Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms

 ,
, 
 ,
, 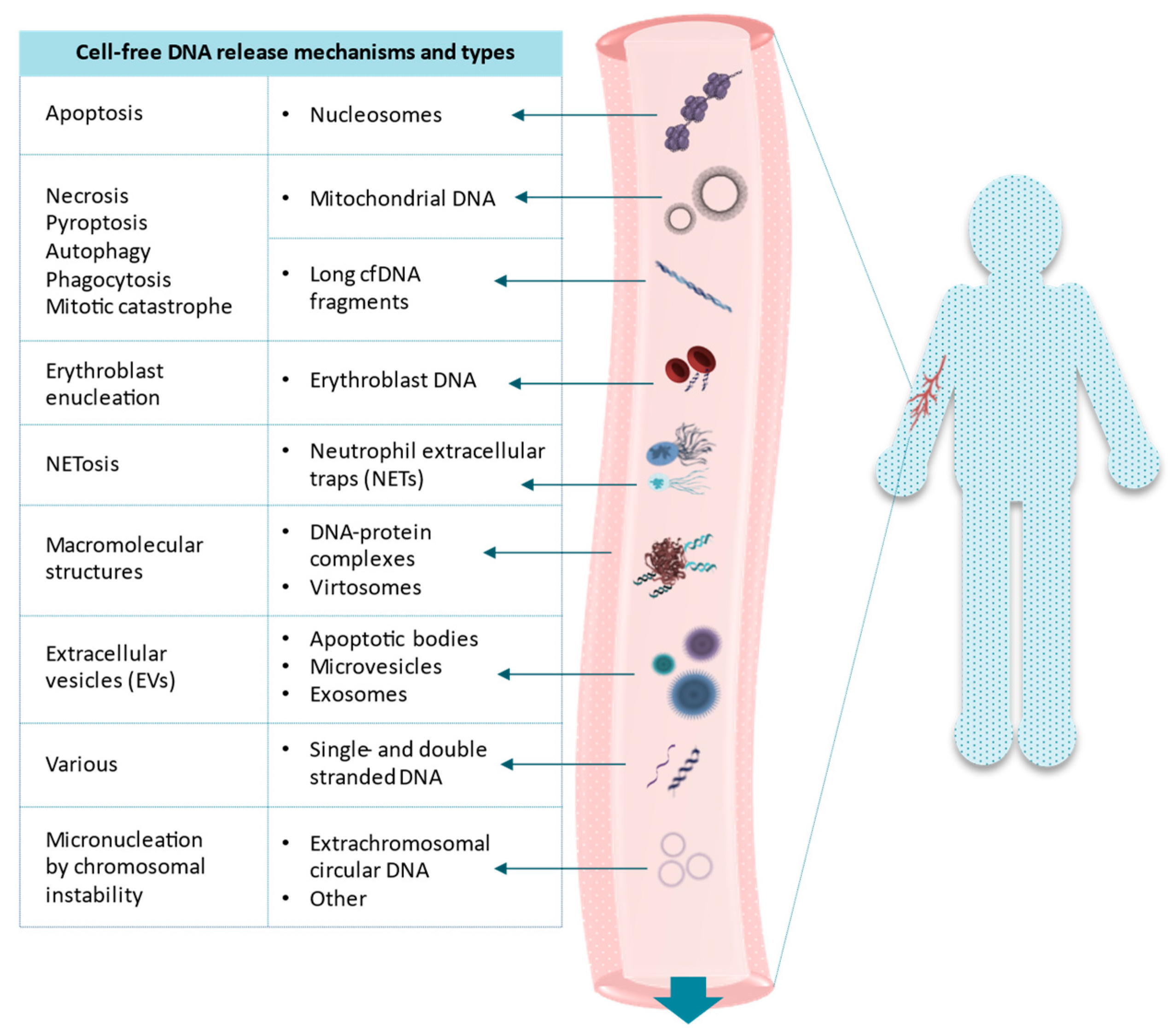{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Apoptosis and Necrosis
3. Erythroblast Enucleation
4. NETosis
5. Macromolecular Structures
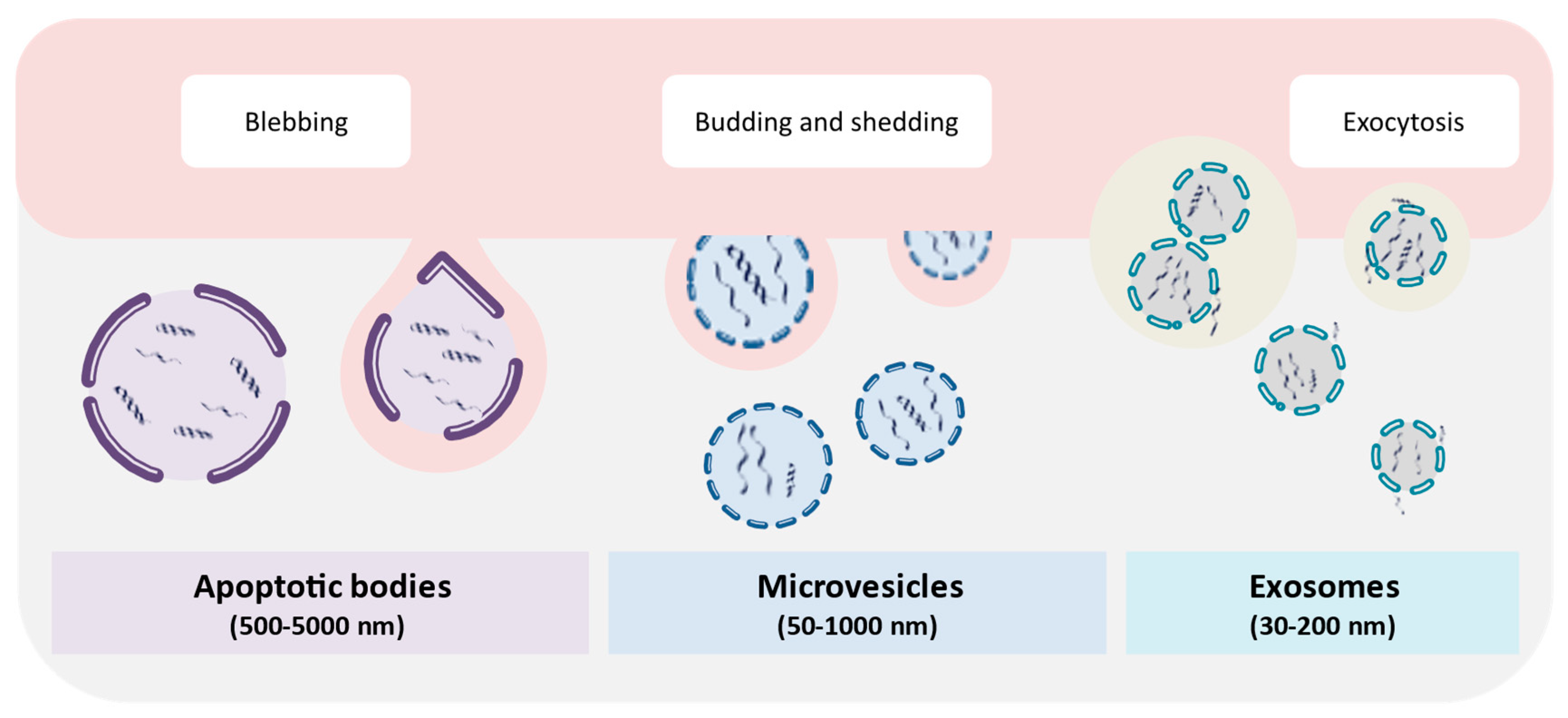
6. Extracellular Vesicles
7. Chromosomal Instability and Micronucleation
7.1. Fundamental Chromosome Segregation Errors During Mitosis
7.2. Other Chromosome Mis-Segregation Events that Can Arise During Mitosis
8. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme. CR Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Hui, L.; Maron, J.L.; Gahan, P.B. Other body fluids as non-invasive sources of cell-free DNA/RNA. Adv. Predict. Prev. Pers. Med. 2015, 5, 295–323. [Google Scholar] [CrossRef]
- Zachariah, R.R.; Schmid, S.; Buerki, N.; Radpour, R.; Holzgreve, W.; Zhong, X. Levels of circulating cell-free nuclear and mitochondrial dna in benign and malignant ovarian tumors. Obstet. Gynecol. 2008, 112, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Radpour, R.; Barekati, Z.; Asadollahi, R.; Bitzer, J.; Wight, E.; Bürki, N.; Diesch, C.; Holzgreve, W.; Zhong, X.Y. Levels of plasma circulating cell free nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol. Cancer 2009, 8. [Google Scholar] [CrossRef]
- Long, Y.; Zhang, Y.; Gong, Y.; Sun, R.; Su, L.; Lin, X.; Shen, A.; Zhou, J.; Caiji, Z.; Wang, X.; et al. Diagnosis of Sepsis with Cell-free DNA by Next-Generation Sequencing Technology in ICU Patients. Arch. Med. Res. 2016, 47, 365–371. [Google Scholar] [CrossRef]
- Kowarsky, M.; Camunas-Soler, J.; Kertesz, M.; De Vlaminck, I.; Koh, W.; Pan, W.; Martin, L.; Neff, N.F.; Okamoto, J.; Wong, R.J.; et al. Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proc. Natl. Acad. Sci. USA 2017, 114, 9623–9628. [Google Scholar] [CrossRef]
- Stroun, M.; Anker, P.; Lyautey, J.; Lederrey, C.; Maurice, P.A. Isolation and characterization of DNA from the plasma of cancer patients. Eur. J. Cancer Clin. Oncol. 1987, 23, 707–712. [Google Scholar] [CrossRef]
- McCoubrey-Hoyer, A.; Okarma, T.B.; Holman, H.R. Partial purification and characterization of plasma DNA and its relation to disease activity in systemic lupus erythematosus. Am. J. Med. 1984, 77, 23–34. [Google Scholar] [CrossRef]
- Rumore, P.M.; Steinman, C.R. Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J. Clin. Investig. 1990, 86, 69–74. [Google Scholar] [CrossRef]
- Giacona, M.B.; Ruben, G.C.; Iczkowski, K.A.; Roos, T.B.; Porter, D.M.; Sorenson, G.D. Cell-free DNA in human blood plasma: Length measurements in patients with pancreatic cancer and healthy controls. Pancreas 1998, 17, 89–97. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Tan, E.M.; Schur, P.H.; Carr, R.I.; Kunkel, H.G. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J. Clin. Investig. 1966, 45, 1732–1740. [Google Scholar] [CrossRef]
- Shapiro, B.; Chakrabarty, M.; Cohn, E.M.; Leon, S.A. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 1983, 51, 2116–2120. [Google Scholar] [CrossRef]
- Leon, S.A.; Ehrlich, G.E.; Shapiro, B.; Labbate, V.A. Free DNA in the serum of rheumatoid arthritis patients. J. Rheumatol. 1977, 4, 139–143. [Google Scholar] [PubMed]
- Davis, G.L.; Davis Iv, J.S. Detection of circulating dna by counterimmunoelectrophoresis (cie). Arthritis Rheum. 1973, 16, 52–58. [Google Scholar] [CrossRef]
- Jiang, P.; Lo, Y.M.D. The Long and Short of Circulating Cell-Free DNA and the Ins and Outs of Molecular Diagnostics. Trends Genet. 2016, 32, 360–371. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Chan, K.C.A.; Jiang, P.; Zheng, Y.W.L.; Liao, G.J.W.; Sun, H.; Wong, J.; Siu, S.S.N.; Chan, W.C.; Chan, S.L.; Chan, A.T.C.; et al. Cancer genome scanning in plasma: Detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef]
- Chu, D.; Park, B.H. Liquid biopsy: Unlocking the potentials of cell-free DNA. Virchows Arch. 2017, 471, 147–154. [Google Scholar] [CrossRef]
- Chan, K.C.A.; Jiang, P.; Chan, C.W.M.; Sun, K.; Wong, J.; Hui, E.P.; Chan, S.L.; Chan, W.C.; Hui, D.S.C.; Ng, S.S.M.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef]
- Suraj, S.; Dhar, C.; Srivastava, S. Circulating nucleic acids: An analysis of their occurrence in malignancies. Biomed. Rep. 2016, 6, 8–14. [Google Scholar] [CrossRef]
- Dennis Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.G.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; Van Den Boom, D.; Bombard, A.T.; Deciu, C.; Grody, W.W.; et al. DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet. Med. 2011, 13, 913–920. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Deciu, C.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; Van Den Boom, D.; Bombard, A.T.; Grody, W.W.; et al. DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: An international collaborative study. Genet. Med. 2012, 14, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Biró, O.; Szemes, T.; Nagy, B. Circulating cell-free nucleic acids: Characteristics and applications. Eur. J. Hum. Genet. 2018, 26, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Aucamp, J.; Bronkhorst, A.J.; Badenhorst, C.P.S.; Pretorius, P.J. The diverse origins of circulating cell-free DNA in the human body: A critical re-evaluation of the literature. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1649–1683. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Sai, S.; Ichikawa, D.; Tomita, H.; Ikoma, D.; Tani, N.; Ikoma, H.; Kikuchi, S.; Fujiwara, H.; Ueda, Y.; Otsuji, E. Quantification of plasma cell-free DNA in patients with gastric cancer. Anticancer Res. 2007, 27, 2747–2751. [Google Scholar] [PubMed]
- Delgado, P.O.; Alves, B.C.A.; Gehrke, F.d.S.; Kuniyoshi, R.K.; Wroclavski, M.L.; Del Giglio, A.; Fonseca, F.L.A. Characterization of cell-free circulating DNA in plasma in patients with prostate cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 983–986. [Google Scholar] [CrossRef]
- Reed, J.C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 2941–2953. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Henry, C.M.; Cullen, S.P. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol. Cell 2012, 46, 387–397. [Google Scholar] [CrossRef]
- Caruso, S.; Poon, I.K.H. Apoptotic cell-derived extracellular vesicles: More than just debris. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Errami, Y.; Naura, A.S.; Kim, H.; Ju, J.; Suzuki, Y.; El-Bahrawy, A.H.; Ghonim, M.A.; Hemeida, R.A.; Mansy, M.S.; Zhang, J.; et al. Apoptotic DNA fragmentation may be a cooperative activity between caspase-activated deoxyribonuclease and the poly(ADP-ribose) polymerase-regulated DNAS1L3, an endoplasmic reticulum-localized endonuclease that translocates to the nucleus during apoptosis. J. Biol. Chem. 2013, 288, 3460–3468. [Google Scholar] [CrossRef]
- Koyama, R.; Arai, T.; Kijima, M.; Sato, S.; Miura, S.; Yuasa, M.; Kitamura, D.; Mizuta, R. DNase γ, DNase I and caspase-activated DNase cooperate to degrade dead cells. Genes Cells 2016, 21, 1150–1163. [Google Scholar] [CrossRef]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef]
- Kitazumi, I.; Tsukahara, M. Regulation of DNA fragmentation: The role of caspases and phosphorylation. FEBS J. 2011, 278, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an in Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Han, D.S.C.; Ni, M.; Chan, R.W.Y.; Chan, V.W.H.; Lui, K.O.; Chiu, R.W.K.; Lo, Y.M.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020, 106, 202–214. [Google Scholar] [CrossRef]
- Jiménez-Alcázar, M.; Napirei, M.; Panda, R.; Köhler, E.C.; Kremer Hovinga, J.A.; Mannherz, H.G.; Peine, S.; Renné, T.; Lämmle, B.; Fuchs, T.A. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J. Thromb. Haemost. 2015, 13, 732–742. [Google Scholar] [CrossRef]
- Watanabe, T.; Takada, S.; Mizuta, R. Cell-free DNA in blood circulation is generated by DNase1L3 and caspase-activated DNase. Biochem. Biophys. Res. Commun. 2019, 516, 790–795. [Google Scholar] [CrossRef]
- Stephan, F.; Marsman, G.; Bakker, L.M.; Bulder, I.; Stavenuiter, F.; Aarden, L.A.; Zeerleder, S. Cooperation of factor VII-activating protease and serum DNase I in the release of nucleosomes from necrotic cells. Arthritis Rheumatol. 2014, 66, 686–693. [Google Scholar] [CrossRef]
- Martin, M.; Leffler, J.; Smoląg, K.I.; Mytych, J.; Björk, A.; Chaves, L.D.; Alexander, J.J.; Quigg, R.J.; Blom, A.M. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016, 23, 903–911. [Google Scholar] [CrossRef]
- Butler, T.M.; Spellman, P.T.; Gray, J. Circulating-tumor DNA as an early detection and diagnostic tool. Curr. Opin. Genet. Dev. 2017, 42, 14–21. [Google Scholar] [CrossRef]
- Yu, S.C.Y.; Lee, S.W.Y.; Jiang, P.; Leung, T.Y.; Chan, K.C.A.; Chiu, R.W.K.; Lo, Y.M.D. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin. Chem. 2013, 59, 1228–1237. [Google Scholar] [CrossRef]
- Celec, P.; Vlková, B.; Lauková, L.; Bábíčková, J.; Boor, P. Cell-free DNA: The role in pathophysiology and as a biomarker in kidney diseases. Expert Rev. Mol. Med. 2018, 20, e1. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Renò, F.; Burattini, S.; Rossi, S.; Luchetti, F.; Columbaro, M.; Santi, S.; Papa, S.; Falcieri, E. Phospholipid rearrangement of apoptotic membrane does not depend on nuclear activity. Histochem. Cell Biol. 1998, 110, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, J.M.; Kim, J.; Hur, J.; Park, S.; Kim, K.; Shin, H.J.; Chwae, Y.J. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc. Natl. Acad. Sci. USA 2018, 115, E11721–E11730. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Van Roosbroeck, K.; Calin, G.A. Cell-to-cell communication: microRNAs as hormones. Mol. Oncol. 2017, 11, 1673–1686. [Google Scholar] [CrossRef]
- Blander, J.M. The many ways tissue phagocytes respond to dying cells. Immunol. Rev. 2017, 277, 158–173. [Google Scholar] [CrossRef]
- Crescitelli, R.; Lässer, C.; Szabó, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzás, E.I.; Lötvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Tixeira, R.; Paone, S.; Mathivanan, S.; Collins, C.; Liem, M.; Goodall, K.J.; Ravichandran, K.S.; Hulett, M.D.; Poon, I.K.H. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Poon, I.K.H. Disassembly of the Dying: Mechanisms and Functions. Trends Cell Biol. 2017, 27, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernández-Delgado, I.; Latorre-Pellicer, A.; Acín-Pérez, R.; Martín-Cófreces, N.B.; Jaso-Tamame, Á.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef]
- Wang, W.; Kong, P.; Ma, G.; Li, L.; Zhu, J.; Xia, T.; Xie, H.; Zhou, W.; Wang, S. Characterization of the release and biological significance of cell-free DNA from breast cancer cell lines. Oncotarget 2017, 8, 43180–43191. [Google Scholar] [CrossRef]
- Serpas, L.; Chan, R.W.Y.; Jiang, P.; Ni, M.; Sun, K.; Rashidfarrokhi, A.; Soni, C.; Sisirak, V.; Lee, W.-S.; Cheng, S.H.; et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc. Natl. Acad. Sci. USA 2019, 116, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auinger, L.; Speicher, M.R. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef]
- Rostami, A.; Lambie, M.; Yu, C.W.; Stambolic, V.; Waldron, J.N.; Bratman, S.V. Senescence, Necrosis, and Apoptosis Govern Circulating Cell-free DNA Release Kinetics. Cell Rep. 2020, 31. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.S. Measurement of Red Cell Lifespan and Aging. Transfus. Med. Hemotherapy 2012, 39, 302–307. [Google Scholar] [CrossRef]
- Ji, P.; Murata-Hori, M.; Lodish, H.F. Formation of mammalian erythrocytes: Chromatin condensation and enucleation. Trends Cell Biol. 2011, 21, 409–415. [Google Scholar] [CrossRef]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef]
- Moras, M.; Lefevre, S.D.; Ostuni, M.A. From Erythroblasts to Mature Red Blood Cells: Organelle Clearance in Mammals. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Keerthivasan, G.; Wickrema, A.; Crispino, J.D. Erythroblast Enucleation. Stem Cells Int. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood 2008, 112, 470–478. [Google Scholar] [CrossRef]
- Gulliver, G. Observations on the size and shapes of the red corpuscles of the blood of vertebrates with drawings of them to a uniform scale and extended and revised tables of measurements. Proc. Zool. Soc. Lond. 1875, 474–495. [Google Scholar]
- Simpson, C.F.; Kling, J.M. The mechanism of denucleation in circulating erythroblasts. J. Cell Biol. 1967, 35, 237–245. [Google Scholar] [CrossRef]
- BESSIS, M. L’ílot érythroblastique, unité fonctionnelle de la moelle osseuse. Rev. Hematol. 1958, 13, 8–11. [Google Scholar]
- Geiduschek, J.B.; Singer, S.J. Molecular changes in the membranes of mouse erythroid cells accompanying differentiation. Cell 1979, 16, 149–163. [Google Scholar] [CrossRef]
- McGrath, K.E.; Kingsley, P.D.; Koniski, A.D.; Porter, R.L.; Bushnell, T.P.; Palis, J. Enucleation of primitive erythroid cells generates a transient population of “pyrenocytes” in the mammalian fetus. Blood 2008, 111, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kawane, K.; Koike, M.; Mori, Y.; Uchiyama, Y.; Nagata, S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 2005, 437, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Kawane, K. Requirement of DNase II for Definitive Erythropoiesis in the Mouse Fetal Liver. Science 2001, 292, 1546–1549. [Google Scholar] [CrossRef] [PubMed]
- Dunaeva, M.; Buddingh’, B.C.; Toes, R.E.M.; Luime, J.J.; Lubberts, E.; Pruijn, G.J.M. Decreased serum cell-free DNA levels in rheumatoid arthritis. Autoimmun. Highlights 2015, 6, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Helmig, S.; Frühbeis, C.; Krämer-Albers, E.-M.; Simon, P.; Tug, S. Release of bulk cell free DNA during physical exercise occurs independent of extracellular vesicles. Eur. J. Appl. Physiol. 2015, 115, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Tug, S.; Helmig, S.; Deichmann, E.R.; Schmeier-Jürchott, A.; Wagner, E.; Zimmermann, T.; Radsak, M.; Giacca, M.; Simon, P. Exercise-induced increases in cell free DNA in human plasma originate predominantly from cells of the haematopoietic lineage. Exerc. Immunol. Rev. 2015, 21, 164–173. [Google Scholar]
- Lui, Y.Y.N.; Chik, K.-W.; Chiu, R.W.K.; Ho, C.-Y.; Lam, C.W.K.; Lo, Y.M.D. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef]
- Wong, F.C.K.; Sun, K.; Jiang, P.; Cheng, Y.K.Y.; Chan, K.C.A.; Leung, T.Y.; Chiu, R.W.K.; Lo, Y.M.D. Cell-free DNA in maternal plasma and serum: A comparison of quantity, quality and tissue origin using genomic and epigenomic approaches. Clin. Biochem. 2016, 49, 1379–1386. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef]
- Lam, W.K.J.; Gai, W.; Sun, K.; Wong, R.S.M.; Chan, R.W.Y.; Jiang, P.; Chan, N.P.H.; Hui, W.W.I.; Chan, A.W.H.; Szeto, C.-C.; et al. DNA of Erythroid Origin Is Present in Human Plasma and Informs the Types of Anemia. Clin. Chem. 2017, 63, 1614–1623. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Estúa-Acosta, G.A.; Zamora-Ortiz, R.; Buentello-Volante, B.; García-Mejía, M.; Garfias, Y. Neutrophil Extracellular Traps: Current Perspectives in the Eye. Cells 2019, 8, 979. [Google Scholar] [CrossRef]
- Branzk, N.; Papayannopoulos, V. Molecular mechanisms regulating NETosis in infection and disease. Semin. Immunopathol. 2013, 35, 513–530. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Hamam, H.J.; Palaniyar, N. Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules 2019, 9, 369. [Google Scholar] [CrossRef]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6, 1–21. [Google Scholar] [CrossRef]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462.e5. [Google Scholar] [CrossRef]
- Farrera, C.; Fadeel, B. Macrophage Clearance of Neutrophil Extracellular Traps Is a Silent Process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef]
- Pisetsky, D.S. The origin and properties of extracellular DNA: From PAMP to DAMP. Clin. Immunol. 2012, 144, 32–40. [Google Scholar] [CrossRef]
- Yangsheng, Y.; Kaihong, S. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J. Clin. Cell. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tydén, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil Extracellular Traps That Are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef]
- Leffler, J.; Stojanovich, L.; Shoenfeld, Y.; Bogdanovic, G.; Hesselstrand, R.; Blom, A.M. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin. Exp. Rheumatol. 2014, 32, 66–70. [Google Scholar]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Doke, M.; Fukamachi, H.; Morisaki, H.; Arimoto, T.; Kataoka, H.; Kuwata, H. Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol. Oral Microbiol. 2017, 32, 288–300. [Google Scholar] [CrossRef]
- Berends, E.T.M.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; Von Köckritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed]
- de Buhr, N.; Neumann, A.; Jerjomiceva, N.; von Köckritz-Blickwede, M.; Baums, C.G. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology 2014, 160, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Storisteanu, D.M.L.; Pocock, J.M.; Cowburn, A.S.; Juss, J.K.; Nadesalingam, A.; Nizet, V.; Chilvers, E.R. Evasion of neutrophil extracellular traps by respiratory pathogens. Am. J. Respir. Cell Mol. Biol. 2017, 56, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.; Völlger, L.; Berends, E.T.M.; Molhoek, E.M.; Stapels, D.A.C.; Midon, M.; Friães, A.; Pingoud, A.; Rooijakkers, S.H.M.; Gallo, R.L.; et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular Traps against degradation by bacterial nucleases. J. Innate Immun. 2014, 6, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361. [Google Scholar] [CrossRef]
- Anderton, H.; Wicks, I.P.; Silke, J. Cell death in chronic inflammation: Breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 2020. [Google Scholar] [CrossRef]
- Katkar, G.D.; Sundaram, M.S.; NaveenKumar, S.K.; Swethakumar, B.; Sharma, R.D.; Paul, M.; Vishalakshi, G.J.; Devaraja, S.; Girish, K.S.; Kemparaju, K. NETosis and lack of DNase activity are key factors in Echis carinatus venom-induced tissue destruction. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 9, 1494. [Google Scholar] [CrossRef]
- Gahan, P.B.; Stroun, M. The virtosome-a novel cytosolic informative entity and intercellular messenger. Cell Biochem. Funct. 2010, 28, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.C.; Boldt, D.; Kornfeld, S.; Skinner, S.A.; Valeri, C.R. Excretion of Deoxyribonucleic Acid by Lymphocytes Stimulated with Phytohemagglutinin or Antigen. Proc. Natl. Acad. Sci. USA 1972, 69, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar] [PubMed]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous extracellular synthesis of DNA released by human blood lymphocytes. Cancer Res. 1976, 36, 2832–2839. [Google Scholar]
- Stroun, M.; Anker, P. Nucleic acids spontaneously released by living frog auricles. Biochem. J. 1972, 128, 100–101. [Google Scholar] [CrossRef]
- Adams, D.H.; Gahan, P.B. The DNA extruded by rat spleen cells in culture. Int. J. Biochem. 1983, 15, 547–552. [Google Scholar] [CrossRef]
- Challen, C.; Adams, D.H. Further studies on the size and composition of the chick embryo fibroblast cytosolic DNA complex. Int. J. Biochem. 1986, 18, 423–429. [Google Scholar] [CrossRef]
- Cataldi, S.; Viola-Magni, M. Components of the cytosolic and released virtosomes from stimulated and non-stimulated human lymphocytes. Biochem. Biophys. Rep. 2016, 6, 236–241. [Google Scholar] [CrossRef]
- Adams, D.H.; Diaz, N.; Gahan, P.B. In vitro stimulation by tumour cell media of [3H]-thymidine incorporation by mouse spleen lymphocytes. Cell Biochem. Funct. 1997, 15, 119–126. [Google Scholar] [CrossRef]
- García-Olmo, D.C.; Domínguez, C.; García-Arranz, M.; Anker, P.; Stroun, M.; García-Verdugo, J.M.; García-Olmo, D. Cell-free nucleic acids circulating in the plasma of colorectal cancer patients induce the oncogenic transformation of susceptible cultured cells. Cancer Res. 2010, 70, 560–567. [Google Scholar] [CrossRef]
- Garcia-Arranz, M.; Garcia-Olmo, D.; Vega-Clemente, L.; Stroun, M.; Gahan, P.B. Non-dividing Cell Virtosomes Affect In Vitro and In Vivo Tumour Cell Replication. In Circulating Nucleic Acids in Serum and Plasma—CNAPS IX. Advances in Experimental Medicine and Biology; Gahan, P., Fleischhacker, M., Schmidt, B., Eds.; Springer: Cham, Switzerland, 2016; pp. 43–45. [Google Scholar]
- Pelc, S.R. Turnover of DNA and function. Nature 1968, 219, 162–163. [Google Scholar] [CrossRef]
- Stroun, M.; Charles, P.; Anker, P.; Pelc, S.R. Metabolic DNA in heart and skeletal muscle and in the intestine of mice. Nature 1967, 216, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Wentzel, J.F.; Aucamp, J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Characterization of the cell-free DNA released by cultured cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 157–165. [Google Scholar] [CrossRef]
- Aucamp, J.; Bronkhorst, A.J.; Peters, D.L.; Van Dyk, H.C.; Van der Westhuizen, F.H.; Pretorius, P.J. Kinetic analysis, size profiling, and bioenergetic association of DNA released by selected cell lines in vitro. Cell. Mol. Life Sci. 2017, 74, 2689–2707. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Pantazi, C.; Kolios, G.; Kakolyris, S.; Chatzaki, E. Circulating cell-free DNA release in vitro: Kinetics, size profiling, and cancer-related gene methylation. J. Cell. Physiol. 2019, 234, 14079–14089. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, G. Prostasomes are mediators of intercellular communication: From basic research to clinical implications. J. Intern. Med. 2012, 271, 400–413. [Google Scholar] [CrossRef]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.J.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Heider, K.; Wan, J.C.M.; Hall, J.; Belic, J.; Boyle, S.; Hudecova, I.; Gale, D.; Cooper, W.N.; Corrie, P.G.; Brenton, J.D.; et al. Detection of ctDNA from Dried Blood Spots after DNA Size Selection. Clin. Chem. 2020, 66, 697–705. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Turner, N.C. Assessing HER2 amplification in plasma cfDNA. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1768, pp. 161–172. [Google Scholar] [CrossRef]
- Bonucci, E. Fine structure of early cartilage calcification. J. Ultrastruct. Res. 1967, 20, 33–50. [Google Scholar] [CrossRef]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.; Park, J.; Gho, Y.S. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin. Cell Dev. Biol. 2015, 40, 97–104. [Google Scholar] [CrossRef]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Olsson, I.; Ronquist, G. Nucleic Acid Association to Human Prostasomes. Arch. Androl. 1990, 24, 1–10. [Google Scholar] [CrossRef]
- Ronquist, K.G.; Ronquist, G.; Carlsson, L.; Larsson, A. Human prostasomes contain chromosomal DNA. Prostate 2009, 69, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Dorward, D.W.; Garon, C.F.; Judd, R.C. Export and intercellular transfer of DNA via membrane blebs of Neisseria gonorrhoeae. J. Bacteriol. 1989, 171, 2499–2505. [Google Scholar] [CrossRef]
- Garon, C.F.; Dorward, D.W.; Corwin, M.D. Structural features of Borrelia burgdorferi--the Lyme disease spirochete: Silver staining for nucleic acids. Scanning Microsc. Suppl. 1989, 3, 109–115. [Google Scholar]
- Soler, N.; Marguet, E.; Verbavatz, J.-M.; Forterre, P. Virus-like vesicles and extracellular DNA produced by hyperthermophilic archaea of the order Thermococcales. Res. Microbiol. 2008, 159, 390–399. [Google Scholar] [CrossRef]
- Kalluri, R.; Lebleu, V.S. Discovery of double-stranded genomic DNA in circulating exosomes. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 275–280. [Google Scholar] [CrossRef]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef]
- Waldenström, A.; Gennebäck, N.; Hellman, U.; Ronquist, G. Cardiomyocyte Microvesicles Contain DNA/RNA and Convey Biological Messages to Target Cells. PLoS ONE 2012, 7, e34653. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Che, S.P.Y.; Kurywchak, P.; Tavormina, J.L.; Gansmo, L.B.; Correa de Sampaio, P.; Tachezy, M.; Bockhorn, M.; Gebauer, F.; Haltom, A.R.; et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol. Ther. 2017, 18, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wu, G.; Jose, P.A.; Zeng, C. Functional transferred DNA within extracellular vesicles. Exp. Cell Res. 2016, 349, 179–183. [Google Scholar] [CrossRef]
- Sharma, A.; Johnson, A. Exosome DNA: Critical regulator of tumor immunity and a diagnostic biomarker. J. Cell. Physiol. 2020, 235, 1921–1932. [Google Scholar] [CrossRef]
- Bitto, N.J.; Chapman, R.; Pidot, S.; Costin, A.; Lo, C.; Choi, J.; D’Cruze, T.; Reynolds, E.C.; Dashper, S.G.; Turnbull, L.; et al. Bacterial membrane vesicles transport their DNA cargo into host cells. Sci. Rep. 2017, 7, 7072. [Google Scholar] [CrossRef]
- Fischer, S.; Cornils, K.; Speiseder, T.; Badbaran, A.; Reimer, R.; Indenbirken, D.; Grundhoff, A.; Brunswig-Spickenheier, B.; Alawi, M.; Lange, C. Indication of Horizontal DNA Gene Transfer by Extracellular Vesicles. PLoS ONE 2016, 11, e0163665. [Google Scholar] [CrossRef]
- Khier, S.; Lohan, L. Kinetics of circulating cell-free DNA for biomedical applications: Critical appraisal of the literature. Future Sci. OA 2018, 4, FSO295. [Google Scholar] [CrossRef]
- Lázaro-Ibáñez, E.; Lässer, C.; Shelke, G.V.; Crescitelli, R.; Jang, S.C.; Cvjetkovic, A.; García-Rodríguez, A.; Lötvall, J. DNA analysis of low- and high-density fractions defines heterogeneous subpopulations of small extracellular vesicles based on their DNA cargo and topology. J. Extracell. Vesicles 2019, 8, 1656993. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Desdín-Micó, G.; Mittelbrunn, M. Role of exosomes in the protection of cellular homeostasis. Cell Adhes. Migr. 2017, 11, 127–134. [Google Scholar] [CrossRef]
- Brahmer, A.; Neuberger, E.; Esch-Heisser, L.; Haller, N.; Jorgensen, M.M.; Baek, R.; Möbius, W.; Simon, P.; Krämer-Albers, E.M. Platelets, endothelial cells and leukocytes contribute to the exercise-triggered release of extracellular vesicles into the circulation. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Wentzel, J.F.; Ungerer, V.; Peters, D.L.; Aucamp, J.; de Villiers, E.P.; Holdenrieder, S.; Pretorius, P.J. Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumor Biol. 2018, 40. [Google Scholar] [CrossRef]
- Grabuschnig, S.; Soh, J.; Heidinger, P.; Bachler, T.; Hirschböck, E.; Rosales Rodriguez, I.; Schwendenwein, D.; Sensen, C.W. Circulating cell-free DNA is predominantly composed of retrotransposable elements and non-telomeric satellite DNA. J. Biotechnol. 2020, 313, 48–56. [Google Scholar] [CrossRef]
- Jefford, C.E.; Irminger-Finger, I. Mechanisms of chromosome instability in cancers. Crit. Rev. Oncol. Hematol. 2006, 59, 1–14. [Google Scholar] [CrossRef]
- Westhorpe, F.G.; Straight, A.F. Functions of the centromere and kinetochore in chromosome segregation. Curr. Opin. Cell Biol. 2013, 25, 334–340. [Google Scholar] [CrossRef]
- Cimini, D.; Degrassi, F. Aneuploidy: A matter of bad connections. Trends Cell Biol. 2005, 15, 442–451. [Google Scholar] [CrossRef]
- Cimini, D.; Moree, B.; Canman, J.C.; Salmon, E.D. Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. J. Cell Sci. 2003, 116, 4213–4225. [Google Scholar] [CrossRef] [PubMed]
- Lampson, M.A.; Renduchitala, K.; Khodjakov, A.; Kapoor, T.M. Correcting improper chromosome-spindle attachments during cell division. Nat. Cell Biol. 2004, 6, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 2001, 153, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Sears, D.A.; Udden, M.M. Howell-Jolly bodies: A brief historical review. Am. J. Med. Sci. 2012, 343, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.; Bolognesi, C.; Kirsch-Volders, M.; Bonassi, S.; Zeiger, E.; Knasmueller, S.; Fenech, M. The micronucleus assay in human buccal cells as a tool for biomonitoring DNA damage: The HUMN project perspective on current status and knowledge gaps. Mutat. Res. 2008, 659, 93–108. [Google Scholar] [CrossRef]
- Ford, J.H.; Schultz, C.J.; Correll, A.T. Chromosome elimination in micronuclei: A common cause of hypoploidy. Am. J. Hum. Genet. 1988, 43, 733–740. [Google Scholar]
- Lindholm, C.; Norppa, H.; Hayashi, M.; Sorsa, M. Induction of micronuclei and anaphase aberrations by cytochalasin B in human lymphocyte cultures. Mutat. Res. 1991, 260, 369–375. [Google Scholar] [CrossRef]
- Ford, J.H.; Correll, A.T. Chromosome errors at mitotic anaphase. Genome 1992, 35, 702–705. [Google Scholar] [CrossRef]
- Catalán, J.; Falck, G.C.; Norppa, H. The X chromosome frequently lags behind in female lymphocyte anaphase. Am. J. Hum. Genet. 2000, 66, 687–691. [Google Scholar] [CrossRef]
- Norppa, H.; Falck, G.C.-M. What do human micronuclei contain? Mutagenesis 2003, 18, 221–233. [Google Scholar] [CrossRef]
- Bailey, S.M.; Murnane, J.P. Telomeres, chromosome instability and cancer. Nucleic Acids Res. 2006, 34, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Björk, J.; Höglund, M.; Mertens, F.; Dal Cin, P.; Akerman, M.; Mandahl, N. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol. 2001, 158, 199–206. [Google Scholar] [CrossRef]
- Gisselsson, D.; Jonson, T.; Petersén, A.; Strömbeck, B.; Dal Cin, P.; Höglund, M.; Mitelman, F.; Mertens, F.; Mandahl, N. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 12683–12688. [Google Scholar] [CrossRef]
- Meeker, A.K.; Hicks, J.L.; Iacobuzio-Donahue, C.A.; Montgomery, E.A.; Westra, W.H.; Chan, T.Y.; Ronnett, B.M.; De Marzo, A.M. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dunham, M.A.; Reddel, R.R. Telomere length dynamics in telomerase-positive immortal human cell populations. Exp. Cell Res. 1998, 239, 370–378. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Hedges, D.J.; Deininger, P.L. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 2007, 616, 46–59. [Google Scholar] [CrossRef]
- Gasior, S.L.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef]
- Ji, W.; Hernandez, R.; Zhang, X.Y.; Qu, G.Z.; Frady, A.; Varela, M.; Ehrlich, M. DNA demethylation and pericentromeric rearrangements of chromosome 1. Mutat. Res. 1997, 379, 33–41. [Google Scholar] [CrossRef]
- Medvedeva, N.G.; Panyutin, I.V.; Panyutin, I.G.; Neumann, R.D. Phosphorylation of histone H2AX in radiation-induced micronuclei. Radiat. Res. 2007, 168, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Stewénius, Y.; Gorunova, L.; Jonson, T.; Larsson, N.; Höglund, M.; Mandahl, N.; Mertens, F.; Mitelman, F.; Gisselsson, D. Structural and numerical chromosome changes in colon cancer develop through telomere-mediated anaphase bridges, not through mitotic multipolarity. Proc. Natl. Acad. Sci. USA 2005, 102, 5541–5546. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, M.N.; Goodwin, E.H. Transmission of radiation-induced acentric chromosomal fragments to micronuclei in normal human fibroblasts. Radiat. Res. 1991, 126, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Warburton, P.E.; Greig, G.M.; Haaf, T.; Willard, H.F. PCR amplification of chromosome-specific alpha satellite DNA: Definition of centromeric STS markers and polymorphic analysis. Genomics 1991, 11, 324–333. [Google Scholar] [CrossRef]
- Fenech, M. Folate, DNA damage and the aging brain. Mech. Ageing Dev. 2010, 131, 236–241. [Google Scholar] [CrossRef]
- Lindberg, H.K.; Wang, X.; Järventaus, H.; Falck, G.C.-M.; Norppa, H.; Fenech, M. Origin of nuclear buds and micronuclei in normal and folate-deprived human lymphocytes. Mutat. Res. 2007, 617, 33–45. [Google Scholar] [CrossRef]
- Stimpson, K.M.; Matheny, J.E.; Sullivan, B.A. Dicentric chromosomes: Unique models to study centromere function and inactivation. Chromosom. Res. Int. J. Mol. Supramol. Evol. Asp. Chromosom. Biol. 2012, 20, 595–605. [Google Scholar] [CrossRef]
- Mackinnon, R.N.; Campbell, L.J. The role of dicentric chromosome formation and secondary centromere deletion in the evolution of myeloid malignancy. Genet. Res. Int. 2011, 2011, 643628. [Google Scholar] [CrossRef]
- McClintock, B. The Fusion of Broken Ends of Chromosomes Following Nuclear Fusion. Proc. Natl. Acad. Sci. USA 1942, 28, 458–463. [Google Scholar] [CrossRef]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Shimizu, N.; Shimura, T.; Tanaka, T. Selective elimination of acentric double minutes from cancer cells through the extrusion of micronuclei. Mutat. Res. 2000, 448, 81–90. [Google Scholar] [CrossRef]
- Shimizu, N.; Shingaki, K.; Kaneko-Sasaguri, Y.; Hashizume, T.; Kanda, T. When, where and how the bridge breaks: Anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp. Cell Res. 2005, 302, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N. Molecular mechanisms of the origin of micronuclei from extrachromosomal elements. Mutagenesis 2011, 26, 119–123. [Google Scholar] [CrossRef]
- Fenech, M. Cytokinesis-block micronucleus assay evolves into a “cytome” assay of chromosomal instability, mitotic dysfunction and cell death. Mutat. Res. 2006, 600, 58–66. [Google Scholar] [CrossRef]
- Shimizu, N.; Itoh, N.; Utiyama, H.; Wahl, G.M. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J. Cell Biol. 1998, 140, 1307–1320. [Google Scholar] [CrossRef]
- Kisurina-Evgenieva, O.P.; Sutiagina, O.I.; Onishchenko, G.E. Biogenesis of Micronuclei. Biochemistry 2016, 81, 453–464. [Google Scholar] [CrossRef]
- Utani, K.; Kohno, Y.; Okamoto, A.; Shimizu, N. Emergence of micronuclei and their effects on the fate of cells under replication stress. PLoS ONE 2010, 5, e10089. [Google Scholar] [CrossRef] [PubMed]
- Utani, K.; Okamoto, A.; Shimizu, N. Generation of micronuclei during interphase by coupling between cytoplasmic membrane blebbing and nuclear budding. PLoS ONE 2011, 6, e27233. [Google Scholar] [CrossRef] [PubMed]
- Sin, S.T.K.; Jiang, P.; Deng, J.; Ji, L.; Cheng, S.H.; Dutta, A.; Leung, T.Y.; Chan, K.C.A.; Chiu, R.W.K.; Lo, Y.M.D. Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc. Natl. Acad. Sci. USA 2020, 117, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Dillon, L.W.; Shibata, Y.; Jazaeri, A.A.; Jones, D.R.; Dutta, A. Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Mol. Cancer Res. 2017, 15, 1197–1205. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, F.; Du, M.; Zhang, P.; Fu, S.; Wang, L. Molecular characterization of cell-free eccDNAs in human plasma. Sci. Rep. 2017, 7, 10968. [Google Scholar] [CrossRef]
- Pampalona, J.; Soler, D.; Genescà, A.; Tusell, L. Telomere dysfunction and chromosome structure modulate the contribution of individual chromosomes in abnormal nuclear morphologies. Mutat. Res. 2010, 683, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Jackson, K.; Weemaes, C. Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J. Rare Dis. 2006, 1, 2. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Qu, G.Z.; Grundy, P.E.; Narayan, A.; Ehrlich, M. Frequent hypomethylation in Wilms tumors of pericentromeric DNA in chromosomes 1 and 16. Cancer Genet. Cytogenet. 1999, 109, 34–39. [Google Scholar] [CrossRef]
- Qu, G.; Dubeau, L.; Narayan, A.; Yu, M.C.; Ehrlich, M. Satellite DNA hypomethylation vs. overall genomic hypomethylation in ovarian epithelial tumors of different malignant potential. Mutat. Res. 1999, 423, 91–101. [Google Scholar] [CrossRef]
- Narayan, A.; Ji, W.; Zhang, X.Y.; Marrogi, A.; Graff, J.R.; Baylin, S.B.; Ehrlich, M. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int. J. Cancer 1998, 77, 833–838. [Google Scholar] [CrossRef]
- Fauth, E.; Scherthan, H.; Zankl, H. Chromosome painting reveals specific patterns of chromosome occurrence in mitomycin C- and diethylstilboestrol-induced micronuclei. Mutagenesis 2000, 15, 459–467. [Google Scholar] [CrossRef]
- Fauth, E.; Scherthan, H.; Zankl, H. Frequencies of occurrence of all human chromosomes in micronuclei from normal and 5-azacytidine-treated lymphocytes as revealed by chromosome painting. Mutagenesis 1998, 13, 235–241. [Google Scholar] [CrossRef]
- Fauth, E.; Zankl, H. Comparison of spontaneous and idoxuridine-induced micronuclei by chromosome painting. Mutat. Res. 1999, 440, 147–156. [Google Scholar] [CrossRef]
- Cimini, D.; Tanzarella, C.; Degrassi, F. Effects of 5-azacytidine on the centromeric region of human fibroblasts studied by CREST staining and in situ hybridization on cytokinesis-blocked cells. Cytogenet. Cell Genet. 1996, 72, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Guttenbach, M.; Schmid, M. Exclusion of specific human chromosomes into micronuclei by 5-azacytidine treatment of lymphocyte cultures. Exp. Cell Res. 1994, 211, 127–132. [Google Scholar] [CrossRef]
- Beck, J.; Urnovitz, H.B.; Riggert, J.; Clerici, M.; Schütz, E. Profile of the Circulating DNA in apparently healthy individuals. Clin. Chem. 2009, 55, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Mittra, I.; Kumar Khare, N.; Venkata Raghuram, G.; Chaubal, R.; Khambatti, F.; Gupta, D.; Gaikwad, A.; Prasannan, P.; Singh, A.; Iyer, A.; et al. Circulating nucleic acids damage DNA of healthy cells by integrating into their genomes. J. Biosci. 2018, 40, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Podgornaya, O.I.; Vasilyeva, I.N.; Bespalov, V.G. Heterochromatic Tandem Repeats in the Extracellular DNA. Adv. Exp. Med. Biol. 2016, 924, 85–89. [Google Scholar] [CrossRef]
- Snyder, G.K.; Sheafor, B.A. Red Blood Cells: Centerpiece in the Evolution of the Vertebrate Circulatory System. Am. Zool. 1999, 39, 189–198. [Google Scholar] [CrossRef]
- Föller, M.; Huber, S.M.; Lang, F. Erythrocyte programmed cell death. IUBMB Life 2008, 60, 661–668. [Google Scholar] [CrossRef]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36. [Google Scholar] [CrossRef]
- ADAMS, D.H.; GAHAN, P.B. Stimulated and Non-stimulated Rat Spleen Cells Release Different DNA-Complexes. Differentiation 1982, 22, 47–52. [Google Scholar] [CrossRef]
- McIntosh, A.A.G.; Adams, D.H. Further studies on the extrusion of cytosol macromolecules by cultured chick embryo fibroblast cells. Int. J. Biochem. 1985, 17, 147–153. [Google Scholar] [CrossRef]
- Khandjian, E.W.; Turian, G. Release of RNA-DNA-protein complex during differentiation of the water mould Allomyces arbuscula. Cell Differ. 1976, 5, 171–188. [Google Scholar] [CrossRef]
- Elzanowska, J.; Semira, C.; Costa-Silva, B. DNA in extracellular vesicles: Biological and clinical aspects. Mol. Oncol. 2020. [Google Scholar] [CrossRef]
- Ermakov, A.V.; Konkova, M.S.; Kostyuk, S.V.; Egolina, N.A.; Efremova, L.V.; Veiko, N.N. Oxidative stress as a significant factor for development of an adaptive response in irradiated and nonirradiated human lymphocytes after inducing the bystander effect by low-dose X-radiation. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 669, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Ermakov, A.V.; Konkova, M.S.; Kostyuk, S.V.; Smirnova, T.D.; Malinovskaya, E.M.; Efremova, L.V.; Veiko, N.N. An extracellular DNA mediated bystander effect produced from low dose irradiated endothelial cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 712, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Konkova, M.S.; Kaliyanov, A.A.; Sergeeva, V.A.; Abramova, M.S.; Kostyuk, S.V. Oxidized Cell-Free DNA Is a Factor of Stress Signaling in Radiation-Induced Bystander Effects in Different Types of Human Cells. Int. J. Genom. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, K.; Miura, T.; Kasai, K.; Fujishima, Y.; Nakata, A.; Yoshida, M. Radiation-Induced Bystander Effect is Mediated by Mitochondrial DNA in Exosome-Like Vesicles. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Brown, P. The Cobas® EGFR Mutation Test v2 assay. Future Oncol. 2016, 12, 451–452. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Roberts, W.L.; Fang, J.C.; Samowitz, W.S.; Heichman, K.A. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9. [Google Scholar] [CrossRef]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2020. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Koffler, D.; Agnello, V.; Winchester, R.; Kunkel, H.G. The occurrence of single-stranded DNA in the serum of patients with systemic lupus erythematosus and other diseases. J. Clin. Investig. 1973, 52, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Beiter, T.; Fragasso, A.; Hudemann, J.; Nieß, A.M.; Simon, P. Short-Term Treadmill Running as a Model for Studying Cell-Free DNA Kinetics In Vivo. Clin. Chem. 2011, 57, 633–636. [Google Scholar] [CrossRef]
- Ullrich, E.; Heidinger, P.; Soh, J.; Villanova, L.; Grabuschnig, S.; Bachler, T.; Hirschböck, E.; Sánchez-Heredero, S.; Ford, B.; Sensen, M.; et al. Evaluation of host-based molecular markers for the early detection of human sepsis. J. Biotechnol. 2020, 310, 80–88. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. https://doi.org/10.3390/ijms21218062
Grabuschnig S, Bronkhorst AJ, Holdenrieder S, Rosales Rodriguez I, Schliep KP, Schwendenwein D, Ungerer V, Sensen CW. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. International Journal of Molecular Sciences. 2020; 21(21):8062. https://doi.org/10.3390/ijms21218062
Chicago/Turabian StyleGrabuschnig, Stefan, Abel Jacobus Bronkhorst, Stefan Holdenrieder, Ingund Rosales Rodriguez, Klaus Peter Schliep, Daniel Schwendenwein, Vida Ungerer, and Christoph Wilhelm Sensen. 2020. "Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms" International Journal of Molecular Sciences 21, no. 21: 8062. https://doi.org/10.3390/ijms21218062
APA StyleGrabuschnig, S., Bronkhorst, A. J., Holdenrieder, S., Rosales Rodriguez, I., Schliep, K. P., Schwendenwein, D., Ungerer, V., & Sensen, C. W. (2020). Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. International Journal of Molecular Sciences, 21(21), 8062. https://doi.org/10.3390/ijms21218062