Are Heat Shock Proteins an Important Link between Type 2 Diabetes and Alzheimer Disease?


 , ,
, , 
{kind=link}
{kind=link}
Abstract
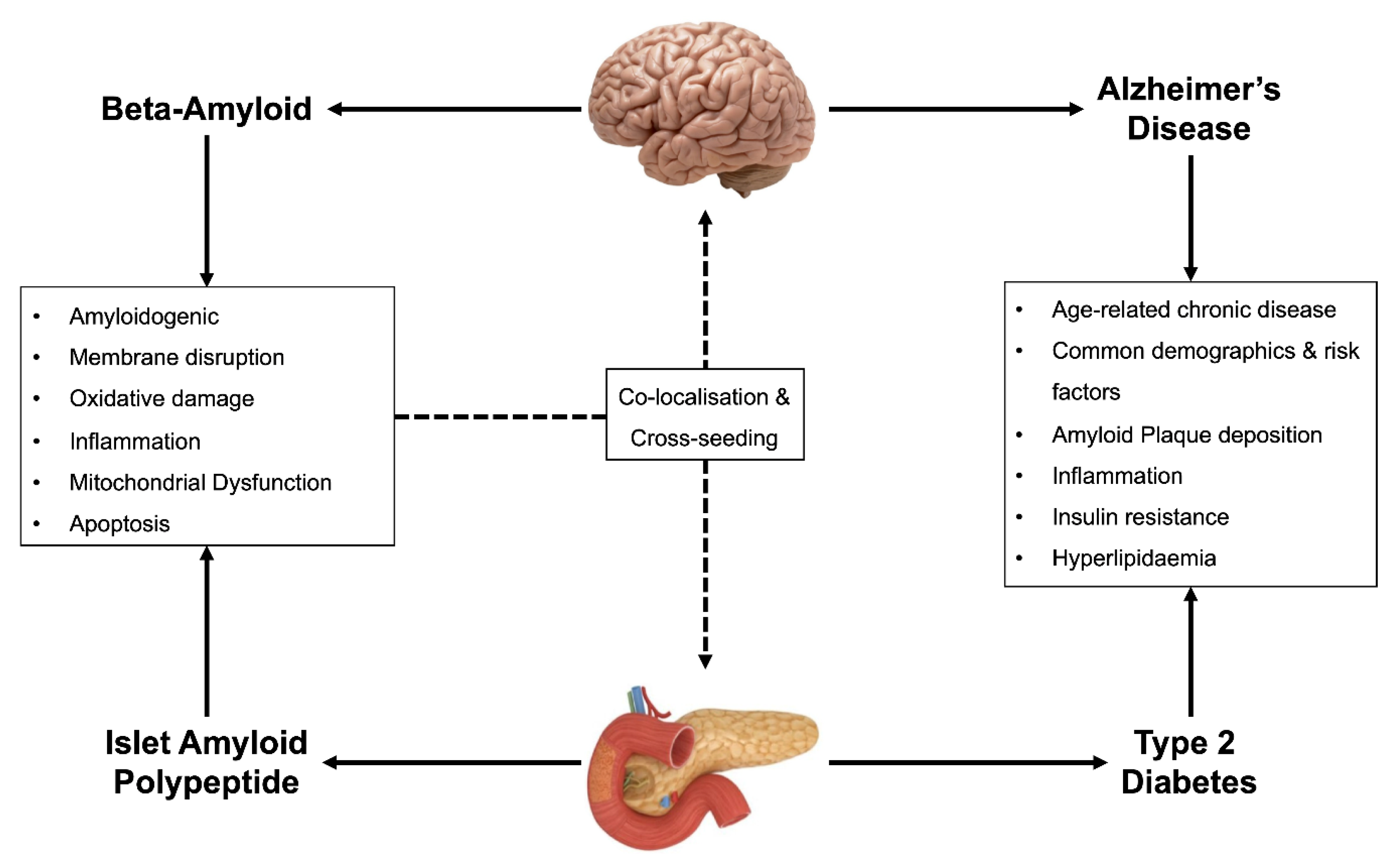
:1. Introduction
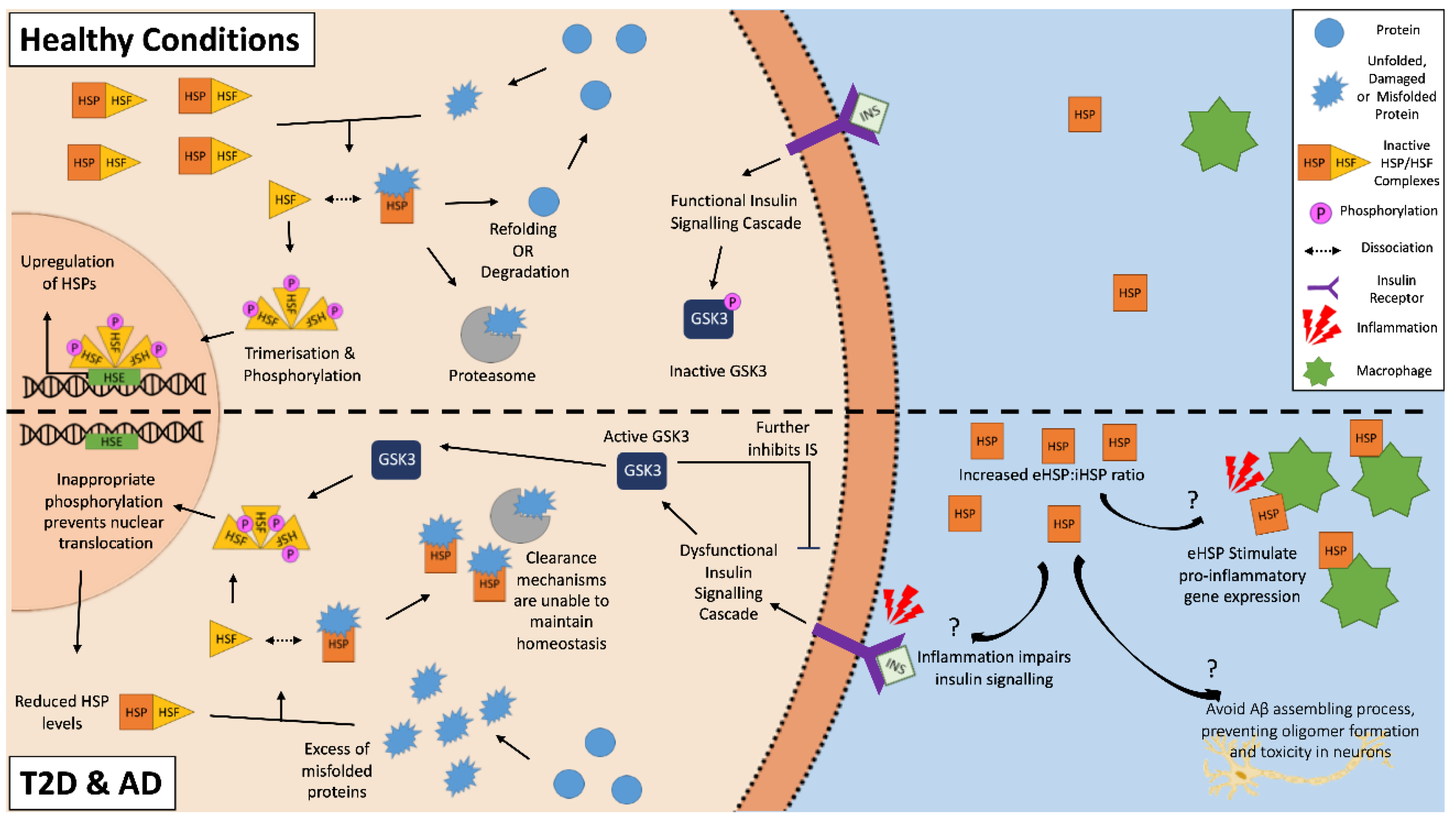
2. Dysregulation of Cellular Homeostasis in T2D and AD
3. Activation and Characterization of Heat Shock Proteins
4. Heat Shock Response in Type 2 Diabetes and Alzheimer’s Disease
4.1. Heat Shock Protein 90
4.2. Heat Shock Protein 70
4.3. Heat Shock Protein 60
4.4. Heat Shock Protein 40
4.5. Small Heat Shock Proteins
5. Extracellular HSPs
6. Conclusions: Heat Shock Proteins as a Therapeutic Target
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ABC | ATP-binding cassette |
| ADP | Adenosine diphosphate |
| Akt | Protein kinase B (PKB) |
| Aβ | β-Amyloid |
| BBB | Blood brain barrier |
| COX IV | Cytochrome c oxidase |
| CNS | Central nervous system |
| eHSP | Extracellular heat shock proteins |
| ER | Endoplasmic Reticulum |
| GGA | Geranylgeranylacetone |
| GRP | Glucose-responsive proteins |
| GSIS | Glucose-stimulated insulin secretion |
| GSK-3 | Glycogen synthase kinase-3 |
| HSC | Heat shock cognate |
| HSFs | Heat shock factors |
| HSR | Heat shock response |
| IAPP | Islet amyloid polypeptide |
| iHSP | Intracellular heat shock proteins |
| JNK | c-Jun N-terminal kinase |
| LRP1 | Low-density lipoprotein receptor-related peptide 1 |
| NBD | Nucleotide binding domain |
| NK | Natural killer |
| pTau | Hyperphosphorylated Tau |
| sHSP | Small HSPs |
| T2D | Type 2 diabetes |
| TLRs | Toll-like receptors |
References
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Akter, K.; Lanza, E.A.; Martin, S.A.; Myronyuk, N.; Rua, M.; Raffa, R.B. Diabetes mellitus and Alzheimer’s disease: Shared pathology and treatment? Br. J. Clin. Pharmacol. 2011, 71, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.S.; Calkins, E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 1959, 183, 1202. [Google Scholar] [CrossRef]
- Epstein, F.H.; Höppener, J.W.M.; Ahrén, B.; Lips, C.J.M. Islet Amyloid and Type 2 Diabetes Mellitus. N. Engl. J. Med. 2000, 343, 411–419. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [Green Version]
- Mirzabekov, T.A.; Lin, M.C.; Kagan, B.L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996, 271, 1988–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Laszlo, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol. Aging 2010, 31, 1503–1515. [Google Scholar] [CrossRef]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berhanu, W.M.; Yaşar, F.; Hansmann, U.H.E. In Silico Cross Seeding of Aβ and Amylin Fibril-like Oligomers. ACS Chem. Neurosci. 2013, 4, 1488–1500. [Google Scholar] [CrossRef] [Green Version]
- Baram, M.; Atsmon-Raz, Y.; Ma, B.; Nussinov, R.; Miller, Y. Amylin-Abeta oligomers at atomic resolution using molecular dynamics simulations: A link between Type 2 diabetes and Alzheimer’s disease. Phys. Chem. Chem. Phys. 2016, 18, 2330–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells. Sci. Rep. 2020, 10, 10356. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimers Dis. JAD 2017, 59, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Valbuena, I.; Valenti-Azcarate, R.; Amat-Villegas, I.; Riverol, M.; Marcilla, I.; de Andrea, C.E.; Sánchez-Arias, J.A.; Del Mar Carmona-Abellan, M.; Marti, G.; Erro, M.E.; et al. Amylin as a potential link between type 2 diabetes and alzheimer disease. Ann. Neurol. 2019, 86, 539–551. [Google Scholar] [CrossRef]
- Kampinga, H.; Hageman, J.; Vos, M.; Kubota, H.; Tanguay, R.; Bruford, E.; Cheetham, M.; Chen, B.; Hightower, L. Guidelines for the nomenclature of the human heat shock proteins. A Compr. J. Stress Biol. Med. 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.M.; Baxter, G.F.; Heads, R.J.; Yellon, D.M.; Downey, J.M.; Cohen, M.V. Infarct limitation of the second window of protection in a conscious rabbit model. Cardiovasc. Res. 1996, 31, 777–783. [Google Scholar] [CrossRef]
- Krause, M.S.; Oliveira, L.P.; Silveira, E.M.S.; Vianna, D.R.; Rossato, J.S.; Almeida, B.S.; Rodrigues, M.F.; Fernandes, A.J.M.; Costa, J.A.B.; Curi, R.; et al. MRP1/GS-X pump ATPase expression: Is this the explanation for the cytoprotection of the heart against oxidative stress-induced redox imbalance in comparison to skeletal muscle cells? Cell Biochem. Funct. 2007, 25, 23. [Google Scholar] [CrossRef]
- Cruzat, V.F.; Pantaleao, L.C.; Donato, J., Jr.; de Bittencourt, P.I., Jr.; Tirapegui, J. Oral supplementations with free and dipeptide forms of L-glutamine in endotoxemic mice: Effects on muscle glutamine-glutathione axis and heat shock proteins. J. Nutr. Biochem. 2014, 25, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Edkins, A.L.; Price, J.T.; Pockley, A.G.; Blatch, G.L. Heat shock proteins as modulators and therapeutic targets of chronic disease: An integrated perspective. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2018, 373, 20160521. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Krause, M. Nutritional regulation of insulin secretion: Implications for diabetes. Clin. Biochem. Rev. 2012, 33, 35–47. [Google Scholar]
- Ripsin, C.M.; Kang, H.; Urban, R.J. Management of blood glucose in type 2 diabetes mellitus. Am. Fam. Physician 2009, 79, 29–36. [Google Scholar]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet Amyloid Polypeptide—Pinpointing amino-acid-residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [Green Version]
- Krampert, M.; Bernhagen, J.; Schmucker, J.; Horn, A.; Schmauder, A.; Brunner, H.; Voelter, W.; Kapurniotu, A. Amyloidogenicity of recombinant human pro-islet amyloid polypeptide (ProIAPP). Chem. Biol. 2000, 7, 855–871. [Google Scholar] [CrossRef]
- Martin, C. The physiology of amylin and insulin—Maintaining the balance between glucose secretion and glucose uptake. Diabetes Educ. 2006, 32, 101S–104S. [Google Scholar] [CrossRef]
- Meier, J.J.; Kayed, R.; Lin, C.Y.; Gurlo, T.; Haataja, L.; Jayasinghe, S.; Langen, R.; Glabe, C.G.; Butler, P.C. Inhibition of human IAPP fibril formation does not prevent beta-cell death: Evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1317–E1324. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.F.; Andersson, A.; Westermark, P.; Westermark, G.T. Intracellular amyloid-like deposits contain unprocessed pro-islet amyloid polypeptide (proIAPP) in beta cells of transgenic mice overexpressing the gene for human IAPP and transplanted human islets. Diabetologia 2006, 49, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurlo, T.; Ryazantsev, S.; Huang, C.-J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for Proteotoxicity in β Cells in Type 2 Diabetes. Am. J. Pathol. 2010, 176, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Miranker, A.D. Concentration-dependent transitions govern the subcellular localization of islet amyloid polypeptide. FASEB J. 2012, 26, 1228–1238. [Google Scholar] [CrossRef] [Green Version]
- Trikha, S.; Jeremic, A.M. Distinct internalization pathways of human amylin monomers and its cytotoxic oligomers in pancreatic cells. PLoS ONE 2013, 8, e73080. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.S. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Radic. Biol. Med. 2006, 41, 177–184. [Google Scholar] [CrossRef]
- Newsholme, P.; Gaudel, C.; Krause, M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv. Exp. Med. Biol. 2012, 942, 235–247. [Google Scholar] [CrossRef]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [Green Version]
- Verdile, G.; Fuller, S.; Atwood, C.S.; Laws, S.M.; Gandy, S.E.; Martins, R.N. The role of beta amyloid in Alzheimer’s disease: Still a cause of everything or the only one who got caught? Pharmacol. Res. 2004, 50, 397–409. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Zhang, F.; Johnson, L.A.; Hall, J.; Edwards, M.; Grammas, P.; Oh, E.; Lyketsos, C.G.; Rissman, R.A. A Precision Medicine Model for Targeted NSAID Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis. Jad 2018, 66, 97–104. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussbaum, J.M.; Seward, M.E.; Bloom, G.S. Alzheimer disease: A tale of two prions. Prion 2013, 7, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Felsky, D.; Roostaei, T.; Nho, K.; Risacher, S.L.; Bradshaw, E.M.; Petyuk, V.; Schneider, J.A.; Saykin, A.; Bennett, D.A.; De Jager, P.L. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat. Commun. 2019, 10, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdier, Y.; Penke, B. Binding sites of amyloid beta-peptide in cell plasma membrane and implications for Alzheimer’s disease. Curr. Protein Pept. Sci. 2004, 5, 19–31. [Google Scholar] [CrossRef]
- Lim, Y.A.; Ittner, L.M.; Lim, Y.L.; Gotz, J. Human but not rat amylin shares neurotoxic properties with Abeta42 in long-term hippocampal and cortical cultures. FEBS Lett. 2008, 582, 2188–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Candeias, E.; Duarte, A.I.; Carvalho, C.; Correia, S.C.; Cardoso, S.; Santos, R.X.; Placido, A.I.; Perry, G.; Moreira, P.I. The impairment of insulin signaling in Alzheimer’s disease. IUBMB Life 2012, 64, 951–957. [Google Scholar] [CrossRef]
- Patel, A.N.; Jhamandas, J.H. Neuronal receptors as targets for the action of amyloid-beta protein (Abeta) in the brain. Expert Rev. Mol. Med. 2012, 14, e2. [Google Scholar] [CrossRef]
- Jimenez-Palomares, M.; Ramos-Rodriguez, J.J.; Lopez-Acosta, J.F.; Pacheco-Herrero, M.; Lechuga-Sancho, A.M.; Perdomo, G.; Garcia-Alloza, M.; Cozar-Castellano, I. Increased Abeta production prompts the onset of glucose intolerance and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1373–E1380. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, B.; Deng, B.; Zhang, F.; Wu, J.; Wang, Y.; Le, Y.; Zhai, Q. Amyloid-beta induces hepatic insulin resistance in vivo via JAK2. Diabetes 2013, 62, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Tabata, H.; Hirayama, J.; Sowa, R.; Furuta, H.; Negoro, T.; Sanke, T.; Nanjo, K. Islet amyloid polypeptide (IAPP/amylin) causes insulin resistance in perfused rat hindlimb muscle. Diabetes Res. Clin. Pract. 1992, 15, 57–61. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Edwards Iii, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Yan, L.M.; Velkova, A.; Kapurniotu, A. Molecular characterization of the hetero-assembly of beta-amyloid peptide with islet amyloid polypeptide. Curr. Pharm. Des. 2014, 20, 1182–1191. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Mizuguchi, M.; Hamaguchi, T.; Yamada, M. Exogenous amyloidogenic proteins function as seeds in amyloid beta-protein aggregation. Biochim. Biophys. Acta 2014, 1842, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijesekara, N.; Ahrens, R.; Sabale, M.; Wu, L.; Ha, K.; Verdile, G.; Fraser, P.E. Amyloid-β and islet amyloid pathologies link Alzheimer’s disease and type 2 diabetes in a transgenic model. FASEB J. 2017, 31, 5409–5418. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, X.; Wallack, M.; Li, H.; Carreras, I.; Dedeoglu, A.; Hur, J.Y.; Zheng, H.; Li, H.; Fine, R.; et al. Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer’s disease. Mol. Psychiatry 2015, 20, 252–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, L.A.; Zhu, H.; Mousa, Y.M.; Wang, E.; Qiu, W.Q.; Kaddoumi, A. Amylin Enhances Amyloid-β Peptide Brain to Blood Efflux Across the Blood-Brain Barrier. J. Alzheimers Dis. JAD 2017, 56, 1087–1099. [Google Scholar] [CrossRef] [Green Version]
- Stutzer, I.; Selevsek, N.; Esterhazy, D.; Schmidt, A.; Aebersold, R.; Stoffel, M. Systematic proteomic analysis identifies beta-site amyloid precursor protein cleaving enzyme 2 and 1 (BACE2 and BACE1) substrates in pancreatic beta-cells. J. Biol. Chem. 2013, 288, 10536–10547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Welch, W.J. The Mammalian Stress Response: Cell Physiology & Biochemistry of Stress Proteins; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1990; p. 55. [Google Scholar]
- Prahlad, V.; Morimoto, R.I. Integrating the stress response: Lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol. 2009, 19, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. Regulation of the mammalian heat shock factor 1. FEBS J. 2017, 284, 1606–1627. [Google Scholar] [CrossRef] [Green Version]
- Metzler, B.; Abia, R.; Ahmad, M.; Wernig, F.; Pachinger, O.; Hu, Y.; Xu, Q. Activation of Heat Shock Transcription Factor 1 in Atherosclerosis. Am. J. Pathol. 2003, 162, 1669–1676. [Google Scholar] [CrossRef] [Green Version]
- Schuetz, T.J.; Gallo, G.J.; Sheldon, L.; Tempst, P.; Kingston, R.E. Isolation of a cDNA for HSF2: Evidence for two heat shock factor genes in humans. Proc. Natl. Acad. Sci. USA 1991, 88, 6911–6915. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Ying, Z.; Jin, X.; Tu, N.; Zhang, Y.; Phillips, M.; Moskophidis, D.; Mivechi, N.F. Essential requirement for both hsf1 and hsf2 transcriptional activity in spermatogenesis and male fertility. Genesis 2004, 38, 66–80. [Google Scholar] [CrossRef]
- Sandqvist, A.; Björk, J.K.; Åkerfelt, M.; Chitikova, Z.; Grichine, A.; Vourc’H, C.; Jolly, C.; Salminen, T.A.; Nymalm, Y.; Sistonen, L. Heterotrimerization of Heat-Shock Factors 1 and 2 Provides a Transcriptional Switch in Response to Distinct Stimuli. Mol. Biol. Cell 2009, 20, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Raboy, B.; Sharon, G.; Parag, H.A.; Shochat, Y.; Kulka, R.G. Effect of stress on protein degradation: Role of the ubiquitin system. Acta Biol. Hung. 1991, 42, 3–20. [Google Scholar]
- Sevin, M.; Girodon, F.; Garrido, C.; de Thonel, A. HSP90 and HSP70: Implication in Inflammation Processes and Therapeutic Approaches for Myeloproliferative Neoplasms. Mediat. Inflamm. 2015, 2015, 970242. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Gurbuxani, S.; Ravagnan, L.; Kroemer, G. Heat shock proteins: Endogenous modulators of apoptotic cell death. Biochem. Biophys. Res. Commun. 2001, 286, 433–442. [Google Scholar] [CrossRef]
- Krause, M.; Bock, P.M.; Takahashi, H.K.; Homem De Bittencourt, P.I., Jr.; Newsholme, P. The regulatory roles of NADPH oxidase, intra- and extra-cellular HSP70 in pancreatic islet function, dysfunction and diabetes. Clin. Sci. 2015, 128, 789–803. [Google Scholar] [CrossRef]
- Yuan, J.; Dunn, P.; Martinus, R.D. Detection of Hsp60 in saliva and serum from type 2 diabetic and non-diabetic control subjects. Cell Stress Chaperones 2011, 16, 689. [Google Scholar] [CrossRef] [Green Version]
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 2012, 10, e1001346. [Google Scholar] [CrossRef] [Green Version]
- Arimon, M.; Grimminger, V.; Sanz, F.; Lashuel, H.A. Hsp104 targets multiple intermediates on the amyloid pathway and suppresses the seeding capacity of Abeta fibrils and protofibrils. J. Mol. Biol. 2008, 384, 1157–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tower, J. Hsps and aging. Trends Endocrinol. Metab. 2009, 20, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbah, N.A.; Rezk, N.A.; Saad, M.S.S. Relationship Between Heat Shock Protein Expression and Obesity With and Without Metabolic Syndrome. Genet. Test. Mol. Biomark. 2019, 23, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Kurucz, I.; Morva, A.; Vaag, A.; Eriksson, K.F.; Huang, X.; Groop, L.; Koranyi, L. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes 2002, 51, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigh, L.; Horvath, I.; Maresca, B.; Harwood, J.L. Can the stress protein response be controlled by ‘membrane-lipid therapy’? Trends Biochem. Sci. 2007, 32, 357–363. [Google Scholar] [CrossRef]
- Hooper, P.L.; Hooper, P.L. Inflammation, heat shock proteins, and type 2 diabetes. Cell Stress Chaperones 2009, 14, 113–115. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Doble, B.; Woodgett, J.R. Glycogen synthase kinase-3 in insulin and Wnt signalling: A double-edged sword? Biochem. Soc. Trans. 2004, 32, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- Kurthy, M.; Mogyorosi, T.; Nagy, K.; Kukorelli, T.; Jednakovits, A.; Talosi, L.; Biro, K. Effect of BRX-220 against peripheral neuropathy and insulin resistance in diabetic rat models. Ann. N. Y. Acad. Sci. 2002, 967, 482–489. [Google Scholar] [CrossRef]
- Hooper, P.L. Insulin Signaling, GSK-3, Heat Shock Proteins and the Natural History of Type 2 Diabetes Mellitus: A Hypothesis. Metab. Syndr. Relat. Disord. 2007, 5, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.; Zhang, L.; Wagner, J.D. Tissue-specific regulation and expression of heat shock proteins in type 2 diabetic monkeys. Cell Stress Chaperones 2009, 14, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, L.; Atack, J.R.; Rapoport, S.I.; Gershfeld, N.L. Evidence for a membrane lipid defect in Alzheimer disease. Mol. Chem. Neuropathol. 1993, 19, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Weijers, R.N.M. Lipid Composition of Cell Membranes and Its Relevance in Type 2 Diabetes Mellitus. Curr. Diabetes Rev. 2012, 8, 390–400. [Google Scholar] [CrossRef]
- Horváth, I.; Multhoff, G.; Sonnleitner, A.; Vígh, L. Membrane-associated stress proteins: More than simply chaperones. Biochim. Biophys. Acta BBA Biomembr. 2008, 1778, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11. [Google Scholar] [CrossRef]
- Mickler, M.; Hessling, M.; Ratzke, C.; Buchner, J.; Hugel, T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol. 2009, 16, 281. [Google Scholar] [CrossRef]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat Shock Proteins 70 and 90 Inhibit Early Stages of Amyloid β-(1–42) Aggregation in Vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [Green Version]
- Dickey, C.A.; Yue, M.; Lin, W.-L.; Dickson, D.W.; Dunmore, J.H.; Lee, W.C.; Zehr, C.; West, G.; Cao, S.; Clark, A.M.K.; et al. Deletion of the Ubiquitin Ligase CHIP Leads to the Accumulation, But Not the Aggregation, of Both Endogenous Phospho- and Caspase-3-Cleaved Tau Species. J. Neurosci. 2006, 26, 6985–6996. [Google Scholar] [CrossRef] [Green Version]
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005, 94, 1254–1263. [Google Scholar] [CrossRef]
- Press, M.; Jung, T.; König, J.; Grune, T.; Höhn, A. Protein aggregates and proteostasis in aging: Amylin and β-cell function. Mech. Ageing Dev. 2019, 177, 46–54. [Google Scholar] [CrossRef]
- Chatterjee Bhowmick, D.; Jeremic, A. Functional proteasome complex is required for turnover of islet amyloid polypeptide in pancreatic β-cells. J. Biol. Chem. 2018, 293, 14210–14223. [Google Scholar] [CrossRef]
- Singh, S.; Trikha, S.; Sarkar, A.; Jeremic, A.M. Proteasome regulates turnover of toxic human amylin in pancreatic cells. Biochem. J. 2016, 473, 2655–2670. [Google Scholar] [CrossRef]
- Zhao, H.; Michaelis, M.L.; Blagg, B.S.J. Hsp90 Modulation for the Treatment of Alzheimer’s Disease. In Advances in Pharmacology; Michaelis, E.K., Michaelis, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 64, pp. 1–25. [Google Scholar]
- Dou, F.; Chang, X.; Ma, D. Hsp90 Maintains the Stability and Function of the Tau Phosphorylating Kinase GSK3β. Int. J. Mol. Sci. 2007, 8, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Gao, J.; Kosinski, P.A.; Elliman, S.J.; Hughes, T.E.; Gromada, J.; Kemp, D.M. Heat shock protein 90 (HSP90) inhibitors activate the heat shock factor 1 (HSF1) stress response pathway and improve glucose regulation in diabetic mice. Biochem. Biophys. Res. Commun. 2013, 430, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, E.; Santa-Maria, I.; Moreno, F.; Lim, F.; Perez, M.; Avila, J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimers Dis. JAD 2009, 17, 319–325. [Google Scholar] [CrossRef]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 Chaperone Machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.-R.; Tan, M.-S.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat Shock Protein 90 in Alzheimer’s Disease. Biomed Res. Int. 2014, 2014, 796869. [Google Scholar] [CrossRef]
- Young, J.C. Mechanisms of the Hsp70 chaperone system. Biochem. Cell Biol. Biochim. Biol. Cell. 2010, 88, 291–300. [Google Scholar] [CrossRef]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [Green Version]
- Ribeil, J.A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sorensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell. Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chilukoti, N.; Sahoo, B.; Maddheshiya, M.; Garai, K. Hsp70 Delays Amyloid Aggregation of Amylin by Inhibiting Primary Nucleation. Biophys. J. 2018, 114, 78a–79a. [Google Scholar] [CrossRef]
- Chung, J.; Nguyen, A.-K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.S.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henstridge, D.C.; Whitham, M.; Febbraio, M.A. Chaperoning to the metabolic party: The emerging therapeutic role of heat-shock proteins in obesity and type 2 diabetes. Mol. Metab. 2014, 3, 781–793. [Google Scholar] [CrossRef]
- Takino, J.-I.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655. [Google Scholar] [CrossRef]
- Hoshino, T.; Murao, N.; Namba, T.; Takehara, M.; Adachi, H.; Katsuno, M.; Sobue, G.; Matsushima, T.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s Disease-Related Phenotypes by Expression of Heat Shock Protein 70 in Mice. J. Neurosci. 2011, 31, 5225–5234. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat Shock Protein 70 Prevents both Tau Aggregation and the Inhibitory Effects of Preexisting Tau Aggregates on Fast Axonal Transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar] [CrossRef] [Green Version]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary, J.C.; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M.; et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinwal, U.K.; Miyata, Y.; Koren, J.; Jones, J.R.; Trotter, J.H.; Chang, L.; O’Leary, J.; Morgan, D.; Lee, D.C.; Shults, C.L.; et al. Chemical Manipulation of Hsp70 ATPase Activity Regulates Tau Stability. J. Neurosci. 2009, 29, 12079–12088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Turner, R.S.; Gaut, J.R. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J. Biol. Chem. 1998, 273, 25552–25555. [Google Scholar] [CrossRef] [Green Version]
- Cappello, F.; Conway de Macario, E.; Marasa, L.; Zummo, G.; Macario, A.J. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef]
- Capello, F.; Gammazza, A.M.; Vilasi, S.; Ortore, M.G.; Biagio, P.L.S.; Campanella, C.; Pace, A.; Piccionello, A.P.; Taglialatela, G.; Macario, E.C.D.; et al. Chaperonotherapy for Alzheimer’s Disease: Focusing on HSP60. In Heat Shock Protein-Based Therapies; Asea, A.A.A., Almasoud, N.N., Krishnan, S., Kaur, P., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 51–77. [Google Scholar]
- Juwono, J.; Martinus, R.D. Does Hsp60 Provide a Link between Mitochondrial Stress and Inflammation in Diabetes Mellitus? J. Diabetes Res. 2016, 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.C.; Kim, S.H.; Cairns, N.; Fountoulakis, M.; Lubec, G. Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2001, 280, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Veereshwarayya, V.; Kumar, P.; Rosen, K.M.; Mestril, R.; Querfurth, H.W. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular beta-amyloid-induced inhibition of complex IV and limit apoptosis. J. Biol. Chem. 2006, 281, 29468–29478. [Google Scholar] [CrossRef] [Green Version]
- Xanthoudakis, S.; Roy, S.; Rasper, D.; Hennessey, T.; Aubin, Y.; Cassady, R.; Tawa, P.; Ruel, R.; Rosen, A.; Nicholson, D.W. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 1999, 18, 2049–2056. [Google Scholar] [CrossRef] [Green Version]
- Samali, A.; Cai, J.; Zhivotovsky, B.; Jones, D.P.; Orrenius, S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 1999, 18, 2040–2048. [Google Scholar] [CrossRef] [Green Version]
- Walls, K.C.; Coskun, P.; Gallegos-Perez, J.L.; Zadourian, N.; Freude, K.; Rasool, S.; Blurton-Jones, M.; Green, K.N.; LaFerla, F.M. Swedish Alzheimer Mutation Induces Mitochondrial Dysfunction Mediated by HSP60 Mislocalization of Amyloid Precursor Protein (APP) and Beta-Amyloid. J. Biol. Chem. 2012, 287, 30317–30327. [Google Scholar] [CrossRef] [Green Version]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2017, 69, 227–237.e224. [Google Scholar] [CrossRef] [PubMed]
- Alderson, T.R.; Kim, J.H.; Markley, J.L. Dynamical structures of Hsp70 and Hsp70–Hsp40 complexes. Structure 2016, 24, 1014–1030. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Cherian, P.; Al-Khairi, I.; Tiss, A.; Khadir, A.; Kavalakatt, S.; Warsame, S.; Dehbi, M.; Behbehani, K.; Abubaker, J. DNAJB3/HSP-40 cochaperone improves insulin signaling and enhances glucose uptake in vitro through JNK repression. Sci. Rep. 2015, 5, 14448. [Google Scholar] [CrossRef] [Green Version]
- Haslbeck, M.; Franzmann, T.; Weinfurtner, D.; Buchner, J. Some like it hot: The structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005, 12, 842–846. [Google Scholar] [CrossRef]
- Lee, G.J.; Roseman, A.M.; Saibil, H.R.; Vierling, E. A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding-competent state. EMBO J. 1997, 16, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.S.; Jakhotia, S.; Reddy, P.Y.; Reddy, G.B. Hyperglycemia induced expression, phosphorylation, and translocation of αB-crystallin in rat skeletal muscle. IUBMB Life 2015, 67, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Raghu, G.; Reddy, S.S.; Pasupulati, A.K.; Suryanarayana, P.; Reddy, G.B. Response of small heat shock proteins in diabetic rat retina. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7674–7682. [Google Scholar] [CrossRef] [Green Version]
- Shimura, H.; Miura-Shimura, Y.; Kosik, K.S. Binding of Tau to Heat Shock Protein 27 Leads to Decreased Concentration of Hyperphosphorylated Tau and Enhanced Cell Survival. J. Biol. Chem. 2004, 279, 17957–17962. [Google Scholar] [CrossRef] [Green Version]
- Mannini, B.; Cascella, R.; Zampagni, M.; van Waarde-Verhagen, M.; Meehan, S.; Roodveldt, C.; Campioni, S.; Boninsegna, M.; Penco, A.; Relini, A.; et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484. [Google Scholar] [CrossRef] [Green Version]
- Bakthisaran, R.; Tangirala, R.; Rao, C.M. Small heat shock proteins: Role in cellular functions and pathology. Biochim. Biophys. Acta BBA Proteins Proteom. 2015, 1854, 291–319. [Google Scholar] [CrossRef] [Green Version]
- Stege, G.J.; Renkawek, K.; Overkamp, P.S.; Verschuure, P.; van Rijk, A.F.; Reijnen-Aalbers, A.; Boelens, W.C.; Bosman, G.J.; de Jong, W.W. The molecular chaperone alphaB-crystallin enhances amyloid beta neurotoxicity. Biochem. Biophys. Res. Commun. 1999, 262, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Pockley, A.G.; Henderson, B. Extracellular cell stress (heat shock) proteins-immune responses and disease: An overview. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Heck, T.G.; Bittencourt, A.; Scomazzon, S.P.; Newsholme, P.; Curi, R.; Homem de Bittencourt, P.I. The Chaperone Balance Hypothesis: The Importance of the Extracellular to Intracellular HSP70 Ratio to Inflammation-Driven Type 2 Diabetes, the Effect of Exercise, and the Implications for Clinical Management. Mediat. Inflamm. 2015, 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Keane, K.; Rodrigues-Krause, J.; Crognale, D.; Egan, B.; De Vito, G.; Murphy, C.; Newsholme, P. Elevated levels of extracellular heat-shock protein 72 (eHSP72) are positively correlated with insulin resistance in vivo and cause pancreatic beta-cell dysfunction and death in vitro. Clin. Sci. 2014, 126, 739–752. [Google Scholar] [CrossRef]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 601–603. [Google Scholar]
- Salari, S.; Seibert, T.; Chen, Y.X.; Hu, T.; Shi, C.; Zhao, X.; Cuerrier, C.M.; Raizman, J.E.; O’Brien, E.R. Extracellular HSP27 acts as a signaling molecule to activate NF-kappaB in macrophages. Cell Stress Chaperones 2013, 18, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Horn, P.; Kalz, A.; Lim, C.L.; Pyne, D.; Saunders, P.; Mackinnon, L.; Peake, J.; Suzuki, K. Exercise-recruited NK cells display exercise-associated eHSP-70. Exerc. Immunol. Rev. 2007, 13, 100–111. [Google Scholar]
- Conrad, C.C.; Marshall, P.L.; Talent, J.M.; Malakowsky, C.A.; Choi, J.; Gracy, R.W. Oxidized proteins in Alzheimer’s plasma. Biochem. Biophys. Res. Commun. 2000, 275, 678–681. [Google Scholar] [CrossRef]
- Lei, H.; Romeo, G.; Kazlauskas, A. Heat shock protein 90alpha-dependent translocation of annexin II to the surface of endothelial cells modulates plasmin activity in the diabetic rat aorta. Circ. Res. 2004, 94, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gezen-Ak, D.; Dursun, E.; Hanagasi, H.; Bilgic, B.; Lohman, E.; Araz, O.S.; Atasoy, I.L.; Alaylioglu, M.; Onal, B.; Gurvit, H.; et al. BDNF, TNFalpha, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J. Alzheimers Dis. JAD 2013, 37, 185–195. [Google Scholar] [CrossRef]
- Zhang, R.; Li, Y.; Hou, X.; Miao, Z.; Wang, Y. Neuroprotective effect of heat shock protein 60 on matrine-suppressed microglial activation. Exp. Ther. Med. 2017, 14, 1832–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruden, G.; Bruno, G.; Chaturvedi, N.; Burt, D.; Schalkwijk, C.; Pinach, S.; Stehouwer, C.D.; Witte, D.R.; Fuller, J.H.; Perin, P.C.; et al. Serum heat shock protein 27 and diabetes complications in the EURODIAB prospective complications study: A novel circulating marker for diabetic neuropathy. Diabetes 2008, 57, 1966–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Gelain, D.P.; de Bittencourt Pasquali, M.A.; Comim, M.C.; Grunwald, M.S.; Ritter, C.; Tomasi, C.D.; Alves, S.C.; Quevedo, J.; Dal-Pizzol, F.; Moreira, J.C. Serum heat shock protein 70 levels, oxidant status, and mortality in sepsis. Shock 2011, 35, 466–470. [Google Scholar] [CrossRef]
- Grunwald, M.S.; Pires, A.S.; Zanotto-Filho, A.; Gasparotto, J.; Gelain, D.P.; Demartini, D.R.; Scholer, C.M.; de Bittencourt, P.I., Jr.; Moreira, J.C. The oxidation of HSP70 is associated with functional impairment and lack of stimulatory capacity. Cell Stress Chaperones 2014, 19, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.R.; Yerbury, J.J.; Poon, S.; Wilson, M.R. Therapeutic targets in extracellular protein deposition diseases. Curr. Med. Chem. 2009, 16, 2855–2866. [Google Scholar] [CrossRef] [Green Version]
- Nakhjavani, M.; Morteza, A.; Khajeali, L.; Esteghamati, A.; Khalilzadeh, O.; Asgarani, F.; Outeiro, T.F. Increased serum HSP70 levels are associated with the duration of diabetes. Cell Stress Chaperones 2010, 15, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Shamaei-Tousi, A.; Stephens, J.W.; Bin, R.; Cooper, J.A.; Steptoe, A.; Coates, A.R.; Henderson, B.; Humphries, S.E. Association between plasma levels of heat shock protein 60 and cardiovascular disease in patients with diabetes mellitus. Eur. Heart J. 2006, 27, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Krause, J.; Krause, M.; O’Hagan, C.; De Vito, G.; Boreham, C.; Murphy, C.; Newsholme, P.; Colleran, G. Divergence of intracellular and extracellular HSP72 in type 2 diabetes: Does fat matter? Cell Stress Chaperones 2012, 17, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Takata, K.; Kitamura, Y.; Tsuchiya, D.; Kawasaki, T.; Taniguchi, T.; Shimohama, S. Heat shock protein-90-induced microglial clearance of exogenous amyloid-beta1-42 in rat hippocampus in vivo. Neurosci. Lett. 2003, 344, 87–90. [Google Scholar] [CrossRef]
- Locke, M.; Noble, E.G. Exercise and Stress Response: The Role of Stress Proteins; CRC Press: New York, NY, USA, 2002; p. 226. [Google Scholar]
- Costa-Beber, L.C.; Hirsch, G.E.; Heck, T.G.; Ludwig, M.S. Chaperone duality: The role of extracellular and intracellular HSP70 as a biomarker of endothelial dysfunction in the development of atherosclerosis. Arch. Physiol. Biochem. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Heck Thiago, G.; Ludwig Mirna, S.; Frizzo Matias, N.; Rasia-Filho Alberto, A.; Homem de Bittencourt, P.I., Jr. Suppressed anti-inflammatory heat shock response in high-risk COVID-19 patients: Lessons from basic research (inclusive bats), light on conceivable therapies. Clin. Sci. 2020, 134, 1991–2017. [Google Scholar] [CrossRef]
- Rivera, I.; Capone, R.; Cauvi, D.M.; Arispe, N.; De Maio, A. Modulation of Alzheimer’s amyloid β peptide oligomerization and toxicity by extracellular Hsp70. Cell Stress Chaperones 2017. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, T.; Nakaya, T.; Araki, W.; Suzuki, K.; Suzuki, T.; Mizushima, T. Endoplasmic reticulum chaperones inhibit the production of amyloid-beta peptides. Biochem. J. 2007, 402, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, M.M.; Otte-Höller, I.; Wesseling, P.; de Waal, R.M.; Boelens, W.C.; Verbeek, M.M. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 2006, 32, 119–130. [Google Scholar] [CrossRef]
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Literáti-Nagy, Z.; Tory, K.; Literáti-Nagy, B.; Kolonics, A.; Török, Z.; Gombos, I.; Balogh, G.; Vígh, L., Jr.; Horváth, I.; Mandl, J.; et al. The HSP co-inducer BGP-15 can prevent the metabolic side effects of the atypical antipsychotics. Cell Stress Chaperones 2012, 17, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Motoshima, H.; Igata, M.; Kawashima, J.; Matsumura, T.; Kai, H.; Araki, E. The role of heat shock response in insulin resistance and diabetes. Diabetes Metab. J. 2014, 38, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.Y.; Wang, H.; Bai, F.; Zhou, X.; Li, S.P.; Ren, L.P.; Sun, R.Q.; Xue, C.C.; Jiang, H.L.; Hu, L.H.; et al. Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target. Br. J. Pharmacol. 2015, 172, 4303–4318. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Li, Y.; Hou, X.; Zhang, R.; Hu, S.; Wang, Y. Oxymatrine inhibits microglia activation via HSP60-TLR4 signaling. Biomed. Rep. 2016, 5, 623–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, A.E.; Von Schulze, A.T.; Geiger, P.C. Exercise, heat shock proteins and insulin resistance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Ludwig, M.S.; Heck, T.G.; Takahashi, H.K. Heat shock proteins and heat therapy for type 2 diabetes: Pros and cons. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.P.; Minett, G.M.; Gibson, O.R.; Kerr, G.K.; Stewart, I.B. Could Heat Therapy Be an Effective Treatment for Alzheimer’s and Parkinson’s Diseases? A Narrative Review. Front. Physiol. 2020, 10. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rowles, J.E.; Keane, K.N.; Gomes Heck, T.; Cruzat, V.; Verdile, G.; Newsholme, P. Are Heat Shock Proteins an Important Link between Type 2 Diabetes and Alzheimer Disease? Int. J. Mol. Sci. 2020, 21, 8204. https://doi.org/10.3390/ijms21218204
Rowles JE, Keane KN, Gomes Heck T, Cruzat V, Verdile G, Newsholme P. Are Heat Shock Proteins an Important Link between Type 2 Diabetes and Alzheimer Disease? International Journal of Molecular Sciences. 2020; 21(21):8204. https://doi.org/10.3390/ijms21218204
Chicago/Turabian StyleRowles, Joanne Elizabeth, Kevin Noel Keane, Thiago Gomes Heck, Vinicius Cruzat, Giuseppe Verdile, and Philip Newsholme. 2020. "Are Heat Shock Proteins an Important Link between Type 2 Diabetes and Alzheimer Disease?" International Journal of Molecular Sciences 21, no. 21: 8204. https://doi.org/10.3390/ijms21218204
APA StyleRowles, J. E., Keane, K. N., Gomes Heck, T., Cruzat, V., Verdile, G., & Newsholme, P. (2020). Are Heat Shock Proteins an Important Link between Type 2 Diabetes and Alzheimer Disease? International Journal of Molecular Sciences, 21(21), 8204. https://doi.org/10.3390/ijms21218204