PDIA3 Expression in Glioblastoma Modulates Macrophage/Microglia Pro-Tumor Activation


 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results
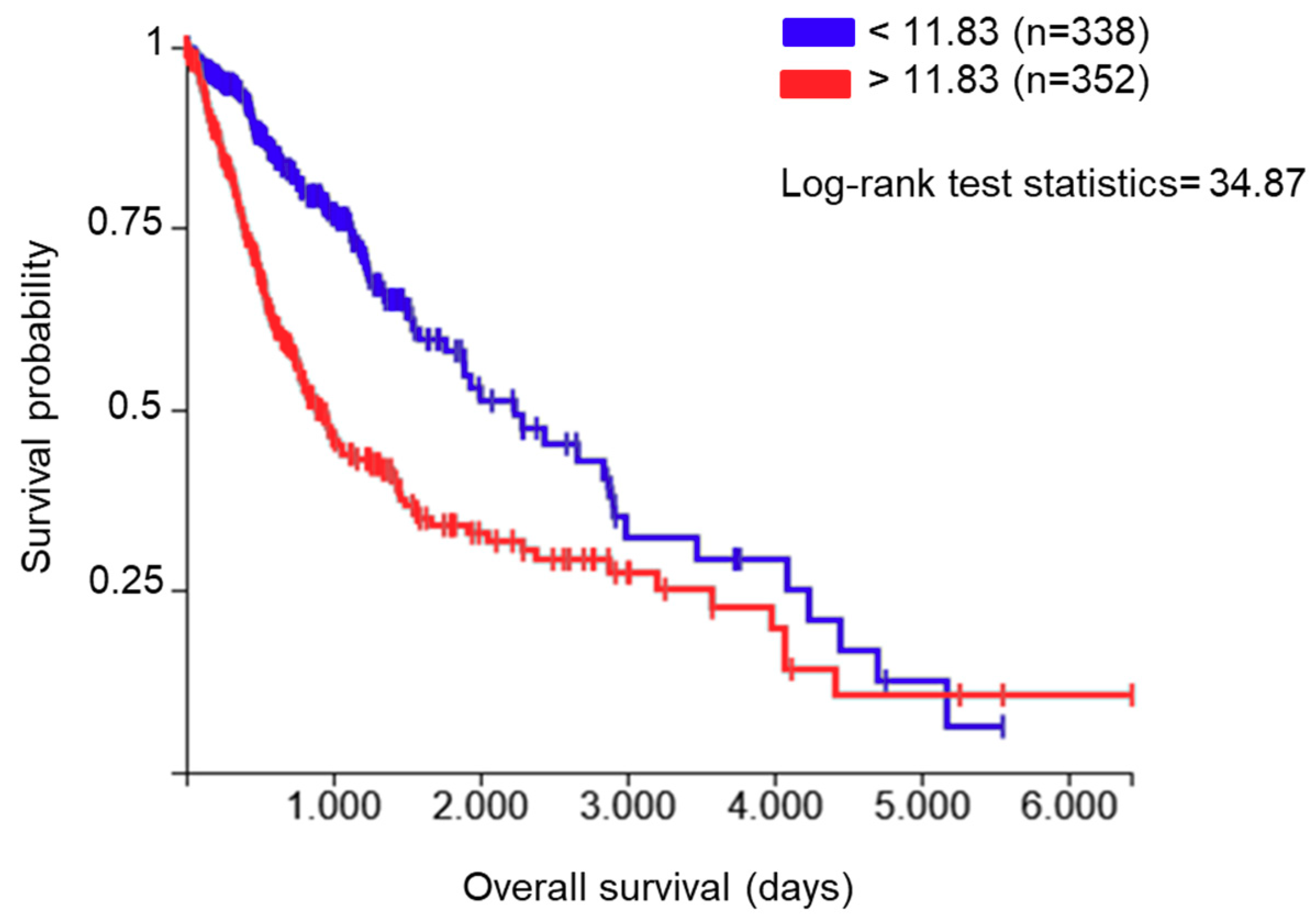
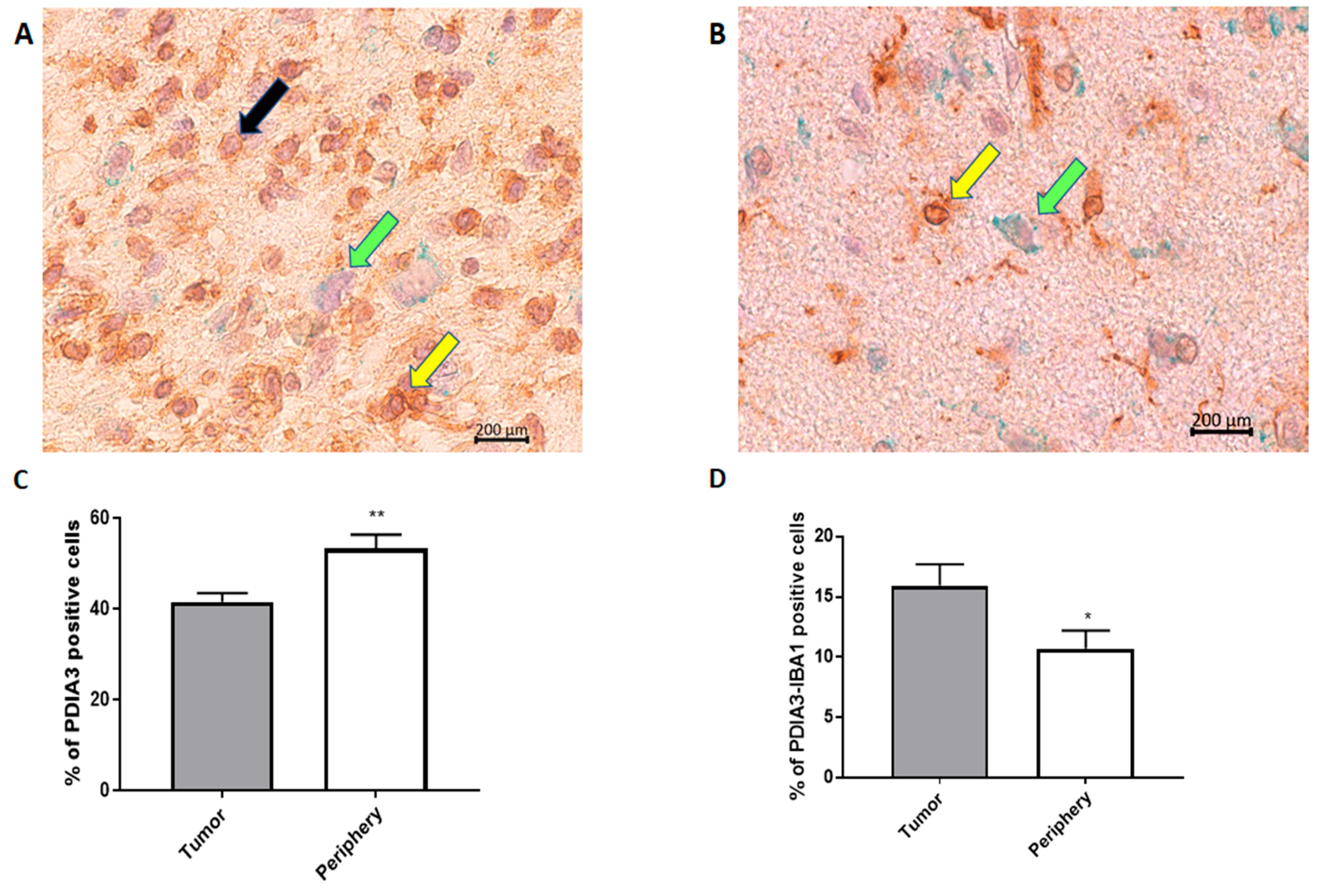
2.1. PDIA3 Expression in GB Specimens
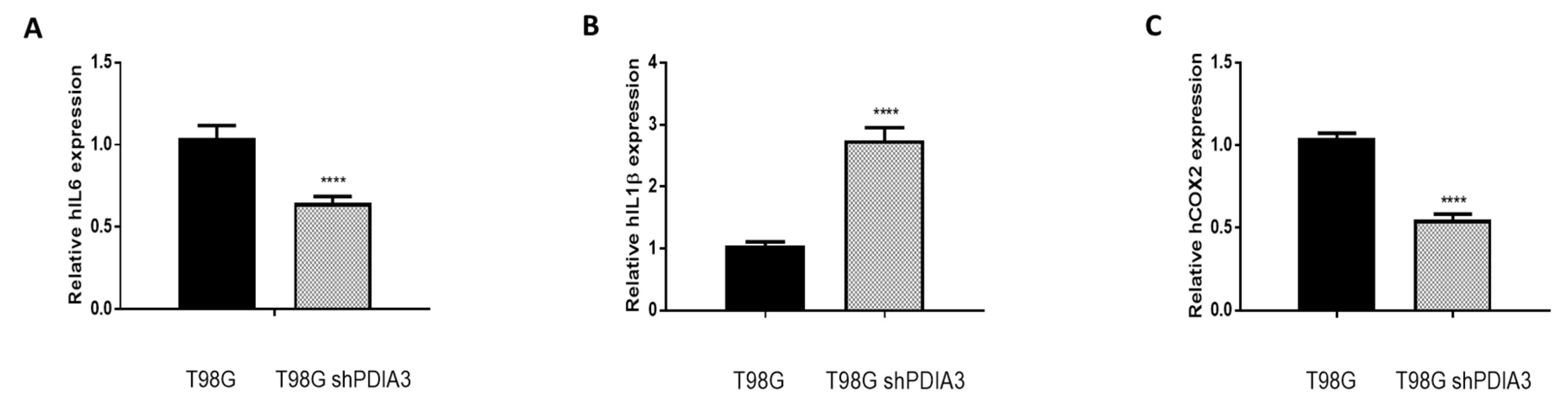
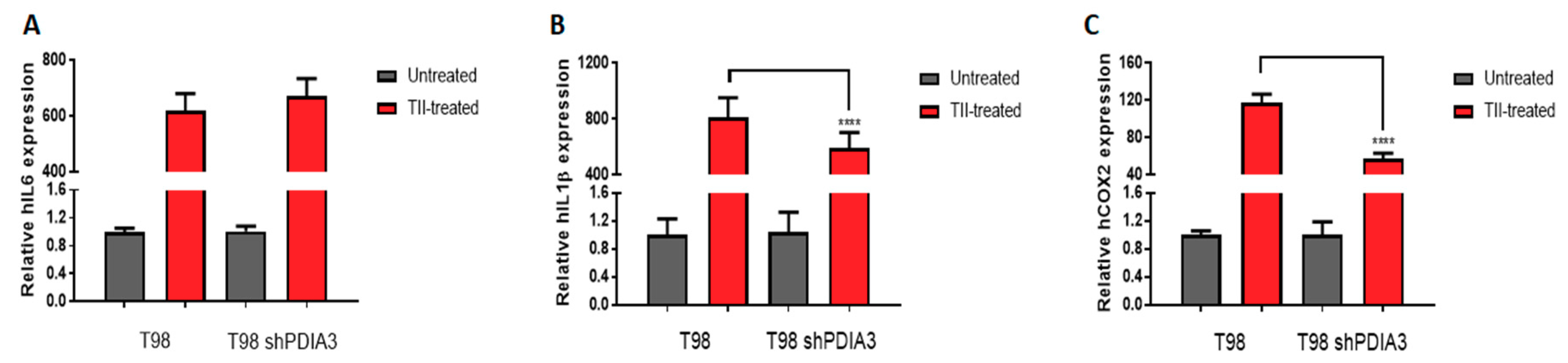
2.2. Influence of PDIA3 Expression on the Inflammatory Response and Viability of Human GB Cells
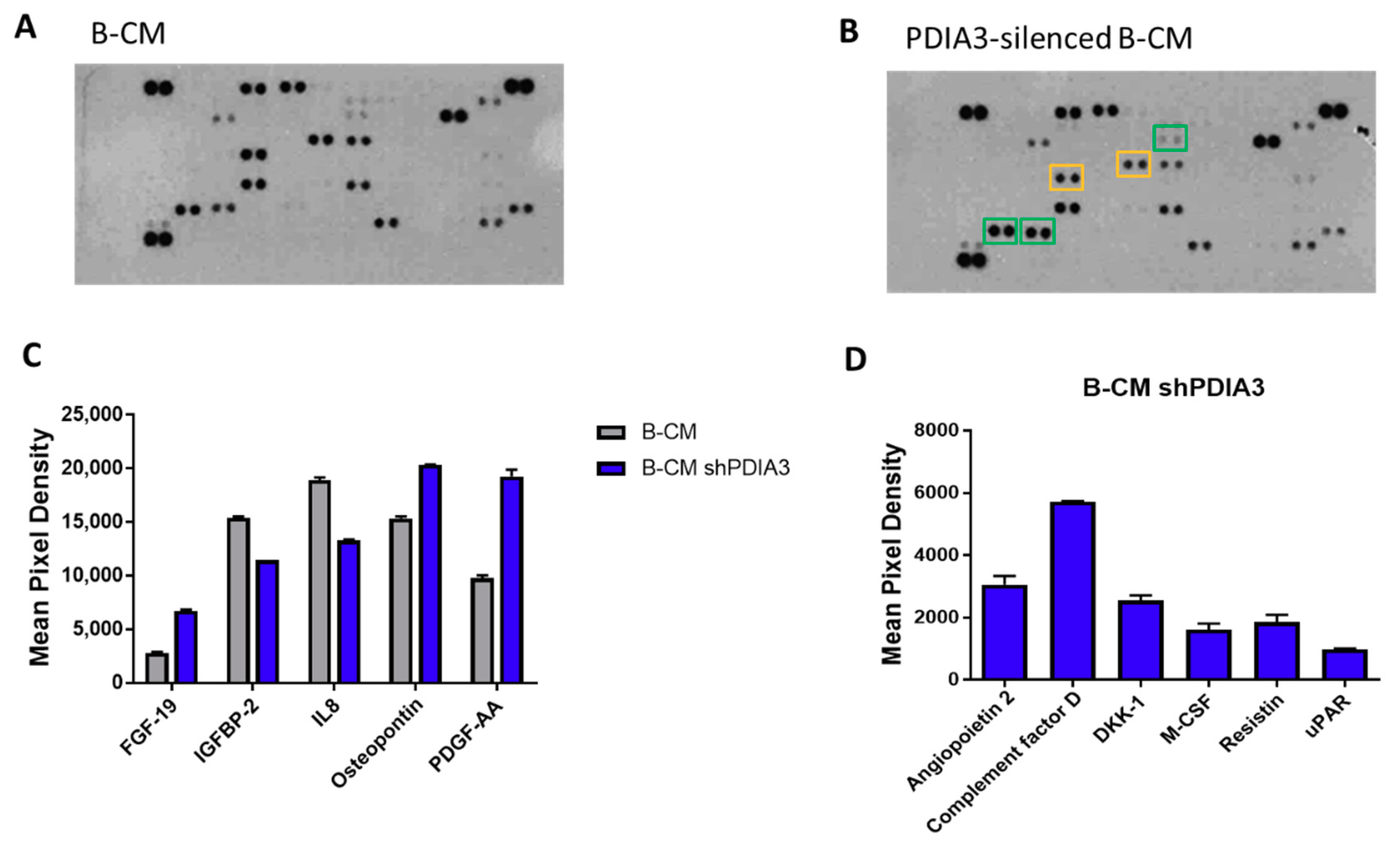
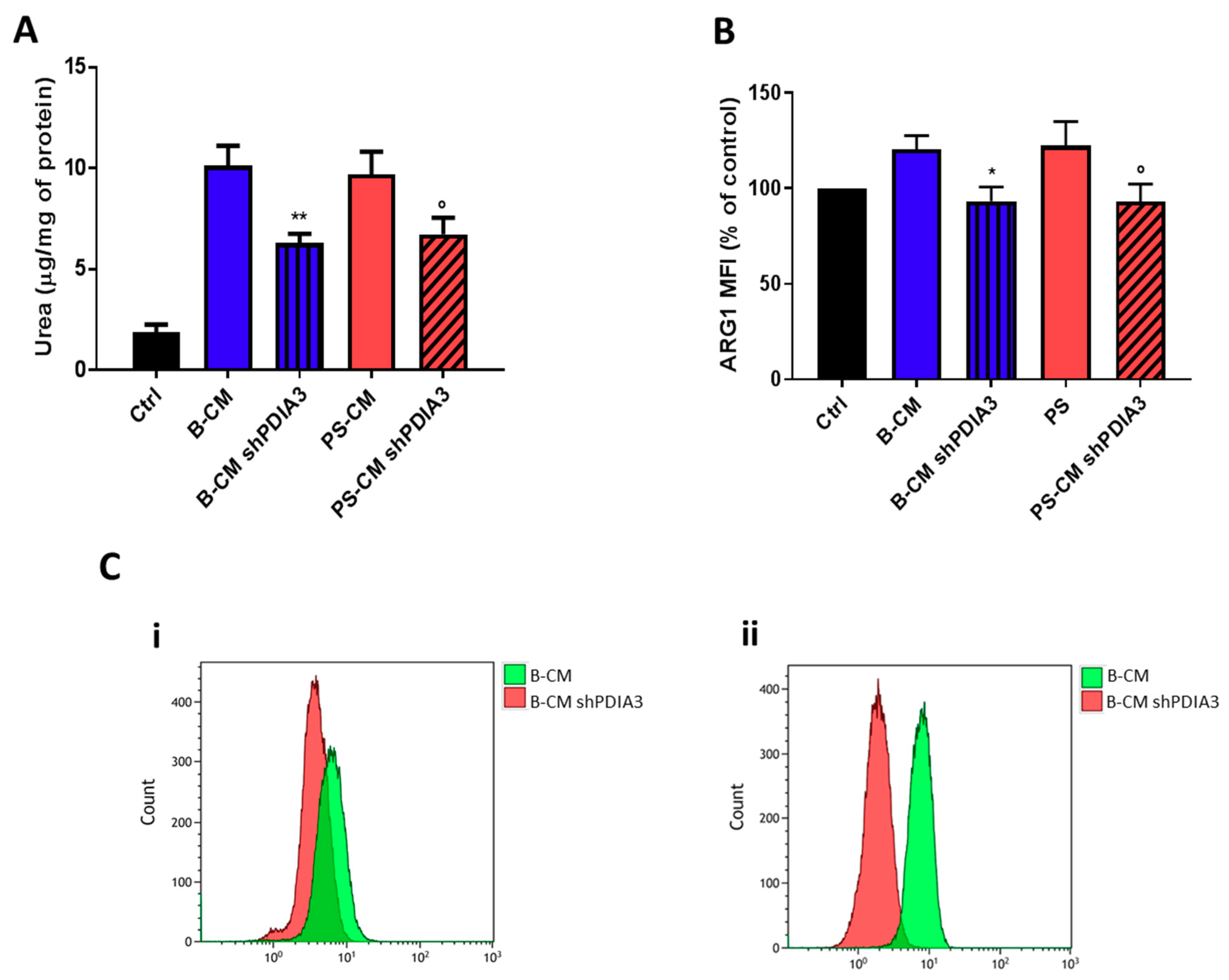
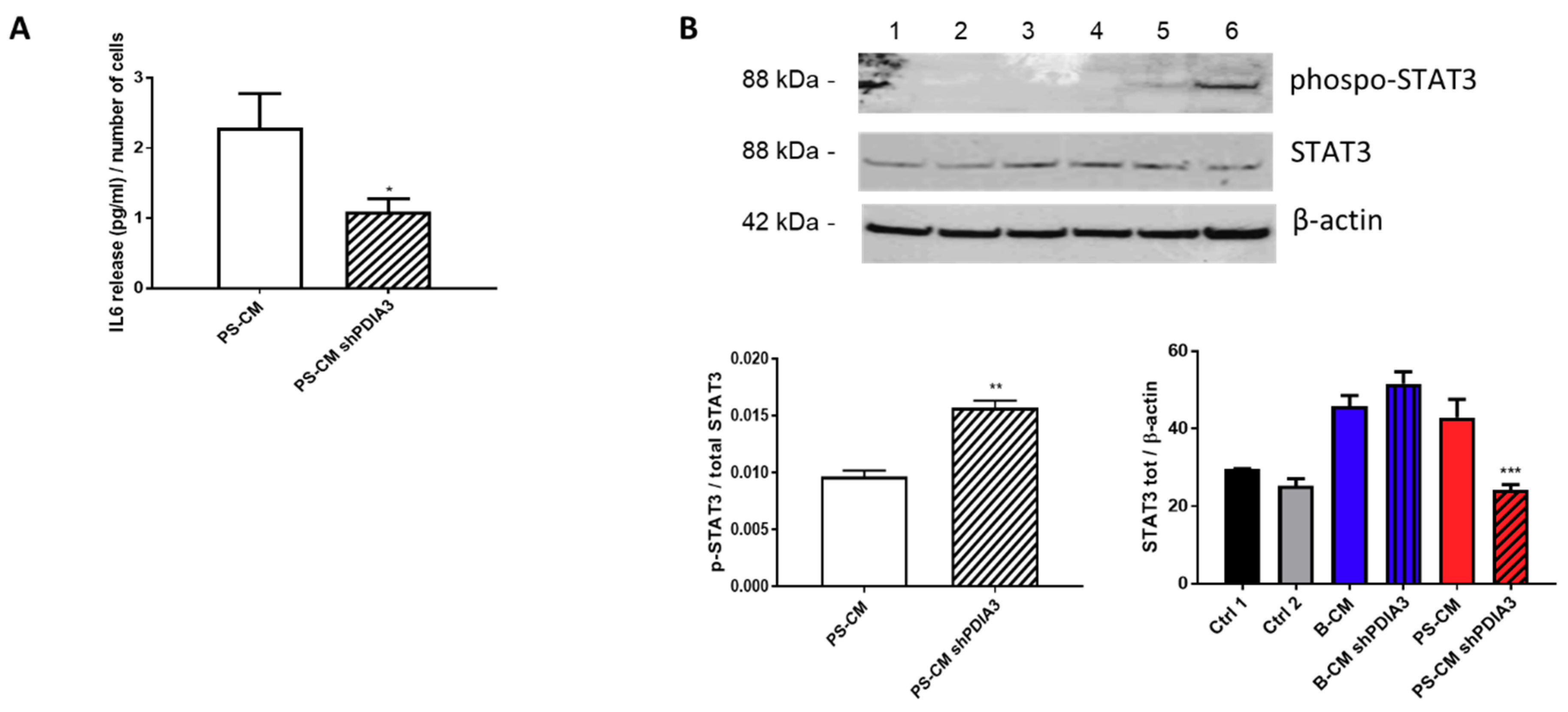
2.3. Effects of PDIA3 Gene Silencing in Microglia-GB Interaction
2.3.1. Effects of CMs from Control and PDIA3-Silenced T98G Cells on CHME-5 Viability and Activation
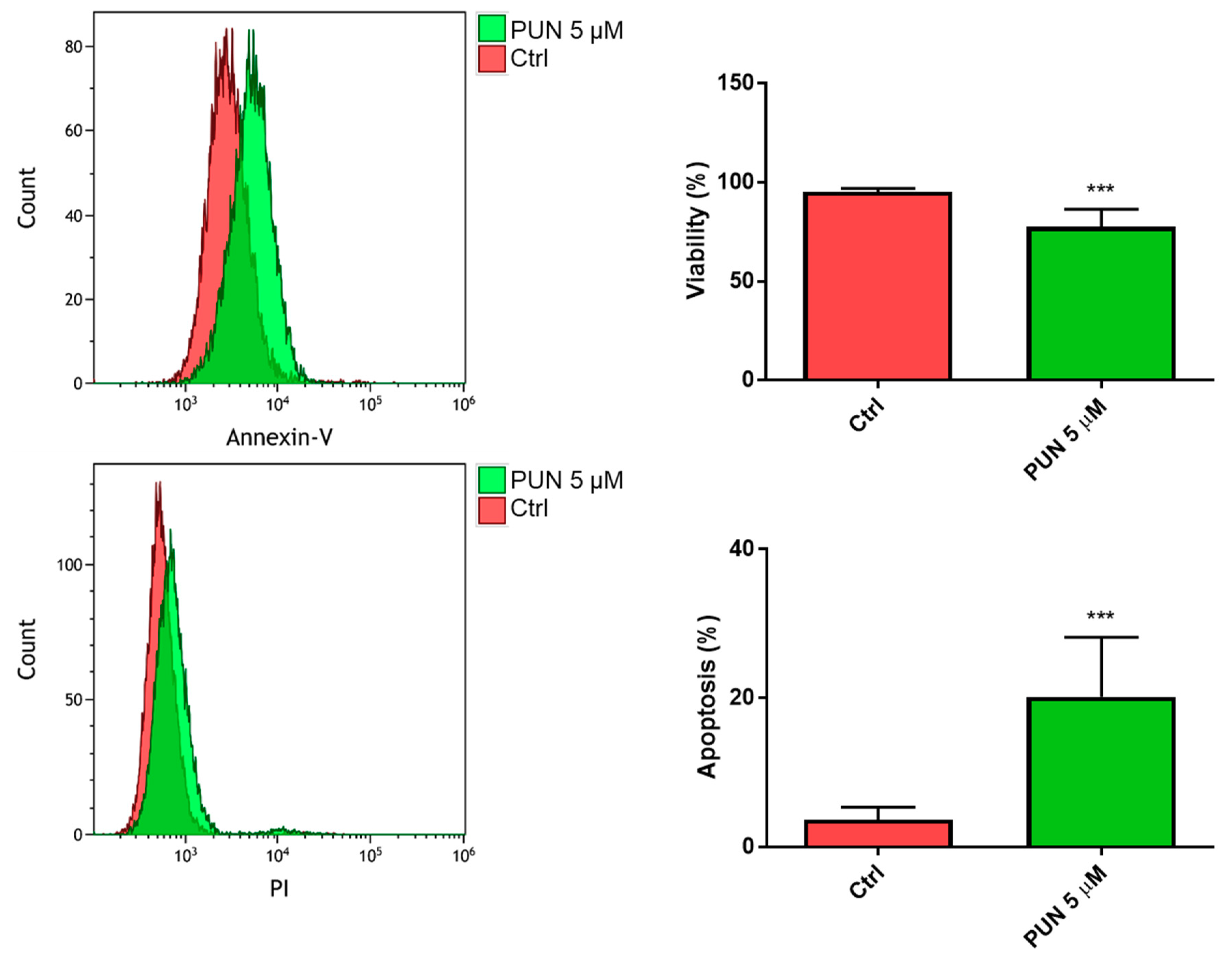
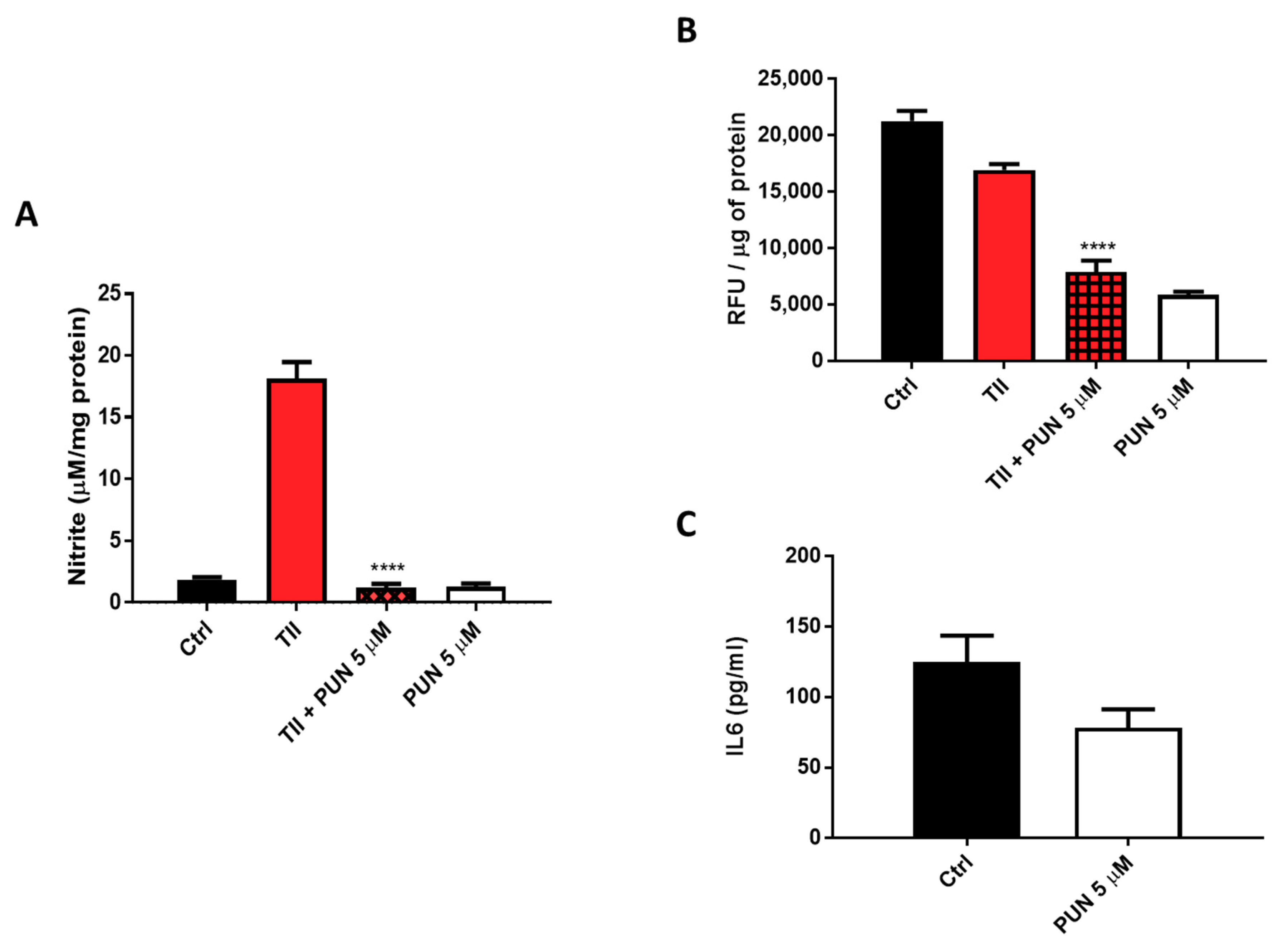
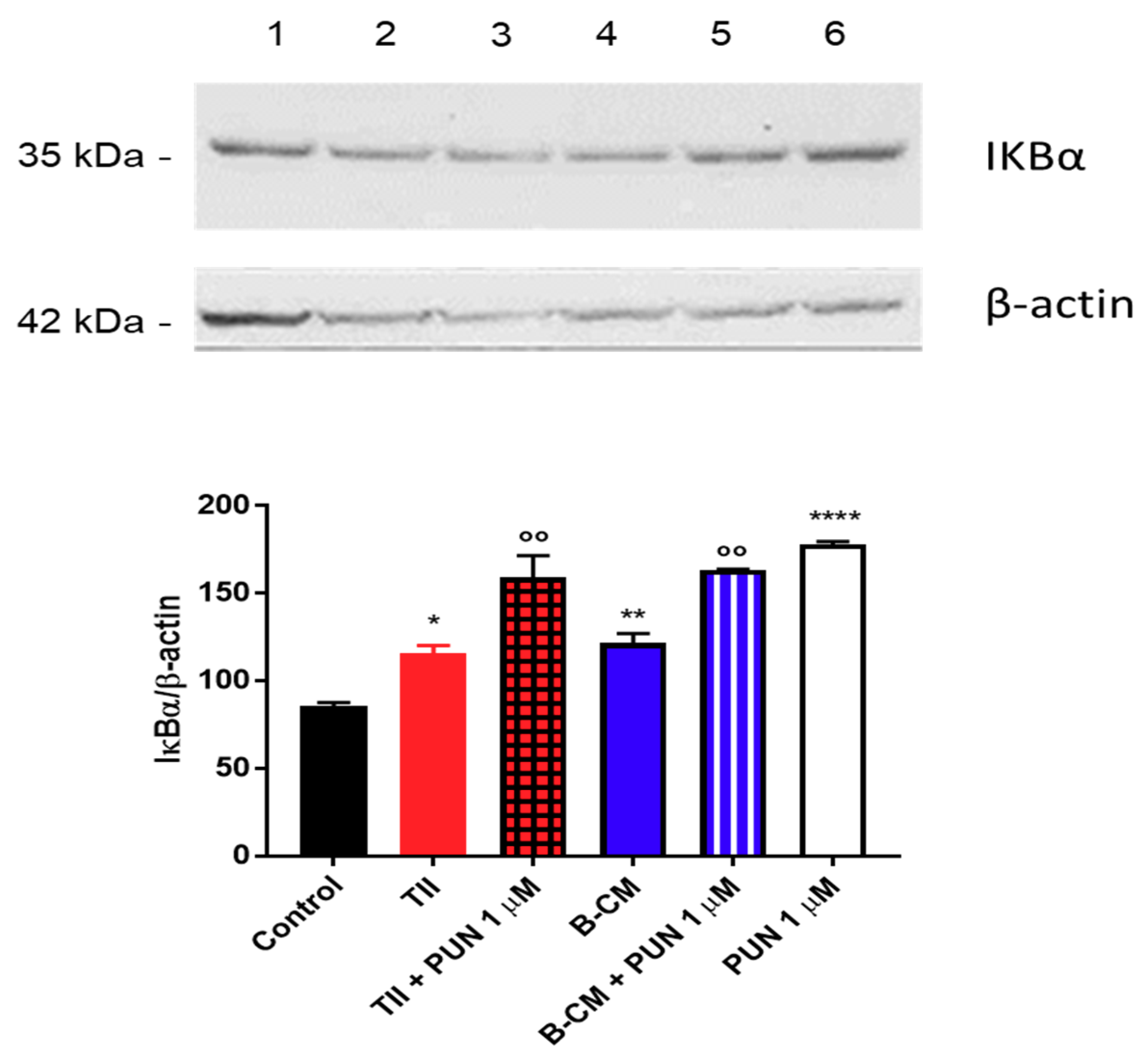
2.3.2. Effects of PUN on Microglia Pro-Inflammatory Activation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Patients and Specimens
4.3. Tissue Preparation
4.4. Immunohistochemistry
4.5. Cell Cultures
4.6. PDIA3 Short Interfering RNA (siRNA) Transfection
4.7. Nitrite Assay
4.8. Urea Assay
4.9. Flow Cytometry Analysis
4.10. Cell Viability and Toxicity
4.11. Reactive Oxygen Species (ROS) Assay
4.12. Real-Time Quantitative RT-PCR (qRT-PCR) Analysis
4.13. IL6 Quantification and Multiple Cytokines Analysis
4.14. Immunoblot Analysis
4.15. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PDIA3 | Protein Disulfide Isomerases 3 |
| GB | Glioblastoma |
| GAMs | Glioma Associated Microglia/macrophages |
| ER | Endoplasmic Reticulum |
| ERp57 | Endoplasmic Reticulum Protein 57 |
| PUN | Punicalagin |
| TCGA | The Cancer Genome Atlas |
| CM | Conditioned Medium |
| IL6 | Interleukin-6 |
| ERp60 | Endoplasmic Reticulum Protein 60 |
| PDI | Protein Disulfide Isomerase |
| TRX | Thioredoxin Domain |
| NFκB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| mTOR | Mammalian Target of Rapamycin |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| CCR5 | C-C chemokine Receptor type 5 |
| ROS | Reactive Oxygen Species |
| TNFα | Tumor Necrosis Factor α |
| siRNA | small interfering RNA |
| COX2 | Cyclooxygenase-2 |
| IL1β | Interleuckin-1 β |
| IFNγ | Interferon gamma |
| B-CM | Basal Conditioned-Medium |
| PS-CM | Prestimulated Conditioned-Medium |
| FGF-19 | Fibroblast Growth Factor 19 |
| PDGF-AA | Platelet-Derived Growth Factor A-A |
| IGFBP2 | Insulin-like Growth Factor-Binding Protein 2 |
| IL8 | Interleukin-8 |
| DKK-1 | Dickkopf-related protein 1 |
| M-CSF | Macrophage Colony-Stimulating Factor |
| uPAR | Urokinase Receptor |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| MCP-3 | Monocyte-Chemotactic Protein 3 |
| CCL20 | C-C Chemokine ligand 20 |
| MCP-1 | Monocyte-Chemotactic Protein 1 |
| CXCL11 | C-X-C motif Chemokine 11 |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| CCL3/CCL4 | C-C Chemokine ligand 3 and 4 |
| ARG1 | Arginase-1 |
| NO | Nitric Oxide |
| IκBα | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| NOS | Nitric Oxide Synthases |
| CCL2 | C-C Chemokine ligand 2 |
| IL10 | Interleukin-10 |
| SDF-1 | Stromal cell-Derived Factor 1 |
| VEGF-A | Vascular Endothelial Growth Factor-A |
| MIF | Macrophage migration Inhibitory Factor |
| IP-10 | Interferon gamma-induced Protein 10 |
| IL24 | Interleukin-24 |
References
- Rajaratnam, V.; Islam, M.M.; Yang, M.; Slaby, R.; Ramirez, H.M.; Mirza, S.P. Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments. Cancers 2020, 12, 937. [Google Scholar] [CrossRef]
- Lisi, L.; Chiavari, M.; Ciotti, G.M.P.; Lacal, P.M.; Navarra, P.; Graziani, G. DNA inhibitors for the treatment of brain tumors. Expert Opin. Drug Metab. Toxicol. 2020, 16, 195–207. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Dello Russo, C.; Lisi, L.; Tentori, L.; Navarra, P.; Graziani, G.; Combs, C.K. Exploiting Microglial Functions for the Treatment of Glioblastoma. Curr. Cancer Drug Targets 2017, 17, 267–281. [Google Scholar] [CrossRef]
- Kvisten, M.; Mikkelsen, V.E.; Stensjøen, A.L.; Solheim, O.; Van Der Want, J.; Torp, S.H. Microglia and macrophages in human glioblastomas: A morphological and immunohistochemical study. Mol. Clin. Oncol. 2019, 11, 31–36. [Google Scholar] [CrossRef]
- Freedman, R.B.; Hirst, T.R.; Tuite, M.F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci. 1994, 19, 331–336. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Wang, C.C. Protein disulfide-isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic. Biol. Med. 2015, 83, 305–313. [Google Scholar] [CrossRef]
- Turano, C.; Gaucci, E.; Grillo, C.; Chichiarelli, S. ERp57/GRP58: A protein with multiple functions. Cell Mol. Biol. Lett. 2011, 16, 539–563. [Google Scholar] [CrossRef]
- Hettinghouse, A.; Liu, R.; Liu, C.J. Multifunctional molecule ERp57: From cancer to neurodegenerative diseases. Pharmacol. Ther. 2018, 181, 34–48. [Google Scholar] [CrossRef]
- Frasconi, M.; Chichiarelli, S.; Gaucci, E.; Mazzei, F.; Grillo, C.; Chinazzi, A.; Altieri, F. Interaction of ERp57 with calreticulin: Analysis of complex formation and effects of vancomycin. Biophys. Chem. 2012, 160, 46–53. [Google Scholar] [CrossRef]
- Kozlov, G.; Gehring, K. Calnexin cycle—Structural features of the ER chaperone system. FEBS J. 2020, 287, 4322–4340. [Google Scholar] [CrossRef]
- Gaucci, E.; Raimondo, D.; Grillo, C.; Cervoni, L.; Altieri, F.; Nittari, G.; Eufemi, M.; Chichiarelli, S. Analysis of the interaction of calcitriol with the disulfide isomerase ERp57. Sci. Rep. 2016, 6, 37957. [Google Scholar] [CrossRef]
- Khanal, R.; Nemere, I. Membrane receptors for vitamin D metabolites. Crit. Rev. Eukaryot. Gene Expr. 2007, 17, 31–47. [Google Scholar] [CrossRef]
- Ramírez-Rangel, I.; Bracho-Valdés, I.; Vázquez-Macías, A.; Carretero-Ortega, J.; Reyes-Cruz, G.; Vázquez-Prado, J. Regulation of mTORC1 complex assembly and signaling by GRp58/ERp57. Mol. Cell Biol. 2011, 31, 1657–1671. [Google Scholar] [CrossRef]
- Chichiarelli, S.; Gaucci, E.; Ferraro, A.; Grillo, C.; Altieri, F.; Cocchiola, R.; Arcangeli, V.; Turano, C.; Eufemi, M. Role of ERp57 in the signaling and transcriptional activity of STAT3 in a melanoma cell line. Arch. Biochem. Biophys. 2010, 494, 178–183. [Google Scholar] [CrossRef]
- Eufemi, M.; Coppari, S.; Altieri, F.; Grillo, C.; Ferraro, A.; Turano, C. ERp57 is present in STAT3-DNA complexes. Biochem. Biophys. Res. Commun. 2004, 323, 1306–1312. [Google Scholar] [CrossRef]
- Guo, G.G.; Patel, K.; Kumar, V.; Shah, M.; Fried, V.A.; Etlinger, J.D.; Sehgal, P.B. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J. Interferon Cytokine Res. 2002, 22, 555–563. [Google Scholar] [CrossRef]
- Parakh, S.; Jagaraj, C.J.; Vidal, M.; Ragagnin, A.M.G.; Perri, E.R.; Konopka, A.; Toth, R.P.; Galper, J.; Blair, I.P.; Thomas, C.J.; et al. ERp57 is protective against mutant SOD1-induced cellular pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2018, 27, 1311–1331. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, P.; Woehlbier, U.; Chian, R.J.; Sapp, P.; Rouleau, G.A.; Leblond, C.S.; Daoud, H.; Dion, P.A.; Landers, J.E.; Hetz, C.; et al. Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 2015, 566, 158–165. [Google Scholar] [CrossRef]
- Padariya, M.; Kalathiya, U.; Houston, D.R.; Alfaro, J.A. Recognition Dynamics of Cancer Mutations on the ERp57-Tapasin Interface. Cancers 2020, 12, 737. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.X.; Nie, Z.Y.; Wen, Y.; Jia, X.J.; Zhang, L.N.; Duan, H.J.; Shi, Y.H. Upregulation of ERp57 promotes clear cell renal cell carcinoma progression by initiating a STAT3/ILF3 feedback loop. J. Exp. Clin. Cancer Res. 2019, 38, 439. [Google Scholar] [CrossRef]
- Wise, R.; Duhachek-Muggy, S.; Qi, Y.; Zolkiewski, M.; Zolkiewska, A. Protein disulfide isomerases in the endoplasmic reticulum promote anchorage-independent growth of breast cancer cells. Breast Cancer Res. Treat. 2016, 157, 241–252. [Google Scholar] [CrossRef]
- Xiao, B.; Wang, J.G.; Han, F.; Shi, Y.X. Effects of calcium-dependent molecular chaperones and endoplasmic reticulum in the amygdala in rats under single-prolonged stress. Mol. Med. Rep. 2018, 17, 1099–1104. [Google Scholar] [CrossRef]
- Choe, M.H.; Min, J.W.; Jeon, H.B.; Cho, D.H.; Oh, J.S.; Lee, H.G.; Hwang, S.G.; An, S.; Han, Y.H.; Kim, J.S. ERp57 modulates STAT3 activity in radioresistant laryngeal cancer cells and serves as a prognostic marker for laryngeal cancer. Oncotarget 2015, 6, 2654–2666. [Google Scholar] [CrossRef]
- Giamogante, F.; Marrocco, I.; Cervoni, L.; Eufemi, M.; Chichiarelli, S.; Altieri, F. Punicalagin, an active pomegranate component, is a new inhibitor of PDIA3 reductase activity. Biochimie 2018, 147, 122–129. [Google Scholar] [CrossRef]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef]
- Lisi, L.; Laudati, E.; Navarra, P.; Dello Russo, C. The mTOR kinase inhibitors polarize glioma-activated microglia to express a M1 phenotype. J. Neuroinflamm. 2014, 11, 125. [Google Scholar] [CrossRef]
- Lisi, L.; Ciotti, G.; Chiavari, M.; Pizzoferrato, M.; Mangiola, A.; Kalinin, S.; Feinstein, D.L.; Navarra, P. Phospho-mTOR expression in human glioblastoma microglia-macrophage cells. Neurochem. Int. 2019, 129, 104485. [Google Scholar] [CrossRef]
- Laudati, E.; Currò, D.; Navarra, P.; Lisi, L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Pia Ciotti, G.M.; Chiavari, M.; Ruffini, F.; Lacal, P.M.; Graziani, G.; Navarra, P. Vascular endothelial growth factor receptor 1 in glioblastoma associated microglia/macrophages. Oncol. Rep. 2020, 43, 2083–2092. [Google Scholar] [CrossRef]
- Seeram, N.P.; Adams, L.S.; Henning, S.M.; Niu, Y.; Zhang, Y.; Nair, M.G.; Heber, D. In vitro antiproliferative, apoptotic and antioxidant activities of punicalagin, ellagic acid and a total pomegranate tannin extract are enhanced in combination with other polyphenols as found in pomegranate juice. J. Nutr. Biochem. 2005, 16, 360–367. [Google Scholar] [CrossRef]
- Liu, M.; Du, L.; He, Z.; Yan, L.; Shi, Y.; Shang, J.; Tang, H. Increased ERp57 Expression in HBV-Related Hepatocellular Carcinoma: Possible Correlation and Prognosis. Biomed. Res. Int. 2017, 2017, 1252647. [Google Scholar] [CrossRef]
- Gaucci, E.; Altieri, F.; Turano, C.; Chichiarelli, S. The protein ERp57 contributes to EGF receptor signaling and internalization in MDA-MB-468 breast cancer cells. J. Cell Biochem. 2013, 114, 2461–2470. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef]
- Weissenberger, J.; Loeffler, S.; Kappeler, A.; Kopf, M.; Lukes, A.; Afanasieva, T.A.; Aguzzi, A.; Weis, J. IL-6 is required for glioma development in a mouse model. Oncogene 2004, 23, 3308–3316. [Google Scholar] [CrossRef] [PubMed]
- Castaño, Z.; San Juan, B.P.; Spiegel, A.; Pant, A.; DeCristo, M.J.; Laszewski, T.; Ubellacker, J.M.; Janssen, S.R.; Dongre, A.; Reinhardt, F.; et al. IL-1β inflammatory response driven by primary breast cancer prevents metastasis-initiating cell colonization. Nat. Cell Biol. 2018, 20, 1084–1097. [Google Scholar] [CrossRef]
- Taketo, M.M. Cyclooxygenase-2 inhibitors in tumorigenesis (part I). J. Natl. Cancer Inst. 1998, 90, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Taketo, M.M. Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J. Natl. Cancer Inst. 1998, 90, 1609–1620. [Google Scholar] [CrossRef]
- Trifan, O.C.; Hla, T. Cyclooxygenase-2 modulates cellular growth and promotes tumorigenesis. J. Cell Mol. Med. 2003, 7, 207–222. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Shono, T.; Tofilon, P.J.; Bruner, J.M.; Owolabi, O.; Lang, F.F. Cyclooxygenase-2 expression in human gliomas: Prognostic significance and molecular correlations. Cancer Res. 2001, 61, 4375–4381. [Google Scholar]
- Xu, W.; Huang, Y.; Zhang, T.; Zhao, L.; Fan, J.; Li, L. Cyclooxygenase-2 gene polymorphisms and susceptibility to hepatocellular carcinoma: A meta-analysis based on 10 case-control studies. J. Cancer Res. Ther. 2018, 14, S105–S113. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Munder, M.; Eichmann, K.; Morán, J.M.; Centeno, F.; Soler, G.; Modolell, M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J. Immunol. 1999, 163, 3771–3777. [Google Scholar]
- Arcuri, C.; Fioretti, B.; Bianchi, R.; Mecca, C.; Tubaro, C.; Beccari, T.; Franciolini, F.; Giambanco, I.; Donato, R. Microglia-glioma cross-talk: A two way approach to new strategies against glioma. Front. Biosci. 2017, 22, 268–309. [Google Scholar] [CrossRef]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef]
- Kondo, R.; Ishino, K.; Wada, R.; Takata, H.; Peng, W.X.; Kudo, M.; Kure, S.; Kaneya, Y.; Taniai, N.; Yoshida, H.; et al. Downregulation of protein disulfide isomerase A3 expression inhibits cell proliferation and induces apoptosis through STAT3 signaling in hepatocellular carcinoma. Int. J. Oncol. 2019, 54, 1409–1421. [Google Scholar] [CrossRef]
- Sun, X.; Wang, J.; Huang, M.; Chen, T.; Chen, J.; Zhang, F.; Zeng, H.; Xu, Z.; Ke, Y. STAT3 promotes tumour progression in glioma by inducing FOXP1 transcription. J. Cell Mol. Med. 2018, 22, 5629–5638. [Google Scholar] [CrossRef]
- Chang, N.; Ahn, S.H.; Kong, D.S.; Lee, H.W.; Nam, D.H. The role of STAT3 in glioblastoma progression through dual influences on tumor cells and the immune microenvironment. Mol. Cell Endocrinol. 2017, 451, 53–65. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V.W. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Wang, S.C.; Hong, J.H.; Hsueh, C.; Chiang, C.S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. 2012, 92, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Sielska, M.; Przanowski, P.; Wylot, B.; Gabrusiewicz, K.; Maleszewska, M.; Kijewska, M.; Zawadzka, M.; Kucharska, J.; Vinnakota, K.; Kettenmann, H.; et al. Distinct roles of CSF family cytokines in macrophage infiltration and activation in glioma progression and injury response. J. Pathol. 2013, 230, 310–321. [Google Scholar] [CrossRef]
- Olajide, O.A.; Kumar, A.; Velagapudi, R.; Okorji, U.P.; Fiebich, B.L. Punicalagin inhibits neuroinflammation in LPS-activated rat primary microglia. Mol. Nutr. Food Res. 2014, 58, 1843–1851. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hwang, C.J.; Lee, H.P.; Kim, C.S.; Son, D.J.; Ham, Y.W.; Hellström, M.; Han, S.B.; Kim, H.S.; Park, E.K.; et al. Inhibitory effect of punicalagin on lipopolysaccharide-induced neuroinflammation, oxidative stress and memory impairment via inhibition of nuclear factor-kappaB. Neuropharmacology 2017, 117, 21–32. [Google Scholar] [CrossRef]
- Rojanathammanee, L.; Puig, K.L.; Combs, C.K. Pomegranate polyphenols and extract inhibit nuclear factor of activated T-cell activity and microglial activation in vitro and in a transgenic mouse model of Alzheimer disease. J. Nutr. 2013, 143, 597–605. [Google Scholar] [CrossRef]
- Wang, S.G.; Huang, M.H.; Li, J.H.; Lai, F.I.; Lee, H.M.; Hsu, Y.N. Punicalagin induces apoptotic and autophagic cell death in human U87MG glioma cells. Acta Pharmacol. Sin. 2013, 34, 1411–1419. [Google Scholar] [CrossRef]
- Atzori, M.G.; Tentori, L.; Ruffini, F.; Ceci, C.; Lisi, L.; Bonanno, E.; Scimeca, M.; Eskilsson, E.; Daubon, T.; Miletic, H.; et al. The anti-vascular endothelial growth factor receptor-1 monoclonal antibody D16F7 inhibits invasiveness of human glioblastoma and glioblastoma stem cells. J. Exp. Clin. Cancer Res. 2017, 36, 106. [Google Scholar] [CrossRef] [PubMed]
- Janabi, N.; Peudenier, S.; Héron, B.; Ng, K.H.; Tardieu, M. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci. Lett. 1995, 195, 105–108. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Cohort (18 Patients) | Demographic Characteristic | Number of Patients (N) | % of Patients |
|---|---|---|---|
| Age | over 70 | 5 | 27.8 |
| 60–70 | 5 | 27.8 | |
| 50–60 | 3 | 16.7 | |
| 40–50 | 3 | 16.7 | |
| under 40 | 1 | 5.6 | |
| NA | 1 | 5.6 | |
| Gender | Male | 13 | 72.2 |
| Female | 5 | 27.8 | |
| WHO grade classes | IV | 18 | 100 |
| Tumor | Primary | 14 | 77.8 |
| Recurrent | 4 | 22.2 | |
| Tumor location | Frontal | 6 | 33.3 |
| Occipital | 2 | 11.1 | |
| Temporal | 5 | 27.8 | |
| Parietal | 1 | 5.6 | |
| Tempo-Parietal | 1 | 5.6 | |
| NA | 3 | 16.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiavari, M.; Ciotti, G.M.P.; Canonico, F.; Altieri, F.; Lacal, P.M.; Graziani, G.; Navarra, P.; Lisi, L. PDIA3 Expression in Glioblastoma Modulates Macrophage/Microglia Pro-Tumor Activation. Int. J. Mol. Sci. 2020, 21, 8214. https://doi.org/10.3390/ijms21218214
Chiavari M, Ciotti GMP, Canonico F, Altieri F, Lacal PM, Graziani G, Navarra P, Lisi L. PDIA3 Expression in Glioblastoma Modulates Macrophage/Microglia Pro-Tumor Activation. International Journal of Molecular Sciences. 2020; 21(21):8214. https://doi.org/10.3390/ijms21218214
Chicago/Turabian StyleChiavari, Marta, Gabriella Maria Pia Ciotti, Francesco Canonico, Fabio Altieri, Pedro Miguel Lacal, Grazia Graziani, Pierluigi Navarra, and Lucia Lisi. 2020. "PDIA3 Expression in Glioblastoma Modulates Macrophage/Microglia Pro-Tumor Activation" International Journal of Molecular Sciences 21, no. 21: 8214. https://doi.org/10.3390/ijms21218214
APA StyleChiavari, M., Ciotti, G. M. P., Canonico, F., Altieri, F., Lacal, P. M., Graziani, G., Navarra, P., & Lisi, L. (2020). PDIA3 Expression in Glioblastoma Modulates Macrophage/Microglia Pro-Tumor Activation. International Journal of Molecular Sciences, 21(21), 8214. https://doi.org/10.3390/ijms21218214