Rhoades, E.; Sachs, J.N. α-Synuclein’s Uniquely Long Amphipathic Helix Enhances its Membrane Binding and Remodeling Capacity. J. Membr. Biol. 2017, 250, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, D.E.; Ugras, S.E.; Daniels, M.J.; Ischiropoulos, H. Dynamic structural flexibility of α-synuclein. Neurobiol. Dis. 2016, 88, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [Green Version]
- Waudby, C.A.; Camilloni, C.; Fitzpatrick, A.W.P.; Cabrita, L.D.; Dobson, C.M.; Vendruscolo, M.; Christodoulou, J. In-Cell NMR Characterization of the Secondary Structure Populations of a Disordered Conformation of α-Synuclein within E. coli Cells. PLoS ONE 2013, 8, e72286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Grazia Spillantini, M.; Anthony Crowther, R.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef] [Green Version]
- Aarsland, D.; Kurz, M.W. The epidemiology of dementia associated with Parkinson disease. J. Neurol. Sci. 2010, 289, 18–22. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.-P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, R.L. Lewy Bodies in Alzheimer’s Disease: A Neuropathological Review of 145 Cases Using α-Synuclein Immunohistochemistry. Brain Pathol. 2006, 10, 378–384. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.W.; Hague, S.M.; Clarimon, J.; Baptista, M.; Gwinn-Hardy, K.; Cookson, M.R.; Singleton, A.B. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 2004, 62, 1835–1838. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.L.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments α-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef] [PubMed]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schüle, B.; et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, E.; Seehra, G.; Sharma, P.; Sidransky, E. GBA1-associated parkinsonism: New insights and therapeutic opportunities. Curr. Opin. Neurol. 2019, 32, 589–596. [Google Scholar] [CrossRef]
- Henderson, M.X.; Cornblath, E.J.; Darwich, A.; Zhang, B.; Brown, H.; Gathagan, R.J.; Sandler, R.M.; Bassett, D.S.; Trojanowski, J.Q.; Lee, V.M.Y. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 2019, 22, 1248–1257. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Ibáñez, P.; Bonnet, A.-M.; Débarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Dürr, A.; Brice, A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Pankratz, N.; Foroud, T. Genetics of Parkinson disease. Genet. Med. 2007, 9, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Maraganore, D.M.; de Andrade, M.; Lesnick, T.G.; Strain, K.J.; Farrer, M.J.; Rocca, W.A.; Pant, P.V.K.; Frazer, K.A.; Cox, D.R.; Ballinger, D.G. High-resolution whole-genome association study of Parkinson disease. Am. J. Hum. Genet. 2005, 77, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpicelli-daley, L.a.; Luk, K.C.; Patel, T.P.; Tanik, S.a.; Dawn, M.; Stieber, A.; Meany, D.F.; Trojanowski, J.Q.; Lee, V.M. Alpha Syn Fibrils. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Froula, J.M.; Castellana-Cruz, M.; Anabtawi, N.M.; Camino, J.D.; Chen, S.W.; Thrasher, D.R.; Freire, J.; Yazdi, A.A.; Fleming, S.; Dobson, C.M.; et al. Correction: Defining α-synuclein species responsible for Parkinson’s disease phenotypes in mice. J. Biol. Chem. 2020, 295, 1142. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Cremades, N.; Dobson, C.M. The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol. Dis. 2018, 109, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T. Amyloid fibril formation requires a chemically discriminating nucleation event: Studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry 1992, 31, 12345–12352. [Google Scholar] [CrossRef]
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.C.; Citron, M.; Biere, A.L. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 1999, 274, 19509–19512. [Google Scholar] [CrossRef] [Green Version]
- Serio, T.R.; Cashikar, A.G.; Kowal, A.S.; Sawicki, G.J.; Moslehi, J.J.; Serpell, L.; Arnsdorf, M.F.; Lindquist, S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000, 289, 1317–1321. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.A.; Cohen, S.I.A.; Dobson, C.M.; Knowles, T.P.J. Nucleation-conversion-polymerization reactions of biological macromolecules with prenucleation clusters. Phys. Rev. E 2014, 89, 32712. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Castillo, E.J.; Koenig, J.L.; Anderson, J.M. Characterization of protein adsorption on soft contact lenses. IV. Comparison of in vivo spoilage with the in vitro adsorption of tear proteins. Biomaterials 1986, 7, 89–96. [Google Scholar] [CrossRef]
- Pronchik, J.; He, X.; Giurleo, J.T.; Talaga, D.S. In vitro formation of amyloid from α-synuclein is dominated by reactions at hydrophobic interfaces. J. Am. Chem. Soc. 2010, 132, 9797–9803. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shah, N.; Thakur, G.; Zhou, F.; Leblanc, R.M. α-Synuclein in α-helical conformation at air-water interface: Implication of conformation and orientation changes during its accumulation/aggregation. Chem. Commun. 2010, 46, 6702–6704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vácha, R.; Linse, S.; Lund, M. Surface effects on aggregation kinetics of amyloidogenic peptides. J. Am. Chem. Soc. 2014, 136, 11776–11782. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.; Linse, S.; Nilsson, H.; Brundin, P.; Sparr, E. Membrane interaction of α-synuclein in different aggregation states. J. Parkinsons. Dis. 2011, 1, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Galvagnion, C.; Brown, J.W.P.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Hashemi, M.; Banerjee, S.; Zagorski, K.; Rochet, J.-C.; Lyubchenko, Y.L. Assembly of α-synuclein aggregates on phospholipid bilayers. Biochim. Biophys. Acta Proteins Proteomics 2019, 1867, 802–812. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pittman, J.M.; Zerweck, J.; Venkata, B.S.; Moore, P.C.; Sachleben, J.R.; Meredith, S.C. β-Amyloid aggregation and heterogeneous nucleation. Protein Sci. 2019, 28, 1567–1581. [Google Scholar] [CrossRef]
- Zuzak, R.; Dorel, R.; Krawiec, M.; Such, B.; Kolmer, M.; Szymonski, M.; Echavarren, A.M.; Godlewski, S. Nonacene Generated by On-Surface Dehydrogenation. ACS Nano 2017, 11, 9321–9329. [Google Scholar] [CrossRef] [PubMed]
- Camino, J.D.; Gracia, P.; Chen, S.W.; Sot, J.; de la Arada, I.; Sebastián, V.; Arrondo, J.L.R.; Goñi, F.M.; Dobson, C.M.; Cremades, N. The extent of protein hydration dictates the preference for heterogeneous or homogeneous nucleation generating either parallel or antiparallel β-sheet α-synuclein aggregates. Chem. Sci. 2020. Advance Article. [Google Scholar] [CrossRef]
- Prestrelski, S.J.; Tedeschi, N.; Arakawa, T.; Carpenter, J.F. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophys. J. 1993, 65, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Chowdhury, P.; Gai, F. Infrared study of the effect of hydration on the amide i band and aggregation properties of helical peptides. J. Phys. Chem. B 2007, 111, 4596–4602. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Chowdhury, P.; Gai, F. Effect of dehydration on the aggregation kinetics of two amyloid peptides. J. Phys. Chem. B 2009, 113, 531–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griebenow, K.; Klibanov, A.M. Lyophilization-induced reversible changes in the secondary structure of proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 10969–10976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, E.J.; Koenig, J.L.; Anderson, J.M.; Lo, J. Protein adsorption on hydrogels. II. Reversible and irreversible interactions between lysozyme and soft contact lens surfaces. Biomaterials 1985, 6, 338–345. [Google Scholar] [CrossRef]
- Castillo, E.J.; Koenig, J.L.; Anderson, J.M.; Jentoft, N. Protein adsorption on soft contact lenses. III. Mucin. Biomaterials 1986, 7, 9–16. [Google Scholar] [CrossRef]
- Dong, A.; Prestrelski, S.J.; Allison, S.D.; Carpenter, J.F. Infrared spectroscopic studies of lyophilization- and temperature-induced protein aggregation. J. Pharm. Sci. 1995, 84, 415–424. [Google Scholar] [CrossRef]
- Zou, Y.; Li, Y.; Hao, W.; Hu, X.; Ma, G. Parallel β-Sheet Fibril and Antiparallel β-Sheet Oligomer: New Insights into Amyloid Formation of Hen Egg White Lysozyme under Heat and Acidic Condition from FTIR Spectroscopy. J. Phys. Chem. B 2013, 117, 4003–4013. [Google Scholar] [CrossRef]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.-M.; Raussens, V. Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef]
- Cerf, E.; Sarroukh, R.; Tamamizu-Kato, S.; Breydo, L.; Derclaye, S.; Dufrêne, Y.F.; Narayanaswami, V.; Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. Antiparallel β-sheet: A signature structure of the oligomeric amyloid β-peptide. Biochem. J. 2009, 421, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Schmidt, H.B.; Rohatgi, R. In Vivo Formation of Vacuolated Multi-phase Report In Vivo Formation of Vacuolated Multi-phase Compartments Lacking Membranes. CellReports 2016, 16, 1228–1236. [Google Scholar]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Hardenberg, M.C.; Sinnige, T.; Casford, S.; Dada, S.; Poudel, C.; Robinson, L.; Fuxreiter, M.; Kaminksi, C.; Kaminski Schierle, G.S.; Nollen, E.A.A.; et al. Observation of an α-synuclein liquid droplet state and its maturation into Lewy body-like assemblies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gaspar, R.; Meisl, G.; Buell, A.K.; Young, L.; Kaminski, C.F.; Knowles, T.P.J.; Sparr, E.; Linse, S. Secondary nucleation of monomers on fibril surface dominates α -synuclein aggregation and provides autocatalytic amyloid amplification. Q. Rev. Biophys. 2017, 50, e6. [Google Scholar] [CrossRef] [Green Version]
- Campioni, S.; Carret, G.; Jordens, S.; Nicoud, L.; Mezzenga, R.; Riek, R. The presence of an air-water interface affects formation and elongation of α-synuclein fibrils. J. Am. Chem. Soc. 2014, 136, 2866–2875. [Google Scholar] [CrossRef]
- Skamris, T.; Marasini, C.; Madsen, K.L.; Foderà, V.; Vestergaard, B. Early Stage Alpha-Synuclein Amyloid Fibrils are Reservoirs of Membrane-Binding Species. Sci. Rep. 2019, 9, 1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.Y.; Knowles, T.P.J.; Terentjev, E.M. Strength of Nanotubes, Filaments, and Nanowires From Sonication-Induced Scission. Adv. Mater. 2009, 21, 3945–3948. [Google Scholar] [CrossRef] [Green Version]
- Tipping, K.W.; Karamanos, T.K.; Jakhria, T.; Iadanza, M.G.; Goodchild, S.C.; Tuma, R.; Ranson, N.A.; Hewitt, E.W.; Radford, S.E. pH-induced molecular shedding drives the formation of amyloid fibril-derived oligomers. Proc. Natl. Acad. Sci. USA 2015, 112, 5691–5696. [Google Scholar] [CrossRef] [Green Version]
- Frankel, R.; Törnquist, M.; Meisl, G.; Hansson, O.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Frohm, B.; Cedervall, T.; Knowles, T.P.J.; et al. Autocatalytic amplification of Alzheimer-associated Aβ42 peptide aggregation in human cerebrospinal fluid. Commun. Biol. 2019, 2, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.I.A.; Arosio, P.; Presto, J.; Kurudenkandy, F.R.; Biverstål, H.; Dolfe, L.; Dunning, C.; Yang, X.; Frohm, B.; Vendruscolo, M.; et al. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 2015, 22, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.J.; Linse, S.; Dobson, C.M. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2014, 111, 7671–7676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Arteni, A.A.; Kumari, P.; Mona, D.; Ringler, P.; Britschgi, M.; Lauer, M.E.; Makky, A.; Verasdock, J.; et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife 2019, 8, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Chen, S.W.; Dobson, C.M. Structural Characteristics of α-Synuclein Oligomers. Int. Rev. Cell Mol. Biol. 2017, 329, 79–143. [Google Scholar]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. Elife 2018, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Zhou, K.; Hughes, M.P.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc. Natl. Acad. Sci. USA 2020, 117, 3592–3602. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Li, Y.; Liu, Z.; Long, H.; Zhao, C.; Luo, F.; Sun, Y.; Tao, Y.; Su, X.; Li, D.; et al. Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nat. Commun. 2020, 11, 2643. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Peduzzo, A.; Linse, S.; Buell, A.K. The properties of α-synuclein secondary nuclei are dominated by the solution conditions rather than the seed fibril strain. ACS Chem. Neurosci. 2020, 11, 909–918. [Google Scholar] [CrossRef]
- Woerman, A.L.; Stöhr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef] [Green Version]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, C.M.G.; Elia, A.E.; Portaleone, S.M.; Cazzaniga, F.A.; Rossi, M.; Bistaffa, E.; De Cecco, E.; Narkiewicz, J.; Salzano, G.; Carletta, O.; et al. Efficient RT-QuIC seeding activity for α-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl. Neurodegener. 2019, 8, 24. [Google Scholar] [CrossRef]
- Fenyi, A.; Leclair-Visonneau, L.; Clairembault, T.; Coron, E.; Neunlist, M.; Melki, R.; Derkinderen, P.; Bousset, L. Detection of alpha-synuclein aggregates in gastrointestinal biopsies by protein misfolding cyclic amplification. Neurobiol. Dis. 2019, 129, 38–43. [Google Scholar] [CrossRef]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Yao, Y.; Dong, X.; Guan, H.; Lu, Q. Cerebrospinal fluid real-time quaking-induced conversion test for sporadic Creutzfeldt-Jakob disease in an 18-year-old woman: A case report. Medicine 2017, 96, e8699. [Google Scholar] [CrossRef]
- Van Rumund, A.; Green, A.J.E.; Fairfoul, G.; Esselink, R.A.J.; Bloem, B.R.; Verbeek, M.M. α-Synuclein real-time quaking-induced conversion in the cerebrospinal fluid of uncertain cases of parkinsonism. Ann. Neurol. 2019, 85, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.D.; Hamed, A.; Settipalle, N.; Jande, S.; Rahman, S.; Szabella, M.E.; Boghossian, J. Real-time Quaking-induced Conversion Assay for the Diagnosis of Sporadic Creutzfeldt-Jakob Disease in a Living Patient. Infect. Dis. Res. Treat. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Jung, B.C.; Lim, Y.-J.; Bae, E.-J.; Lee, J.S.; Choi, M.S.; Lee, M.K.; Lee, H.-J.; Kim, Y.S.; Lee, S.-J. Amplification of distinct α-synuclein fibril conformers through protein misfolding cyclic amplification. Exp. Mol. Med. 2017, 49, e314. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Davidson, N.R.; Demircioğlu, D.; Fonseca, N.A.; He, Y.; Kahles, A.; Lehmann, K.-V.; Liu, F.; Shiraishi, Y.; Soulette, C.M.; et al. Genomic basis for RNA alterations in cancer. Nature 2020, 578, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iljina, M.; Garcia, G.A.; Horrocks, M.H.; Tosatto, L.; Choi, M.L.; Ganzinger, K.A.; Abramov, A.Y.; Gandhi, S.; Wood, N.W.; Cremades, N.; et al. Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc. Natl. Acad. Sci. USA 2016, 113, E1206–E1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by a-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T.J. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Hu, X.; Khant, H.; Ludtke, S.J.; Chiu, W.; Schmid, M.F.; Frieden, C.; Lee, J.-M. Interprotofilament interactions between Alzheimer’s Abeta1-42 peptides in amyloid fibrils revealed by cryoEM. Proc. Natl. Acad. Sci. USA 2009, 106, 4653–4658. [Google Scholar] [CrossRef] [Green Version]
- Celej, M.S.; Jares-Erijman, E.A.; Jovin, T.M. Fluorescent N-Arylaminonaphthalene Sulfonate Probes for Amyloid Aggregation of α-Synuclein. Biophys. J. 2008, 94, 4867–4879. [Google Scholar] [CrossRef] [Green Version]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014, 9, 2309–2317. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T.J. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T.J. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Paslawski, W.; Mysling, S.; Thomsen, K.; Jørgensen, T.J.D.; Otzen, D.E. Co-existence of two different α-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry. Angew. Chem. Int. Ed. Engl. 2014, 53, 7560–7563. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Nielsen, S.B.; Buell, A.K.; Kaspersen, J.D.; Arosio, P.; Vad, B.S.; Paslawski, W.; Christiansen, G.; Valnickova-Hansen, Z.; Andreasen, M.; et al. The role of stable α-synuclein oligomers in the molecular events underlying amyloid formation. J. Am. Chem. Soc. 2014, 136, 3859–3868. [Google Scholar] [CrossRef]
- Giehm, L.; Svergun, D.I.; Otzen, D.E.; Vestergaard, B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. USA 2011, 108, 3246–3251. [Google Scholar] [CrossRef] [Green Version]
- Gallea, J.I.; Celej, M.S. Structural insights into amyloid oligomers of the Parkinson disease-related protein α-synuclein. J. Biol. Chem. 2014, 289, 26733–26742. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.-P.; Fink, A.L.; Uversky, V.N. Structural characteristics of alpha-synuclein oligomers stabilized by the flavonoid baicalein. J. Mol. Biol. 2008, 383, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-J.; Baek, S.M.; Ho, D.-H.; Suk, J.-E.; Cho, E.-D.; Lee, S.-J. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp. Mol. Med. 2011, 43, 216. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.L.L.; Leong, S.L.; Ali, F.E.; Kenche, V.B.; Hill, A.F.; Gras, S.L.; Barnham, K.J.; Cappai, R. Dopamine and the dopamine oxidation product 5,6-dihydroxylindole promote distinct on-pathway and off-pathway aggregation of alpha-synuclein in a pH-dependent manner. J. Mol. Biol. 2009, 387, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G.; Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66, S74–S78. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.F.; Knowles, T.P.J.; Dobson, C.M.; Macphee, C.E.; Welland, M.E. Characterization of the nanoscale properties of individual amyloid fibrils. Proc. Natl. Acad. Sci. USA 2006, 103, 15806–15811. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.-C.; Minakaki, G.; Menges, S.; Salvi, R.; Savitskiy, S.; Kazman, A.; Vicente Miranda, H.; Mielenz, D.; Klucken, J.; Winkler, J.; et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci. Rep. 2019, 9, 544. [Google Scholar] [CrossRef] [Green Version]
- Volles, M.J.; Lansbury, P.T.J. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- Stefanovic, A.N.D.; Stöckl, M.T.; Claessens, M.M.A.E.; Subramaniam, V. α-Synuclein oligomers distinctively permeabilize complex model membranes. FEBS J. 2014, 281, 2838–2850. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.-M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Lindersson, E.; Beedholm, R.; Højrup, P.; Moos, T.; Gai, W.; Hendil, K.B.; Jensen, P.H. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 2004, 279, 12924–12934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Bousset, L.; Renner, M.; Redeker, V.; Savistchenko, J.; Triller, A.; Melki, R. Differential Membrane Binding and Seeding of Distinct α-Synuclein Fibrillar Polymorphs. Biophys. J. 2020, 118, 1301–1320. [Google Scholar] [CrossRef] [Green Version]
- Peralta Ramos, J.M.; Iribarren, P.; Bousset, L.; Melki, R.; Baekelandt, V.; Van der Perren, A. Peripheral Inflammation Regulates CNS Immune Surveillance Through the Recruitment of Inflammatory Monocytes Upon Systemic α-Synuclein Administration. Front. Immunol. 2019, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Gustot, A.; Gallea, J.I.; Sarroukh, R.; Celej, M.S.; Ruysschaert, J.-M.; Raussens, V. Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem. J. 2015, 471, 323–333. [Google Scholar] [CrossRef]
- Kordower, J.H.; Brundin, P. Lewy body pathology in long-term fetal nigral transplants: Is Parkinson’s disease transmitted from one neural system to another? Neuropsychopharmacology 2009, 34, 254. [Google Scholar] [CrossRef]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.V.; Gorenberg, E.L.; Nagy, M.; Thrasher, D.; Fenton, W.A.; Volpicelli-Daley, L.; Horwich, A.L.; Chandra, S.S. Hsp110 mitigates α-synuclein pathology in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 24310–24316. [Google Scholar] [CrossRef] [PubMed]
- Tittelmeier, J.; Sandhof, C.A.; Ries, H.M.; Druffel-Augustin, S.; Mogk, A.; Bukau, B.; Nussbaum-Krammer, C. The HSP110/HSP70 disaggregation system generates spreading-competent toxic α-synuclein species. EMBO J. 2020, 39, e103954. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Xu, Y.; Deng, Y.; Qing, H. The phosphorylation of α-synuclein: Development and implication for the mechanism and therapy of the Parkinson’s disease. J. Neurochem. 2015, 135, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef]
- Oueslati, A.; Paleologou, K.E.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Mimicking phosphorylation at serine 87 inhibits the aggregation of human α-synuclein and protects against its toxicity in a rat model of Parkinson’s disease. J. Neurosci. 2012, 32, 1536–1544. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of α-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Invest. 2009, 119, 3257–3265. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Kuwahara, T.; Tonegawa, R.; Ito, G.; Mitani, S.; Iwatsubo, T. Phosphorylation of α-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 7098–7109. [Google Scholar] [CrossRef] [Green Version]
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fredenburg, R.A.; Lansbury, P.T.J.; Fernandez, C.O.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, O.S.; Li, S.; Sullivan, L.F.; Chen, W.; Kondrikova, G.; Manfredsson, F.P.; Mandel, R.J.; Muzyczka, N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2008, 105, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Ferreira, R.; Kovacik, L.; Ni, D.; Stahlberg, H. New insights on the structure of alpha-synuclein fibrils using cryo-electron microscopy. Curr. Opin. Neurobiol. 2020, 61, 89–95. [Google Scholar] [CrossRef]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohäker, T.; Jung, B.C.; Liou, S.H.; Fernandez, C.O.; Riedel, D.; Becker, S.; Halliday, G.M.; Bennati, M.; Kim, W.S.; Lee, S.J.; et al. Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gracia, P.; Camino, J.D.; Volpicelli-Daley, L.; Cremades, N. Multiplicity of α-Synuclein Aggregated Species and Their Possible Roles in Disease. Int. J. Mol. Sci. 2020, 21, 8043. https://doi.org/10.3390/ijms21218043
Gracia P, Camino JD, Volpicelli-Daley L, Cremades N. Multiplicity of α-Synuclein Aggregated Species and Their Possible Roles in Disease. International Journal of Molecular Sciences. 2020; 21(21):8043. https://doi.org/10.3390/ijms21218043
Chicago/Turabian StyleGracia, Pablo, José D. Camino, Laura Volpicelli-Daley, and Nunilo Cremades. 2020. "Multiplicity of α-Synuclein Aggregated Species and Their Possible Roles in Disease" International Journal of Molecular Sciences 21, no. 21: 8043. https://doi.org/10.3390/ijms21218043