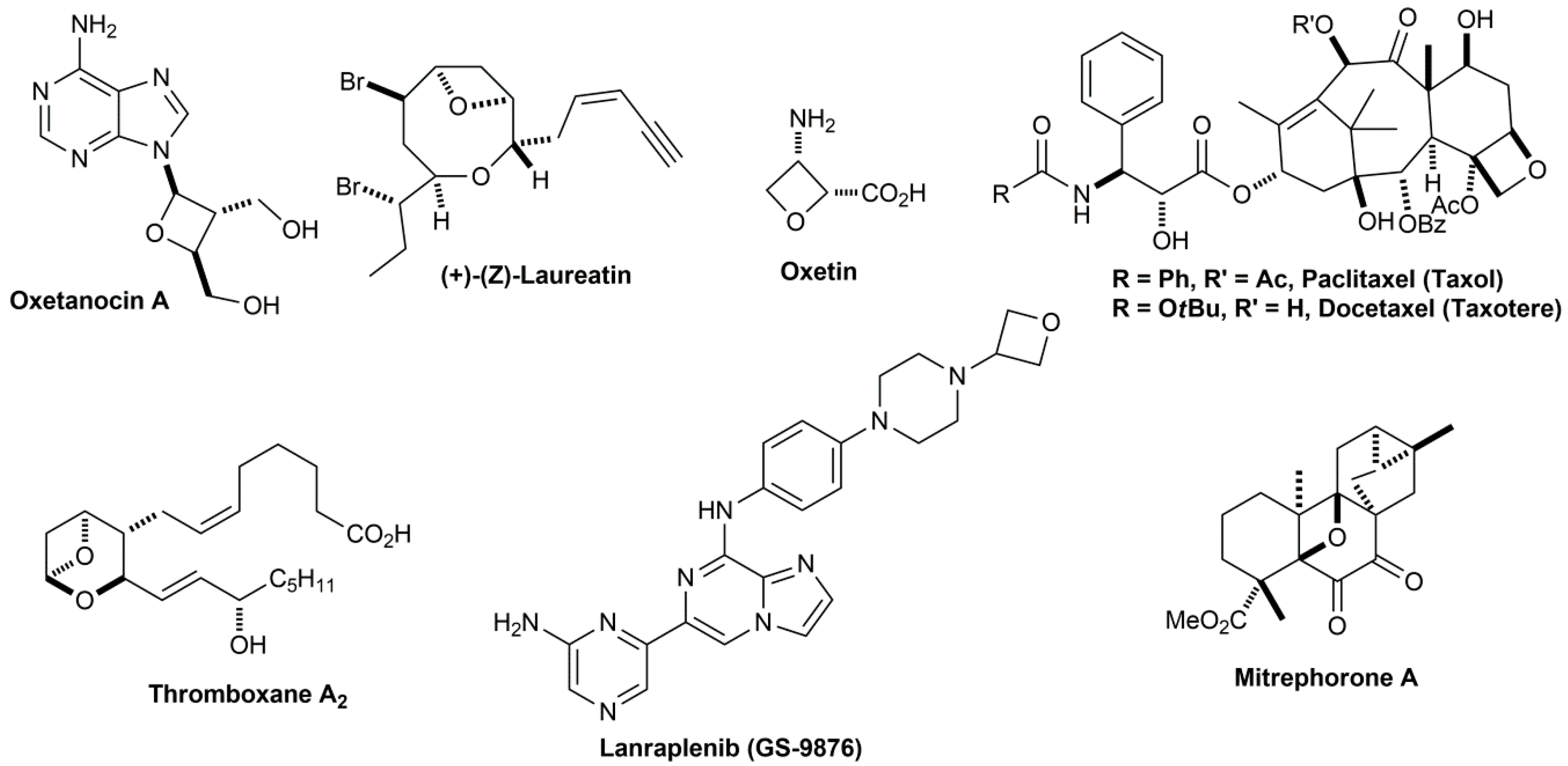
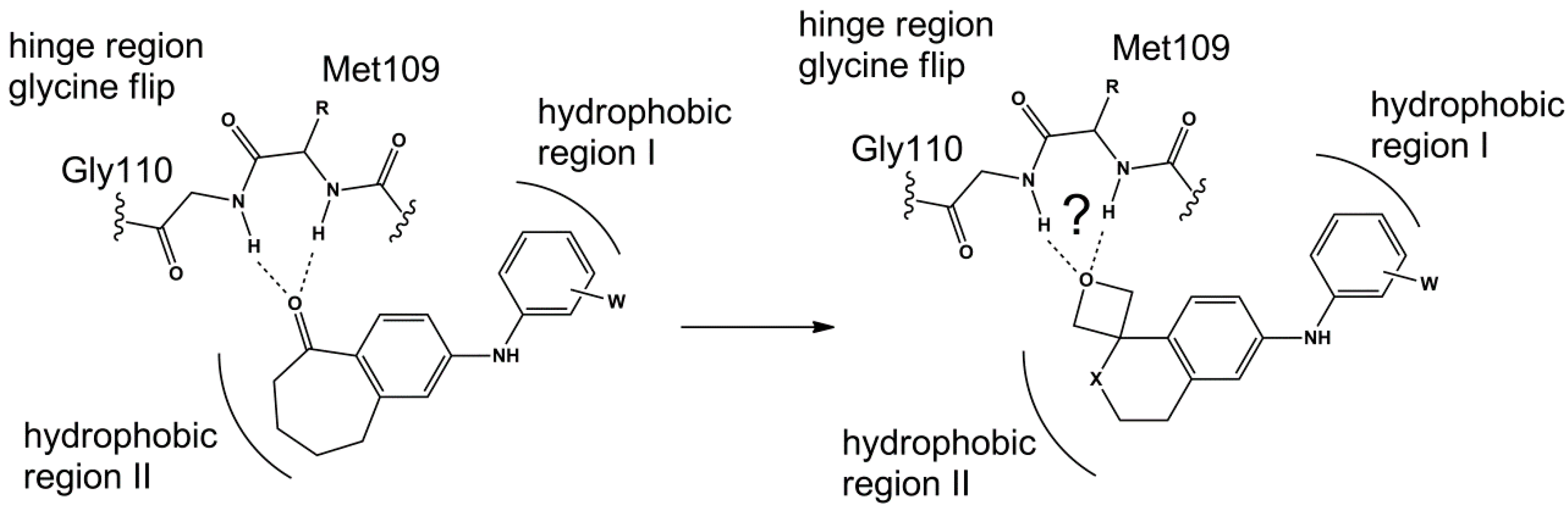
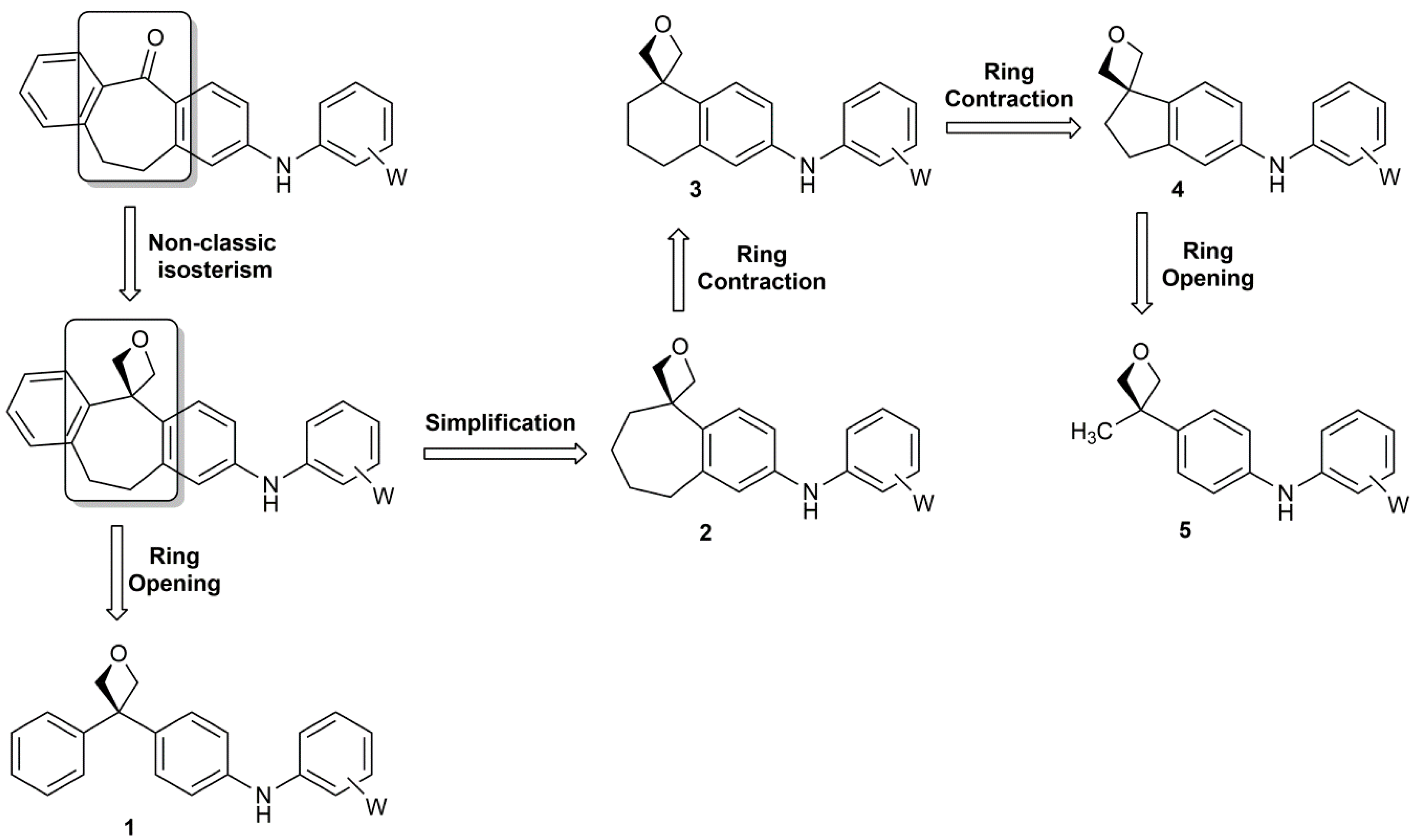
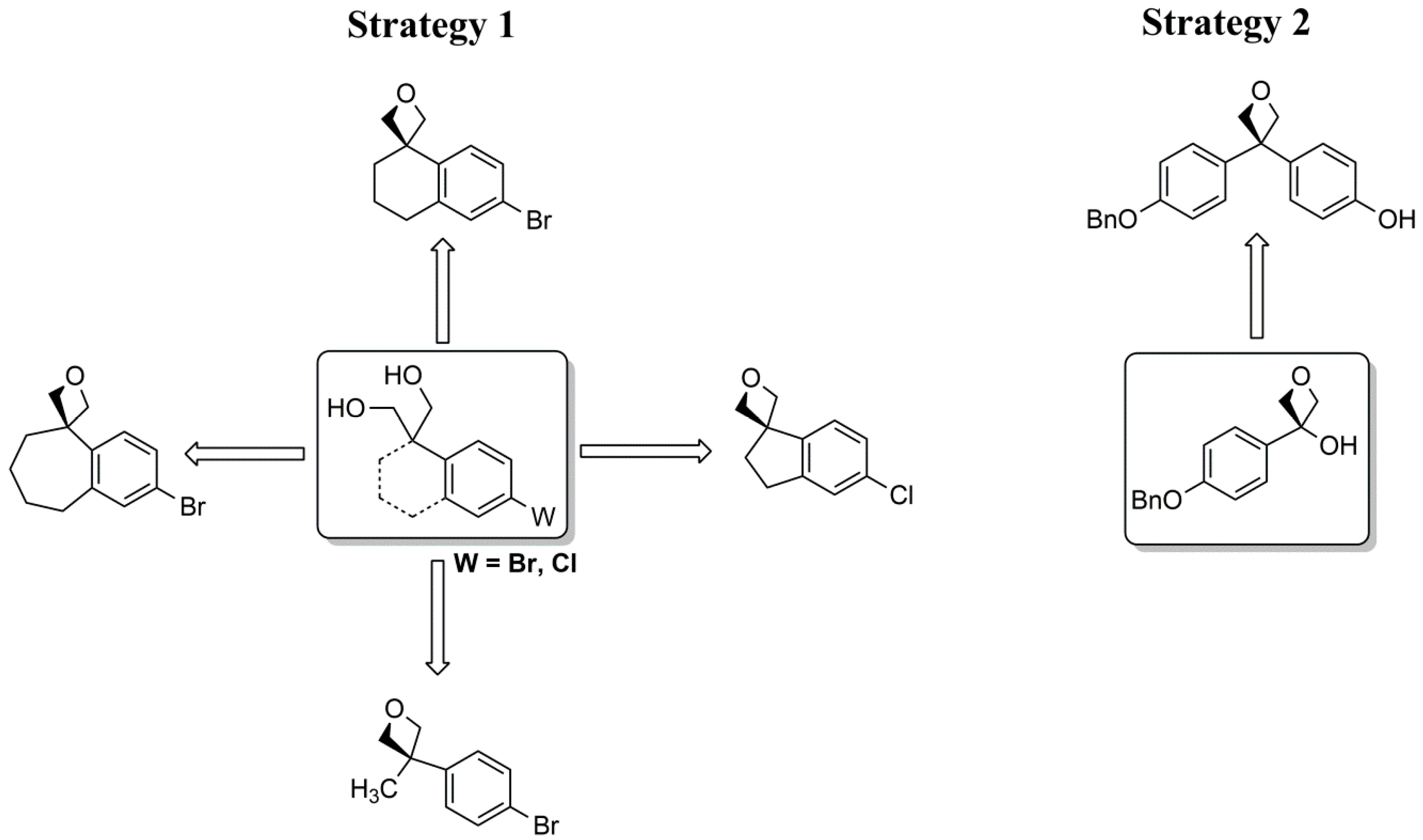
3. Materials and Methods
All commercially available reagents and solvents were used without further purification. 1H and 13C nuclear magnetic resonance (NMR) spectra were determined in DMSO-d6, CDCl3 and Acetone-d6 solutions using a Bruker Avance 200 (1H = 200 MHz; 13C = 50 MHz) or a Bruker Avance 400 (1H = 400 MHz; 13C = 100 MHz) spectrometer. The chemical shifts are given in parts per million (ppm) from solvent residual peaks and the coupling constant values (J) are given in Hz. Signal multiplicities are represented by: s (singlet), d (doublet), dd (double doublet), t (triplet), m (multiplet) and br (broad signal). The evaluation of spectra was performed using the MestReNova program. Infrared spectra were obtained using a Thermo Scientific Nicolet’s Avatar iS10 spectrometer equipped with a smart endurance diamond ATR unit for direct measurements. ESI mass spectra were obtained from a TLC-MS interface CAMAG in negative mode [M − H+]. EI-MS mass spectra were obtained from the Mass Spectrometry Department of the Institute of Organic Chemistry, University of Tübingen: Finnigan MAT, TSQ 70 Tripel quadrupol; ion source temperature at 200 °C; temperature of evaporation: 30–300 °C; ionization energy: 70 eV. The compounds were purified through column chromatography, using silica gel as stationary phase (Geduran® Si 60, 0.063–0.200 mm). The purity of compounds was determined by HPLC (Merck Hitachi L-6200 intelligent pump, Merck Hitachi AS-2000 auto sampler, Merck Hitachi L-4250 UV vis detector) using a ZORBAX Eclipse XDB C8 column (5 mm), employing a gradient of 0.01 M KH2PO4 (pH 2.3) and methanol as solvent system with a flow rate of 1.5 mL/min and detection at 254 nm.
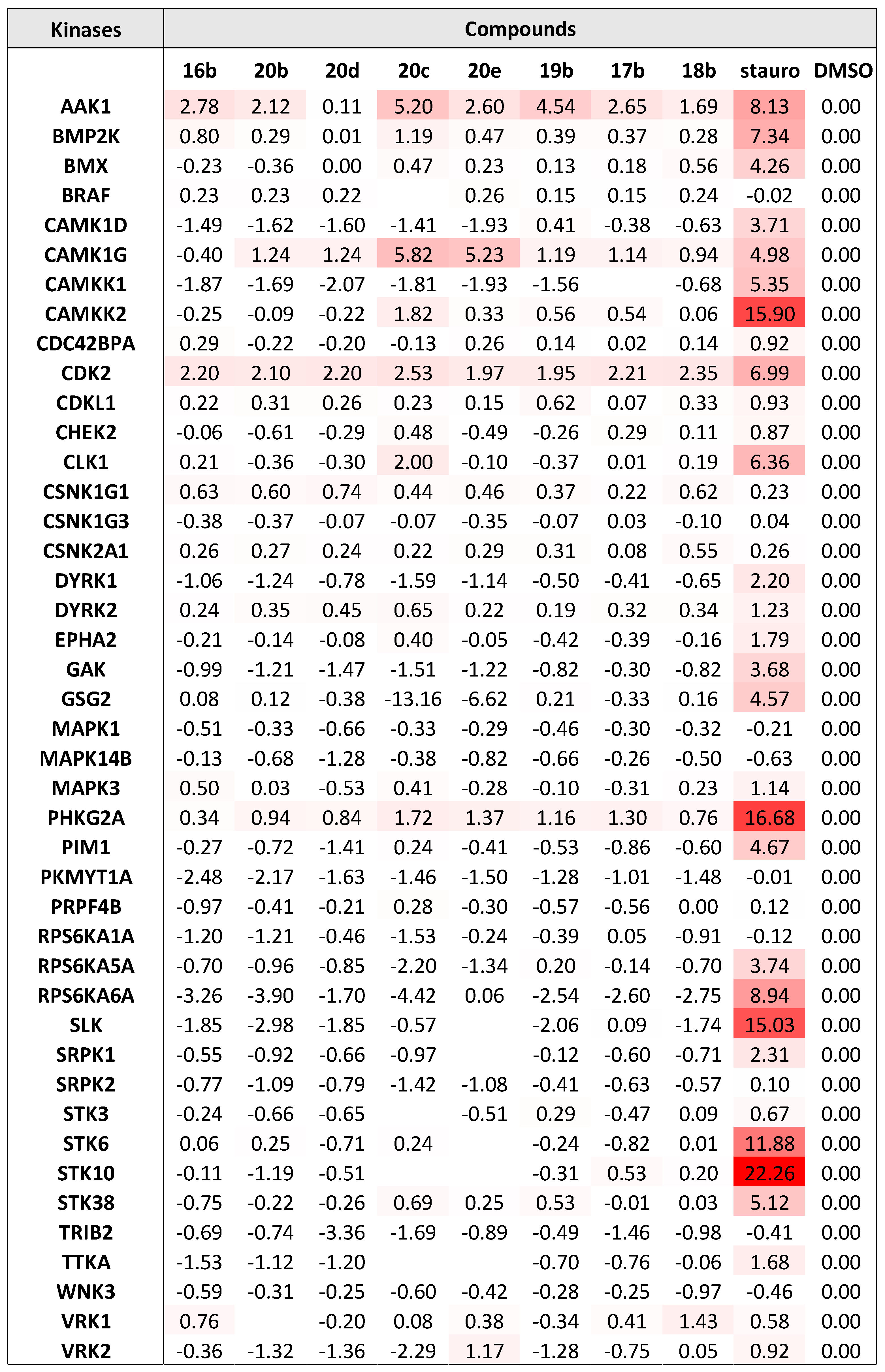
Kinase selectivity assay: Starting from 100 µM protein stocks, all the kinases present in the panel were diluted to 1 µM in buffer 100 mM K2HPO4 pH 7.5 containing 150 mM NaCl, 10% glycerol and 1X dye (Applied Biosystems catalog 4461806). The protein/dye mixture was transferred column-wise to a 384-well PCR microplate with 20 µL per well. Compounds at 10 mM concentration in DMSO (from a separate plate arranged in rows) were added next, in 20 nL volume, using a liquid handling device setup with a pin head to make a final 10 µM compound in the assay plate. The final DMSO concentration in all wells is 0.2%, including the reference with DMSO only. Thermal shift data was measured in a qPCR instrument (Applied Biosystems QuantStudio 6) programmed to equilibrate the plate at 25 °C for 5 min followed by ramping the temperature to 95 °C at a rate of 0.05 °C per second. Data was processed on Protein Thermal shift software (Applied Biosystems) fitting experimental curves to a Boltzmann function to calculate differential thermal shifts (dTm) referenced to protein/dye in 0.2% DMSO. The kinases were cloned, expressed (bacteria or insect cell expression) and purified at the laboratory of SGC.
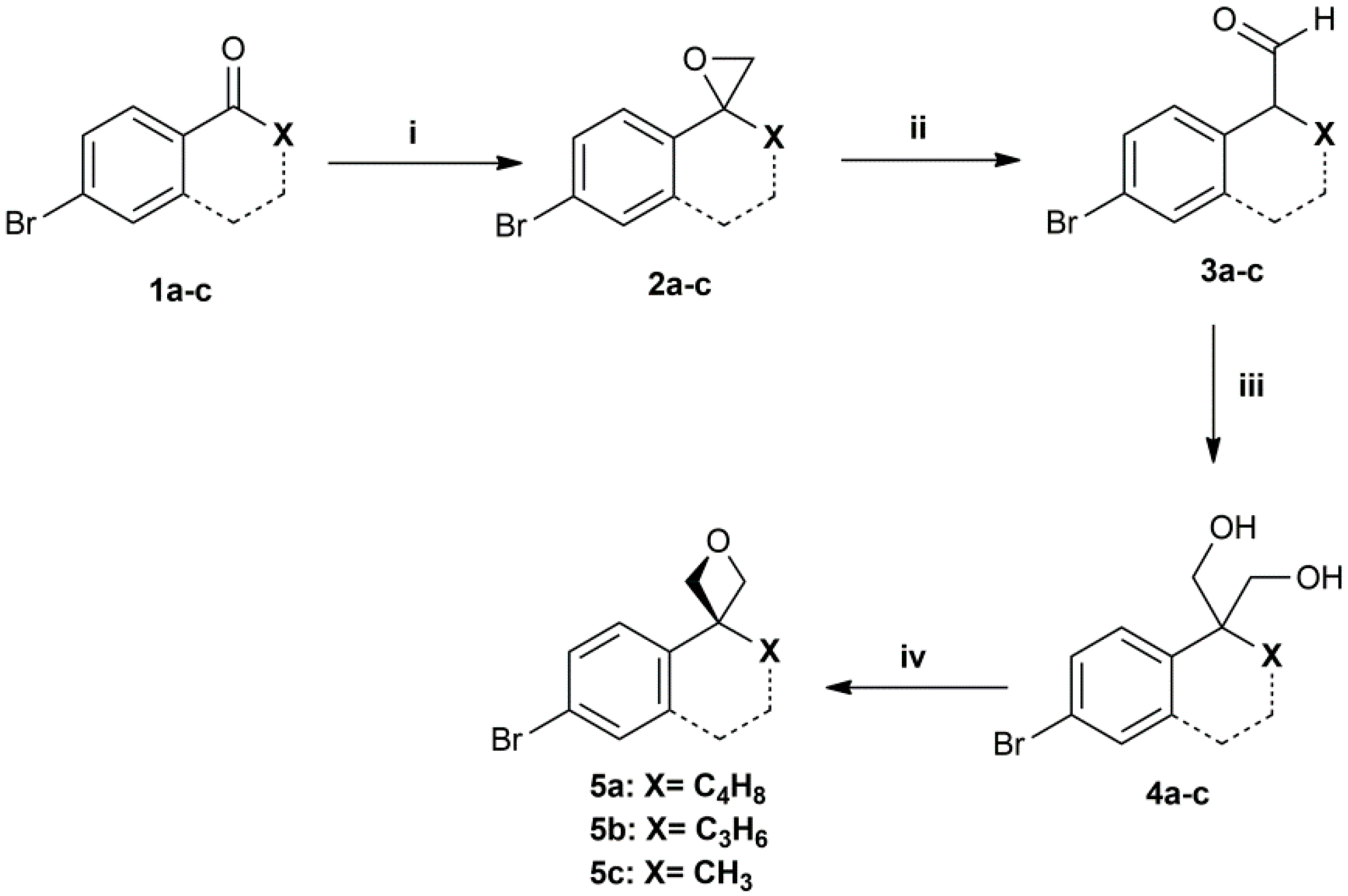
3.1. General Procedure A—Corey-Chaykovsky Epoxidation
4.0 eq. of NaH (60% dispersion in mineral oil) was placed in a three-necked flask under argon and then DMSO (0.6 M) and the ketone were added, after which 2.25 eq. of trimethylsulfonium iodide was also added, all at once. The reaction mixture was stirred at ambient temperature. After controlling through TLC, the mixture was poured over ice and extracted with diethyl ether and ethyl acetate (1:1). The extracts were combined, washed with brine and afterwards dried over Na
2SO
4. The solvent was removed under vacuum, and the residue was used without purification for the next step. Adapted from Winter [
19].
2-bromo-6,7,8,9-tetrahydrospiro[benzo[7]annulene-5,2′-oxirane] (2a): Yellow oil. 95% yield. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.62–1.80 (m, 2H), 1.80–2.15 (m, 2H), 2.74 (d, 1H, J = 6.0 Hz), 2.82–2.87 (m, 2H), 2.98 (d, 1H, J = 6.0 Hz), 7.30 (m, 3H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 27.0, 28.6, 35.9, 36.0, 53.9, 60.8, 121.2, 126.9, 129.1, 131.8, 140.1, 143.3.
6-bromo-3,4-dihydro-2H-spiro[naphthalene-1,2′-oxirane] (2b): Yellow solid. 90% yield. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.75–1.85 (m, 2H), 2.06–2.11 (m, 2H), 2.83–2.87 (m, 2H), 2.97 (s, 2H), 6.92–6.96 (d, 2H), 7.27–7.29 (s, 1H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 21.9, 29.6, 31.7, 56.4, 58.9, 121.5, 125.4, 129.6, 131.4, 134.9, 141.7.
2-(4-bromophenyl)-2-methyloxirane (2c): Yellow solid. 90% yield. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.70 (s, 3H), 2.74–2.77 (d, 1H), 2.97–2.99 (d, 1H), 7.22–7.26 (d, J = 8.0 Hz, 2H), 7.44–7.48 (d, J = 8.0 Hz, 2H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 21.7, 56.5, 57.1, 121.6, 127.2, 131.6, 140.4.
3.2. General Procedure B—Meinwald Rearrangement
The crude epoxide was dissolved in benzene (0.3 M) and 0.3 eq. of ZnI
2 was added; the reaction mixture was heated at reflux under argon. After controlling through TLC, it was then diluted with ethyl acetate and washed with water. The organic layer was dried with Na
2SO
4, and the solvent was evaporated to afford the pure aldehyde, which was used without purification for the next step. Adapted from Snyder et al. [
20].
2-bromo-6,7,8,9-tetrahydro-5H-benzo[7]annulene-5-carbaldehyde (3a): Brown oil. 98% yield. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.59–2.25 (m, 6H, CH2), 2.68 (t, 2H, CH2), 3.71 (d, 1H, CH), 6.95 (d, 1H, J = 8.0 Hz), 7.31 (m, 2H), 9.89 (s, 1H).
6-bromo-1,2,3,4-tetrahydronaphthalene-1-carbaldehyde (3b): Brown oil. 95% yield. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.92 (m, 2H), 2.24 (m, 2H), 2.72–2.78 (t, 2H), 3.58 (t, 1H), 6.99–7.03 (d, 1H), 7.30–7.34 (m, 2H), 9.64 (s, 1H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 20.2, 22.9, 29.1, 51.3, 121.2, 129.4, 129.9, 131.3, 132.6, 140.3, 201.4.
2-(4-bromophenyl)propanal (3c): Brown oil. 98% yield. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.41–1.45 (d, 3H), 3.55–3.66 (q, 1H), 7.06–7.11 (d, 2H), 7.48–7.52 (d, 2H), 9.65 (s, 1H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 14.7, 52.5, 121.7, 130.1, 132.3, 136.8, 200.5.
3.3. General Procedure C—Crossed Cannizzaro Reaction
A mixture of 35% formalin, KOH 50% aqueous solution and the aldehyde in ethylene glycol (0.4 M) was heated at reflux overnight. Water was added to the resulting mixture, and the organic layer was extracted with CH
2Cl
2, washed with brine, and dried over Na
2SO
4. After concentration of the solution, hexane was added immediately with vigorous stirring, and then the mixture was cooled to 0 °C. White precipitate was collected, washed with hexane, and dried under air to obtain the diol. Adapted from Sato et al. [
21].
(2-bromo-6,7,8,9-tetrahydro-5H-benzo[7]annulene-5,5-diyl)dimethanol (4a): White solid. 61% yield. HPLC: 6.948 min (83%) at 254 nm. IR (ATR): 3276, 2931, 2882, 2855, 1556, 1445 cm−1. ESI-MS: Calculated: 284.0 for C13H17BrO2. Found: 283 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.26 (s, 2H, CH2), 1.79 (m, 4H, CH2), 2.31 (s, 2H, CH2), 2.90 (br, 2H), 3.88 (d, 2H, J = 10 Hz), 4.04 (d, 2H, J = 12 Hz), 7.27–7.30 (m, 3H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 23.9, 27.3, 30.9, 35.9, 48.6, 68.5, 120.7, 129.3, 129.9, 134.4, 140.2, 144.8.
(6-bromo-1,2,3,4-tetrahydronaphthalene-1,1-diyl)dimethanol (4b): White solid. 45% yield. HPLC: 6.950 min (96%). IR (ATR): 3269, 2928, 2868, 2360, 1479 cm−1. ESI-MS: Calculated: 270.03 for C12H15BrO2. Found: 269 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.77–1.92 (m, 4H), 1.93 (s, 2H), 2.72–2.78 (t, 2H), 3.71–3.76 (d, 2H, J = 10 Hz), 3.87–3.92 (d, 2H, J = 10 Hz), 7.27 (s, 3H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 18.9, 27.7, 30.5, 43.3, 69.7, 120.6, 128.9, 129.2, 132.4, 136.9, 141.2.
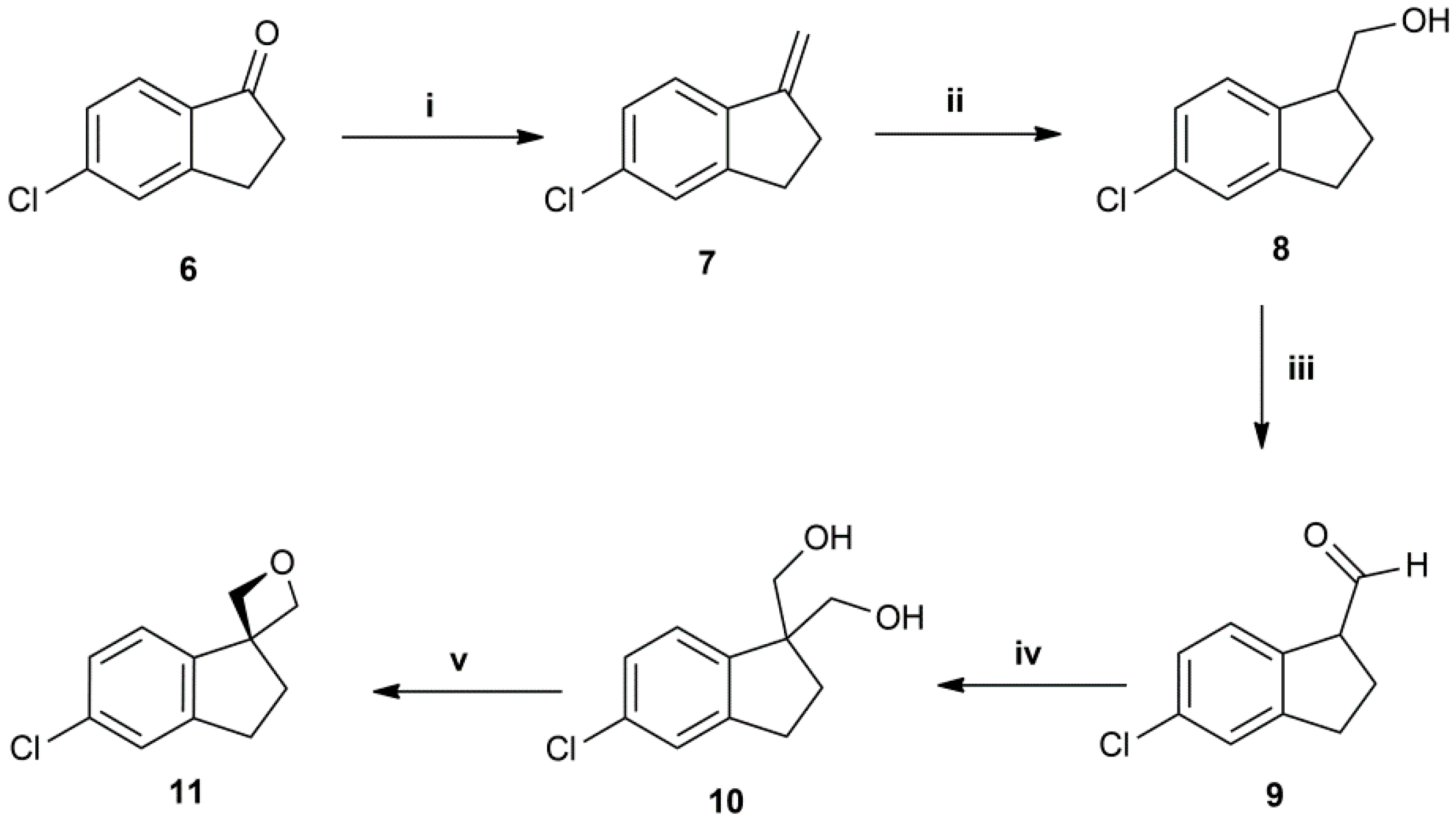
(5-chloro-2,3-dihydro-1H-indene-1,1-diyl)dimethanol (10): White solid. 30% yield. HPLC: 5.580 min (98%). IR (ATR): 3280, 2933, 2877, 2843, 1451, 1200, 824 cm−1. ESI-MS: Calculated: 212.0 for C11H13ClO2. Found: 211 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.04–2.12 (m, 4H), 2.88–2.96 (t, 2H), 3.72–3.87 (q, 4H), 7.12–7.25 (m, 3H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 30.2, 31.4, 54.2, 68.2, 125.2, 125.4, 126.8, 133.6, 143.2, 146.8.
2-(4-bromophenyl)-2-methylpropane-1,3-diol (4c): White solid. 67% yield. HPLC: 5.280 min (100%). ESI-MS: Calculated: 244.01/246.01 for C10H13BrO2. Found: 243/245 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.25 (s, 3H), 2.22 (s, 2H), 3.78–3.83 (d, 2H), 3.91–3.97 (d, 2H), 7.29–7.33 (d, 2H), 7.46–7.51 (d, 2H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 20.9, 44.5, 70.0, 120.8, 128.7, 131.8, 142.3.
3.4. General Procedure D—Wittig Reaction
Methyltriphenylphosphonium bromide (1.287 g, 3.601 mmol) was suspended in 15 mL of dry THF at 0 °C. KO
tBu (0.404 g, 3.601 mmol) was added to the suspension and a bright yellow color was observed. The mixture was stirred at 0 °C for 30 min. Then 5-chloro-2,3-dihydro-1H-inden-1-one (0.200 g, 1.200 mmol) was added neat and the reaction mixture was stirred at 0 °C to r.t. for 4 h (the solution turned green and then dark brown). Water was added to the mixture and the product was extracted with CH
2Cl
2 and dried with Na
2SO
4, filtrated and concentrated in vacuum. The residue was purified by column chromatography using
n-hexane. Adapted from Liwosz and Chemler [
24].
5-chloro-1-methylene-2,3-dihydro-1H-indene (7): Incolor oil. 83% yield. HPLC: 9.720 min (99.4%). 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.77–2.85 (m, 2H), 2.93–3.00 (m, 2H), 5.04–5.07 (t, 1H), 5.42–5.44 (t, 1H), 7.15–7.19 (d, J = 8.0 Hz, 1H), 7.23 (s, 1H), 7.38–7.42 (d, J = 8.0 Hz, 1H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 13.1, 37.6, 119.7, 124.1, 126.3, 129.2, 130.7, 139.5, 144.8, 146.1.
3.5. General Procedure E—Brown Hydroboration—Oxidation
A solution of 1.6 g (9.537 mmol) of 5-chloro-1-methylene-2,3-dihydro-1H-indene in 10 mL of THF was treated with a solution of 19.0 mL of BH
3-THF complex at ice-bath temperature. The reaction mixture was stirred until disappearance of the alkene. It was then cooled in a water-ice bath, and carefully treated with methanol followed by addition of 31.8 mL (95.365 mmol) of NaOH 3.0 M and 25.3 mL (247.959 mmol) of 30% H
2O
2. The resulting mixture was stirred at room temperature, poured into water, acidified with 10% HCl, and extracted with ether. The organic layer was dried with Na
2SO
4 and the solvent was evaporated to afford the pure alcohol after purification by column in silica gel (20% EtOAc:
n-hexane). Adapted from Zimmerman and Nesterov [
26].
(5-chloro-2,3-dihydro-1H-inden-1-yl)methanol (8): Yellow oil. 80% yield. HPLC: 9.720 min (99.4%). IR (ATR): 3323, 2940, 2866, 1598, 1471, 1067, 870 cm−1. 1H-NMR (400 MHz, CDCl3) δ in ppm: 1.62 (br s, 1H), 1.91–1.98 (sextet, 1H), 2.23–2.32 (sextet, 1H), 2.82–2.99 (m, 2H), 3.27–3.34 (quintet, 1H), 3.76–3.77 (d, 2H), 7.13–7.15 (d, 1H), 7.19–7.21 (m, 2H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 28.7, 31.4, 47.1, 65.9, 125.1, 125.3, 126.5, 132.8, 142.5, 146.8.
3.6. General Procedure F—Dess–Martin Reaction
To a solution of 0.470 g (2.573 mmol) of (5-chloro-2,3-dihydro-1H-inden-1-yl)methanol in 20 mL of CH
2Cl
2 (0.13 M) 1.6 g (3.860 mmol) of Dess–Martin periodate was added. The reaction was allowed to stir at room temperature for 3 h. TLC and HPLC analysis showed formation of the desired product. The solution was diluted with CH
2Cl
2 and a 10% Na
2S
2O
3 solution to consume the excess Dess–Martin reagent was added. The mixture was stirred until the two layers separated. The CH
2Cl
2 layer was collected, washed with saturated Na
2CO
3 solution and dried with Na
2SO
4, filtered and evaporated to give the crude aldehyde, which was used in the next step without purification. Adapted from Thongsornkleeb and Danheiser [
27].
5-chloro-2,3-dihydro-1H-indene-1-carbaldehyde (9): Yellow oil. 98% yield. 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.36–2.45 (m, 2H), 2.95–3.03 (t, 2H), 3.88–3.95 (t, 1H), 7.20 (s, 3H), 9.64 (s, 1H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 25.8, 31.7, 57.3, 125.5, 126.0, 127.1, 134.0, 137.0, 146.8, 200.2.
3.7. General Procedure G—Nucleophilic Addition to the Carbonyl Group
Compound
9 was synthesized according to General Procedure H: 2,6 mL (6.513 mmol) of
n-BuLi was added dropwise over 5 min to a solution of 1.71 g (6.513 mmol) of 1-(benzyloxy)-4-bromobenzene in 20 mL of THF at −78 °C. The reaction mixture was stirred at −78 °C for a further 30 min. Then, 0.36 g (5.010 mmol) of oxetan-3-one was added dropwise to the reaction mixture. After a further 45 min at −78 °C the reaction mixture was warmed to room temperature then quenched with water. The layers were separated and the aqueous portion extracted with diethylether. The organic extracts were combined, washed with brine, dried over Na
2SO
4, filtered and concentrated in vacuum. Purification by column chromatography (40% EtOAc:
n-hexane) afforded the product. Adapted from Croft et al. [
28].
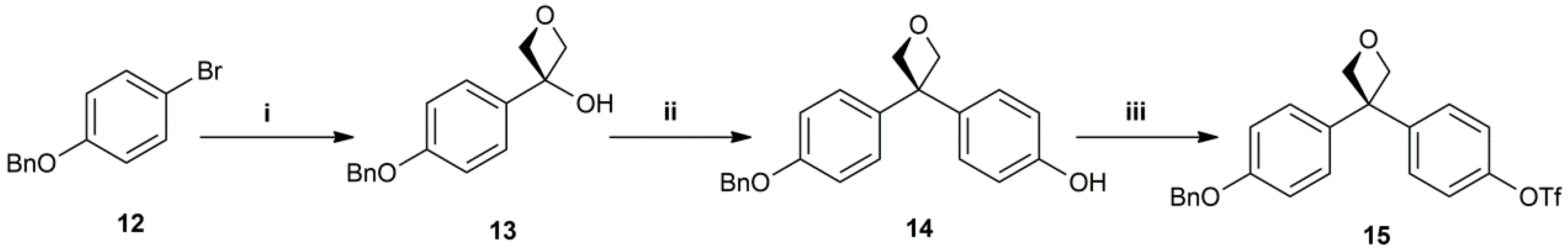
3-(4-(benzyloxy)phenyl)oxetan-3-ol (13): White solid. 70% yield. HPLC: 6.898 min (98%). 1H-NMR (400 MHz, CDCl3) δ in ppm: 2.92 (br, 1H), 4.86–4.91 (m, 4H), 5.09 (s, 2H), 7.00–7.02 (d, 2H, J = 8.0 Hz), 7.33–7.35 (d, 2H, J = 8.0 Hz), 7.38–7.45 (m, 4H), 7.47–7.49 (d,2H, J = 8.0 Hz); 13C-NMR (100 MHz, CDCl3) δ in ppm: 70.2, 75.8, 85.7, 115.2, 126.1, 127.6, 128.2, 128.8, 134.9, 136.9, 158.6.
3.8. General Procedure H—Oxetane Formation via Williamson Etherification
The diol was dissolved in THF (0.1 M) and then 4 eq. of CCl
4 was added. The solution was cooled to −48 °C and then 1.1 eq. of Tris(dimethylamino)phosphine in 1 mL of THF was added slowly to this solution during 5 min. The solution turned slightly turbid. The flask was transferred to an ice-bath and then agitated until it reached room temperature. After controlling using HPLC, the solvent was evaporated and then methanol was added followed by 4 eq. of a 5.4 M solution of sodium methoxide. The mixture was refluxed at 90 °C during 1 h. The reaction was then diluted with ethyl acetate and washed with water. The organic layer was dried with Na
2SO
4 and the solvent was evaporated to afford a product after purification by column in silica gel (5% EtOAc:
n-hexane). Adapted from Castro and Selve [
23].
2-bromo-6,7,8,9-tetrahydrospiro[benzo[7]annulene-5,3′-oxetane] (5a): Yellow oil. 47% yield. HPLC: 8.497 min (93%). EI-MS: Calculated 266.03 for C13H15BrO. Found: 235.9 [M-CH2O]+●. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.53–1.59 (t, 2H), 1.99–2.01 (t, 2H), 2.12–2.15 (d, 2H), 2.4–2.45 (d, 2H), 4.70–4.73 (d, J = 6.0 Hz, 2H), 4.97–5.00 (d, J = 6.0 Hz, 2H), 6.92–6.96 (d, 1H), 7.21–7.33 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ in ppm: 26.4, 26.9, 34.9, 36.5, 47.9, 80.4, 120.0, 129.2, 132.5, 143.4, 144.4.
6-bromo-3,4-dihydro-2H-spiro[naphthalene-1,3′-oxetane] (5b): Yellow oil. 48% yield. HPLC: 8.230 min (98%). EI-MS: Calculated 252.01 for C12H13BrO. Found: 221.9 [M-CH2O]+●. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.65–1.77 (m, 2H), 2.14–2.20 (t, 2H), 2.69–2.75 (t, 2H), 4.62–4.65 (d, J = 6.0 Hz, 2H), 4.75–4.78 (d, J = 6.0 Hz, 2H), 7.21–7.22 (s, 1H), 7.38–7.43 (d, 1H), 7.77–7.81 (d, 1H). 13C-NMR (50 MHz, CDCl3) δ in ppm: 20.0, 29.9, 35.3, 42.2, 85.4, 120.4, 128.4, 129.9, 131.7, 138.7, 139.2.
5-chloro-2,3-dihydrospiro[indene-1,3′-oxetane] (11): Yellow oil. 47% yield. HPLC: 7.664 min (97%). EI-MS: Calculated 194.0 for C11H11ClO. Found: 163.9 [M-CH2O]+●. 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.43–2.50 (t, J = 6.0 Hz, 2H), 2.83–2.90 (t, J = 6.0 Hz, 2H), 4.80 (s, 4H), 7.17 (s, 1H), 7.18–7.29 (d, 1H), 7.56–7.60 (d, 1H). 13C-NMR (50 MHz, CDCl3) δ in ppm: 30.4, 37.9, 50.9, 84.2, 123.8, 124.8, 127.4, 133.4, 144.7, 145.4.
3-(4-bromophenyl)-3-methyloxetane (5c): Yellow solid. 35% yield. HPLC: 7.390 min (98%). EI-MS: Calculated 226.00 for C10H11BrO. Found: 195.9 [M-CH2O]+●. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.71 (s, 3H), 4.62–4.64 (d, 2H), 4.90–4.93 (d, 2H), 7.07–7.11 (d, J = 8.0 Hz, 2H), 7.46–7.50 (d, J = 8.0 Hz, 2H). 13C-NMR (50 MHz, CDCl3) δ in ppm: 27.7, 43.3, 83.64, 120.3, 127.0, 131.8, 145.6.
3.9. General Procedure I—Oxetane Formation via Friedel-Crafts Reaction
Quantities of 0.008 g (0.028 mmol) of lithium bis(trifluoromethanesulfonimide) and 0.006 g (0.014 mmol) of tetrabutylammonium hexafluorophosphate were added to a solution of 0.066 g (0.258 mmol) of 3-(4-(benzyloxy)phenyl)oxetan-3-ol and 0.12 g (1.288 mmol) of phenol in 0.5 mL of chloroform. The reaction mixture was stirred at 40 °C for 1 h then quenched with sat. aq. NaHCO
3. CH
2Cl
2 was added and the layers were separated. The organic extracts were combined, dried over Na
2SO
4, filtered and concentrated in vacuum. Purification by column chromatography (30% EtOAc:
n-hexane) afforded the oxetane. Adapted from Croft et al. [
28].
4-(3-(4-(benzyloxy)phenyl)oxetan-3-yl)phenol (14): White solid. 60% yield. HPLC: 8.286 min (93%). ESI-MS: Calculated: 332.14 for C22H20O3. Found: 331.1 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 5.06 (s, 2H), 5.21 (s, 4H), 6.78–6.82 (d, J = 8.0 Hz, 2H), 6.94–6.98 (d, J = 8.0 Hz, 2H), 7.05–7.15 (m, 4H), 7.36–7.42 (m, 5H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 50.5, 70.2, 85.2, 114.9, 115.5, 127.6, 127.8, 127.9, 128.2, 128.8, 137.1, 138.3, 138.5, 154.4, 157.6.
3.10. General Procedure J—Triflate Formation
First, 0.2 mL (2.708 mmol) of pyridine, then 0.3 mL (2.031 mmol) of triflic anhydride were added to a solution of 4-(3-(4-(benzyloxy)phenyl)oxetan-3-yl)phenol in 3.0 mL of CH
2Cl
2. The reaction mixture was stirred at room temperature. After controlling through TLC and HPLC, the work-up was done adding water followed by CH
2Cl
2. The layers were separated and the aqueous portion was extracted with CH
2Cl
2. The organic extracts were combined, dried over Na
2SO
4, filtered and concentrated in vacuum. Purification by column chromatography (20% EtOAc:
n-hexane) afforded the triflate. Adapted from Thompson et al. [
30].
4-(3-(4-(benzyloxy)phenyl)oxetan-3-yl)phenyl trifluoromethanesulfonate (15): White solid. 90% yield. HPLC: 10.332 min (100%). ESI-MS: Calculated: 464.09 for C23H19F3O5S. Found: 463.0 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 5.06 (s, 2H), 5.21 (s, 4H), 6.78–6.82 (d, J = 8.0 Hz, 2H), 6.94–6.98 (d, J = 8.0 Hz, 2H),7.05–7.15 (m, 4H), 7.36–7.42 (m, 5H).
3.11. General Procedure K—Buchwald-Hartwig Reaction via Halides
A mixture of 1.0 eq. of aryl halide, 1.0 eq. of amine, 3.0 eq. of Cs
2CO
3 as the base, 0.5 eq. of X-Phos as the ligand and 0.1 eq. of Pd(OAc)
2 as the catalyst in dry 1,4-dioxane and absolute tert-butanol (4:1) (0.05 M) was stirred at reflux under atmosphere of argon. After controlling through TLC and HPLC, the mixture was allowed to cool to room temperature. It was diluted with water and subsequently extracted with ethyl acetate. The extracts were combined, washed with brine and afterwards dried over Na
2SO
4. The solvent was removed under vacuum, and the residue was purified by column chromatography, affording the desired product. Adapted from Martz et al. [
31].
N-phenyl-6,7,8,9-tetrahydrospiro[benzo[7]annulene-5,3′-oxetan]-2-amine (16a): Brown solid. 86% yield. HPLC: 9.015 min (96%). ESI-MS: Calculated: 279.1 for C19H21NO. Found: 278.0 [M − H]−. 1H-NMR (400 MHz, CDCl3) δ in ppm: 1.59 (m, 2H, CH2), 2.01 (m, 2H, CH2), 2.18 (t, 2H, J = 8.0 Hz, CH2), 2.42 (t, 2H, J = 8.0 Hz, CH2), 4.73 (d, 2H), 5.04 (d, 2H), 5.78 (br, 1H), 6.82 (s, 1H), 6.91–7.00 (m, 3H), 7.08 (d, 2H, J = 8.0 Hz),7.27 (t, 2H, J = 8.0 Hz); 13C-NMR (100 MHz, CDCl3) δ in ppm: 26.5, 27.3, 35.5, 37.1, 47.7, 80.7, 115.1, 117.9, 119.4, 121.0,126.2, 129.5, 137.1, 141.8, 143.4.
N-(2,4-difluorophenyl)-6,7,8,9-tetrahydrospiro[benzo[7]annulene-5,3′-oxetan]-2-amine (16b): Brown solid. 73% yield. HPLC: 8.542 min (98%). ESI-MS: Calculated: 315.1 for C19H19F2NO. Found: 314.2 [M − H]−. 1H-NMR (400 MHz, CDCl3) δ in ppm: 1.49 (m, 2H), 1.91 (m, 2H), 2.08 (t, 2H), 2.31 (t, 2H), 4.63–4.64 (d, 2H, J = 4.0 Hz), 4.93–4.94 (d, 2H, J = 4.0 Hz), 5.46 (br, 1H), 6.66 (s, 1H), 6.70–6.82 (m, 3H), 6.89 (d, 1H, J = 8.0 Hz), 7.18 (s, 1H); 13C-NMR (100 MHz, CDCl3) δ in ppm: 26.5, 27.3, 37.1, 47.7, 80.7, 104.1 (dd, J1 = 23.6 Hz, J2 = 23.7 Hz), 110.9 (dd, J1 = 3.64 Hz, J2 = 3.71 Hz), 114.9, 119.3, 119.5 (dd, J1 = 2.28 Hz, J2 = 3.17 Hz), 126.4, 127.9 (dd, J1 = 2.35 Hz, J2 = 2.36 Hz), 137.6, 141.4, 143.6.
N-(2-(trifluoromethyl)phenyl)-6,7,8,9-tetrahydrospiro[benzo[7]annulene-5,3′-oxetan]-2-amine (16c): Yellow solid. 77% yield. HPLC: 10.008 min (96%). ESI-MS: Calculated: 347.1 for C20H20F3NO. Found: 346.1 [M − H]−. 1H-NMR (400 MHz, CDCl3) δ in ppm: 1.58 (m, 2H), 1.99 (m, 2H), 2.17 (t, 2H), 2.42 (t, 2H), 4.73 (d, 2H), 5.02 (d, 2H), 6.02 (br, 1H), 6.84 (s, 1H), 6.90–6.96 (m, 2H), 7.01 (d, 1H, J = 8.0 Hz), 7.32–7.39 (m, 2H), 7.54 (d, 1H, J = 8.0 Hz); 13C-NMR (100 MHz, CDCl3) δ in ppm: 26.5, 27.2, 35.4, 36.9, 47.8, 80.7, 117.2, 117.5, 117.7 (d, J = 10.6 Hz), 119.6, 122.0, 123.6, 126.4, 127.0 (dd, J1 = 5.5 Hz, J2 = 5.4 Hz), 132.8, 139.0, 140.1, 142.5, 143.7.
N-(4-methoxyphenyl)-6,7,8,9-tetrahydrospiro[benzo[7]annulene-5,3′-oxetan]-2-amine (16d): Yellow solid. 86% yield. HPLC: 7.976 min (98%). ESI-MS: Calculated: 309.1 for C20H23NO2. Found: 308.0 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.58 (m, 2H), 1.98 (m, 2H), 2.17 (t, 2H), 2.43 (t, 2H), 3.80 (s, 3H), 4.67 (d, 2H, J = 6.0 Hz), 4.99 (d, 2H, J = 6.0 Hz), 5.45 (br, 1H), 6.63–6.74 (m, 3H), 6.84–6.93 (m, 3H), 7.04 (t, 2H); 13C-NMR (100 MHz, CDCl3) δ in ppm: 26.6, 27.3, 35.6, 37.2, 47.9, 55.7, 112.8, 114.8, 117.3, 122.2, 126.2, 135.6, 136.0, 143.3, 143.9, 155.4.
N-phenyl-3,4-dihydro-2H-spiro[naphthalene-1,3′-oxetan]-6-amine (17a): Brown solid. 92% yield. HPLC: 7.967 min (97%). ESI-MS: Calculated: 265.1 for C18H19NO. Found: 264.0 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.75–1.84 (m, 2H), 2.24 (t, 2H, J = 6.0 Hz), 2.75 (t, 2H, J = 6.0 Hz), 4.66 (d, 2H, J = 6.0 Hz), 4.87 (d, 2H, J = 6.0 Hz), 6.84 (s, 1H), 6.94–7.10 (m, 4H), 7.33 (t, 2H), 7.83 (d, 2H, J = 8.0 Hz); 13C-NMR (50 MHz, CDCl3) δ in ppm: 20.3, 30.4, 35.9, 42.1, 85.8, 116.8, 117.4, 117.9, 121.1, 127.7, 129.5, 132.1, 138.1, 141.7, 143.2.
N-(2,4-difluorophenyl)-3,4-dihydro-2H-spiro[naphthalene-1,3′-oxetan]-6-amine (17b): Brown solid. 51% yield. HPLC: 8.586 min (98%). ESI-MS: Calculated: 301.1 for C18H17F2NO. Found: 299.9 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.70–1.79 (m, 2H), 2.19 (t, 2H, J = 6.0 Hz), 2.70 (t, 2H, J = 6.0 Hz), 4.61 (d, 2H, J = 6.0 Hz), 4.80 (d, 2H, J = 6.0 Hz), 6.70–6.99 (m, 4H), 7.19–7.31 (m, 1H), 7.79 (d, 2H, J = 10 Hz); 13C-NMR (50 MHz, CDCl3) δ in ppm: 20.3, 30.4, 35.8, 42.1, 85.8, 103.9 (dd, J1 = 23.5 Hz, J2 = 23.5 Hz), 110.8 (dd, J1 = 3.76 Hz, J2 = 3.79 Hz), 116.5, 117.3, 119.6 (dd, J1 = 3.22 Hz, J2 = 3.30 Hz), 127.9, 132.7, 138.3, 141.3, 151.0 (d, J = 11.8 Hz), 154.5 (d, J = 11.1 Hz), 155.9 (d, J = 11.8 Hz), 159.3 (d, J = 11.3 Hz).
N-(2-(trifluoromethyl)phenyl)-3,4-dihydro-2H-spiro[naphthalene-1,3′-oxetan]-6-amine (17c): White solid. 82% yield. HPLC: 9.943 min (98%). ESI-MS: Calculated: 333.1 for C19H18F3NO. Found: 332.2 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.71–1.74 (m, 2H), 2.20 (t, 2H, J = 6.0 Hz), 2.72 (t, 2H, J = 6.0 Hz), 4.63 (d, 2H, J = 6.0 Hz), 4.82 (d, 2H, J = 6.0 Hz), 6.03 (br, 1H), 6.83 (s, 1H), 6.94 (t, 1H, J = 8.0 Hz), 7.06 (d, 1H, J = 8.0 Hz), 7.32–7.43 (m, 2H), 7.54 (d, 1H, J = 8.0 Hz), 7.84 (d, 1H, J = 8.0 Hz); 13C-NMR (50 MHz, CDCl3) δ in ppm: 20.3, 30.3, 35.8, 42.2, 85.8, 117.3, 117.9, 118.0, 118.9, 119.8, 120.1, 126.9 (dd, J1 = 5.5 Hz, J2 = 5.5 Hz), 127.9, 132.7, 134.1, 138.3, 140.1, 142.2.
N-(2-methoxyphenyl)-3,4-dihydro-2H-spiro[naphthalene-1,3′-oxetan]-6-amine (17d): Yellow solid. 82% yield. HPLC: 9.221 min (95%). ESI-MS: Calculated: 295.1 for C19H21NO2. Found: 294.4 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.72–1.83 (m, 2H), 2.24 (t, 2H, J = 6.0 Hz), 2.76 (t, 2H, J = 6.0 Hz), 3.93 (s, 3H), 4.66 (d, 2H, J = 6.0 Hz), 4.87 (d, 2H, J = 6.0 Hz), 6.92 (m, 4H), 7.13 (d, 1H), 7.32 (m, 1H), 7.84 (d, 1H, J = 8.0 Hz); 13C-NMR (50 MHz, CDCl3) δ in ppm: 20.4, 30.4, 35.9, 42.1, 55.7, 85.8, 110.7, 114.9, 117.5, 118.2, 120.0, 120.9, 127.7, 132.3, 133.0, 138.0, 141.3, 148.4.
N-phenyl-2,3-dihydrospiro[indene-1,3′-oxetan]-5-amine (18a): Brown solid. 71% yield. HPLC: 8.231 min (95%). ESI-MS: Calculated: 251.1 for C17H17NO. Found: 250.1 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.52 (t, 2H, J = 8.0 Hz), 2.89 (d, 2H, J = 8.0 Hz), 4.83–4.92 (m, 4H), 5.82 (br, 1H), 6.94–7.12 (m, 5H), 7.28–7.36 (m, 2H), 7.57 (d, 2H, J = 8.0 Hz); 13C-NMR (50 MHz, CDCl3) δ in ppm: 30.6, 38.4, 50.9, 84.6, 113.8, 117.4, 177.9, 121.1, 123.6, 129.5, 138.8, 143.0, 143.5, 144.9.
N-(2,4-difluorophenyl)-2,3-dihydrospiro[indene-1,3′-oxetan]-5-amine (18b): Brown solid. 76% yield. HPLC: 8.670 min (100%). ESI-MS: Calculated: 287.1 for C17H15F2NO. Found: 286.1 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.47 (t, 2H, J = 6.0 Hz), 2.89 (d, 2H, J = 6.0 Hz), 4.79–4.87 (m, 4H), 6.76–6.98 (m, 4H), 7.17–7.29 (m, 1H), 7.53 (d, 2H, J = 8.0 Hz); 13C-NMR (100 MHz, CDCl3) δ in ppm: 30.6, 38.3, 50.9, 84.6, 103.9, 104.4 (J = 2.87 Hz), 110.8 (J1 = 3.83, J2 = 3.97 Hz), 113.6, 117.3, 119.6 (J1 = 2.86, J2 = 3.34 Hz), 123.7, 139.3, 142.7, 145.1.
N-(2-(trifluoromethyl)phenyl)-2,3-dihydrospiro[indene-1,3′-oxetan]-5-amine (18c): White solid. 95% yield. HPLC: 9.588 min (100%). ESI-MS: Calculated: 319.1 for C18H16F3NO. Found: 318.1 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.49 (t, 2H, J = 8.0 Hz), 2.86 (d, 2H, J = 8.0 Hz), 4.80–4.88 (m, 4H), 6.06 (br, 1H), 6.90–6.97 (m, 2H), 7.04 (d, 2H, J = 8.0 Hz), 7.29–7.41 (m, 2H), 7.58 (t, 2H, J = 8.0 Hz); 13C-NMR (50 MHz, CDCl3) δ in ppm: 30.6, 38.2, 50.9, 84.5, 116.5, 117.9, 119.8, 123.7, 127.1, 132.8, 140.7, 141.4, 142.5, 145.1.
N-(2-methoxyphenyl)-2,3-dihydrospiro[indene-1,3′-oxetan]-5-amine (18d): Yellow solid. 80% yield. HPLC: 8.718 min (98%). ESI-MS: Calculated: 281.1 for C18H19NO2. Found: 280.1 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 2.48 (t, 2H, J = 8.0 Hz), 2.86 (d, 2H, J = 8.0 Hz), 3.89 (s, 3H), 4.79–4.89 (m, 4H), 6.15 (br, 1H), 6.85–6.91 (m, 3H), 7.02 (s, 1H), 7.07 (d, 1H), 7.28 (m, 1H), 7.54 (d, 2H, J = 8.0 Hz); 13C-NMR (50 MHz, CDCl3) δ in ppm: 30.6, 38.4, 50.9, 55.7, 84.6, 110.6, 114.5, 114.8, 118.1, 119.9, 120.9, 123.5, 133.3, 138.9, 142.6, 144.8, 148.3.
4-(3-methyloxetan-3-yl)-N-phenylaniline (19a): Brown solid. 83% yield. HPLC: 7.967 min (96%). ESI-MS: Calculated: 239.1 for C16H17NO. Found: 238.0 [M − H]−. 1H-NMR (400 MHz, CDCl3) δ in ppm: 1.75 (s, 3H, CH3), 4.65–4.66 (d, 2H, J = 4.0 Hz, CH2), 4.98–4.99 (d, 2H, J = 4.0 Hz, CH2), 5.76 (br, 1H), 6.94–6.97 (t, 1H, J = 8.0 Hz), 7.08–7.11 (m, 4H), 7.16–7.18 (d, 2H, J = 8.0 Hz), 7.28–7.36 (t, 2H, J = 8.0 Hz); 13C-NMR (100 MHz, CDCl3) δ in ppm: 27.7, 43.0, 84.2, 117.8, 118.2, 121.1, 126.2, 129.5, 139.1, 141.6, 143.4.
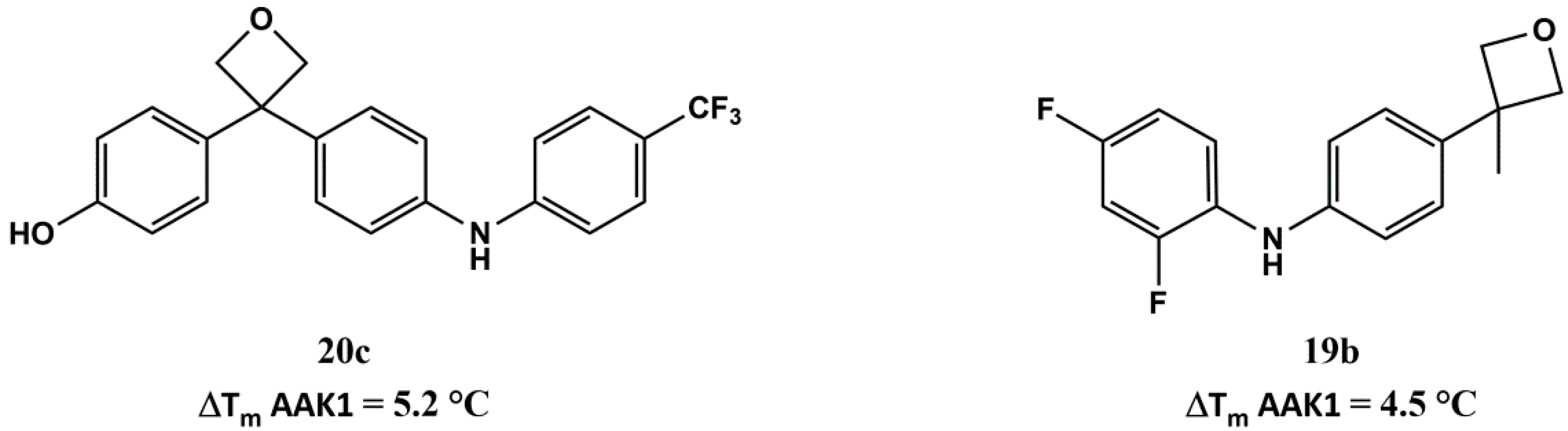
2,4-difluoro-N-(4-(3-methyloxetan-3-yl)phenyl)aniline (19b): Brown solid. 71% yield. HPLC: 7.689 min (100%). ESI-MS: Calculated: 275.1 for C16H15F2NO. Found: 274.0 [M − H]−. 1H-NMR (200 MHz, CDCl3) δ in ppm: 1.73 (s, 3H), 4.62 (d, 2H, J = 6.0 Hz), 4.94 (d, 2H, J = 6.0 Hz), 6.75–6.95 (m, 2H), 6.99 (d, 2H, J = 10 Hz), 7.14 (d, 2H, J = 8.0 Hz), 7.23–7.28 (d, 1H); 13C-NMR (50 MHz, CDCl3) δ in ppm: 27.7, 42.9, 84.1, 103.9 (dd, J1 = 23.5 Hz, J2 = 23.5 Hz), 110.8 (dd, J1 = 3.8 Hz, J2 = 3.8 Hz), 117.9, 119.3 (dd, J1 = 3.4 Hz, J2 = 3.3 Hz), 126.3, 127.8 (dd, J1 = 3.4 Hz, J2 = 3.1 Hz), 139.6, 141.2, 150.9 (d, J = 11.9 Hz), 154.5 (d, J = 11.1 Hz), 155.8 (d, J = 11.7 Hz), 159.3 (d, J = 11.1 Hz).
N-(4-(3-methyloxetan-3-yl)phenyl)-2-(trifluoromethyl)aniline (19c): White solid. 88% yield. HPLC: 9.306 min (100%). ESI-MS: Calculated: 307.1 for C17H16F3NO. Found: 306.0 [M − H]−. 1H-NMR (400 MHz, CDCl3) δ in ppm: 1.78 (s, 3H, CH3), 4.67–4.70 (d, 2H, J = 6.0 Hz, CH2), 4.99–5.02 (d, 2H, J = 6.0 Hz, CH2), 6.10 (br, 1H), 6.94–7.02 (t, 1H, J = 8.0 Hz), 7.13–7.25 (q, 4H), 7.30–7.41 (m, 2H), 7.59–7.63 (d, 2H, J = 8.0 Hz); 13C-NMR (100 MHz, CDCl3) δ in ppm: 27.7, 43.1, 84.0, 114.3, 117.7, 119.8, 120.7, 126.4, 127.1, 132.8, 140.0, 140.7, 141.1, 142.4.
2-methoxy-N-(4-(3-methyloxetan-3-yl)phenyl)aniline (19d): Yellow solid. 81% yield. HPLC: 8.251 min (98%). EI-MS: Calculated: 269.1 for C17H19NO2. Found: 269.1. 1H-NMR (400 MHz, CDCl3) δ in ppm: 1.73 (s, 3H, CH3), 3.90 (s, 3H, OCH3), 4.62–4.64 (d, 2H, J = 8.0 Hz, CH2), 4.96–4.97 (d, 2H, J = 4.0 Hz, CH2), 6.85–6.90 (m, 3H), 7.15 (s, 4H), 7.27–7.29 (d, 1H); 13C-NMR (100 MHz, CDCl3) δ in ppm: 27.7, 43.0, 55.7, 84.2, 110.7, 114.7, 118.9, 120.0, 120.9, 126.1, 133.2, 139.2, 141.2, 148.4.
3.12. General Procedure L—Buchwald–Hartwig Reaction via Triflates—Benzyl Ether Deprotection
A mixture of aryl triflate, amine, Cs
2CO
3 as the base, BINAP as the ligand and Pd(OAc)
2 as the catalyst in dry 1,4-dioxane and absolute tert-butanol (4:1) (0.03 M) was stirred at 110 °C under an atmosphere of argon. After controlling through TLC and HPLC, the mixture was allowed to cool to room temperature. It was diluted with water and subsequently extracted with ethyl acetate. The extracts were combined, washed with brine and afterwards dried over Na
2SO
4. The solvent was removed under vacuum, and the residue was purified by column chromatography, affording the product. Adapted from Åhman and Buchwald [
33].
Then the benzyl ether deprotection was performed as follows: 0.6 eq. of palladium on carbon (10%
w/w) was added to a solution of oxetane in ethanol/acetonitrile. The reaction mixture was stirred under an atmosphere of hydrogen at 25 °C until HPLC analysis indicated the end of reaction. The reaction mixture was filtered through celite, washed through with ethanol and then concentrated in vacuum to afford the final oxetane. Adapted from Croft et al. [
28].
4-(3-(4-(phenylamino)phenyl)oxetan-3-yl)phenol (20a): White solid. 85% yield. HPLC: 7.618 min (96%). ESI-MS: Calculated: 317.1 for C21H19NO2. Found: 316.2 [M − H]−. 1H-NMR (400 MHz, d-acetone) δ in ppm: 5.13 (s, 4H), 6.82–6.85 (t, 3H), 7.10–7.17 (m, 8H), 7.20–7.24 (t, 2H), 7.41 (br, 1H); 13C-NMR (100 MHz, d-acetone) δ in ppm: 51.3, 84.9, 116.1, 117.9, 118.1, 120.9, 128.0, 129.9, 138.5, 139.5, 142.9, 144.7, 156.7.
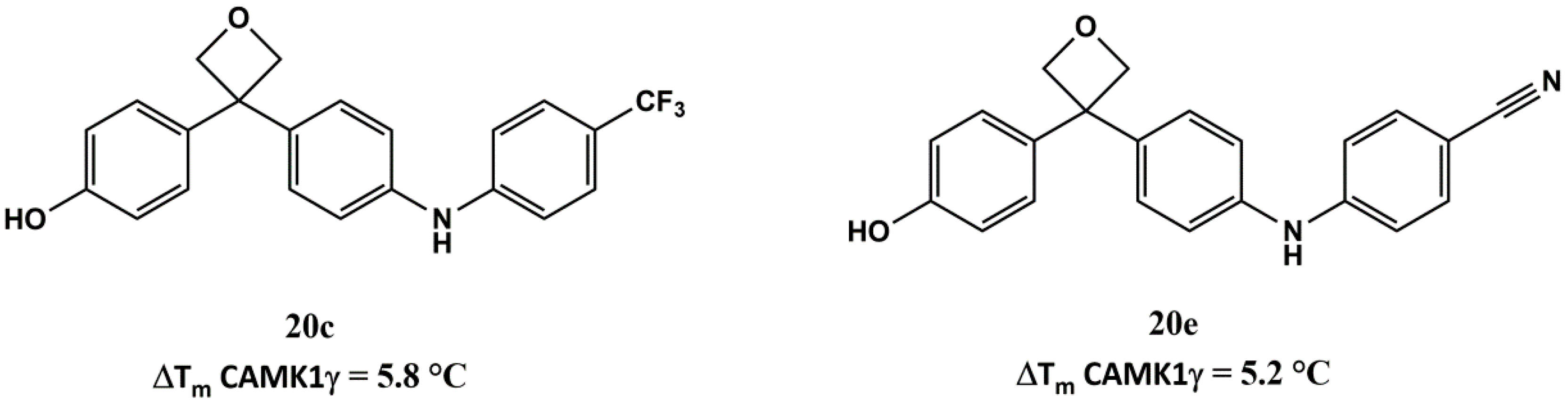
4-(3-(4-((2,4-difluorophenyl)amino)phenyl)oxetan-3-yl)phenol (20b): Brown solid. 66% yield. HPLC: 7.930 min (98%). ESI-MS: Calculated: 353.1 for C21H17F2NO2. Found: 352.0 [M − H]−. 1H-NMR (400 MHz, d-acetone) δ in ppm: 5.12 (s, 4H), 6.81–6.83 (d, 2H, J = 8.0 Hz), 6.90–6.95 (t, 1H), 7.0–7.02 (d, 2H, J = 8.0 Hz), 7.11–7.13 (d, 2H, J = 8.0 Hz), 7.15–7.17 (d, 2H, J = 8.0 Hz), 7.32–7.38 (m, 1H), 8.29 (br, 1H); 13C-NMR (100 MHz, d-acetone) δ in ppm: 51.6, 85.0, 104.9, 105.1 (d, J = 3.0 Hz), 105.4, 111.9 (d, J = 4.0 Hz), 112.1 (d, J = 4.0 Hz), 116.3, 117.9, 122.1 (d, J = 3.0 Hz), 122.2 (d, J = 3.0 Hz), 128.3, 128.5, 138.7, 140.0, 143.3, 156.9.
4-(3-(4-((4-(trifluoromethyl)phenyl)amino)phenyl)oxetan-3-yl)phenol (20c): White solid. 71% yield. HPLC: 7.967 min (96%). ESI-MS: Calculated: 385.1 for C22H18F3NO2. Found: 383.9 [M − H]−. 1H-NMR (200 MHz, d-acetone) δ in ppm: 5.15 (s, 4H), 6.82–6.86 (d, 2H, J = 8.0 Hz), 7.11–7.29 (m, 8H), 7.49–7.53 (d, 2H, J = 8.0 Hz), 7.93 (br, 1H), 8.34 (br, 1H); 13C-NMR (50 MHz, d-acetone) δ in ppm: 51.5, 84.8, 115.7, 116.1, 120.5, 121.0, 127.3, 127.4, 128.2, 128.4, 138.3, 140.9, 141.8, 148.8, 156.8.
4-(3-(4-((4-methoxyphenyl)amino)phenyl)oxetan-3-yl)phenol (20d): Yellow solid. 54% yield. HPLC: 7.159 min (99%). ESI-MS: Calculated: 347.1 for C22H21NO3. Found: 345.9 [M − H]−. 1H-NMR (400 MHz, d-acetone) δ in ppm: 3.75 (s, 3H), 5.11 (s, 4H), 6.80–6.89 (t, 4H), 6.93–6.97 (d, 2H, J = 8.0 Hz), 7.07–7.13 (m, 7H); 13C-NMR (100 MHz, d-acetone) δ in ppm: 27.7, 43.0, 84.2, 117.8, 118.2, 121.1, 126.2, 129.5, 139.1, 141.6, 143.4.
4-((4-(3-(4-hydroxyphenyl)oxetan-3-yl)phenyl)amino)benzonitrile (20e): Yellow solid. 70% yield. HPLC: 6.883 min (96%). ESI-MS: Calculated: 342.1 for C22H18N2O2. Found: 341.1 [M − H]−. 1H-NMR (200 MHz, d-acetone) δ in ppm: 5.15 (s, 4H), 6.81–6.86 (d, 2H, J = 10 Hz), 7.11–7.16 (d, 2H, J = 10 Hz), 7.20–7.31 (dd, 4H, J = 10 Hz, J = 8.0 Hz), 7.52–7.56 (d, 2H, J = 8.0 Hz), 8.12 (br, 1H), 8.32 (br, 1H); 13C-NMR (50 MHz, d-acetone) δ in ppm: 51.6, 84.8, 101.5, 115.7, 116.2, 120.4, 121.5, 128.4, 128.5, 134.5, 138.3, 140.1, 142.7, 149.7, 156.9.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









