A?-Induced Damage Memory in hCMEC/D3 Cells Mediated by Sirtuin-1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
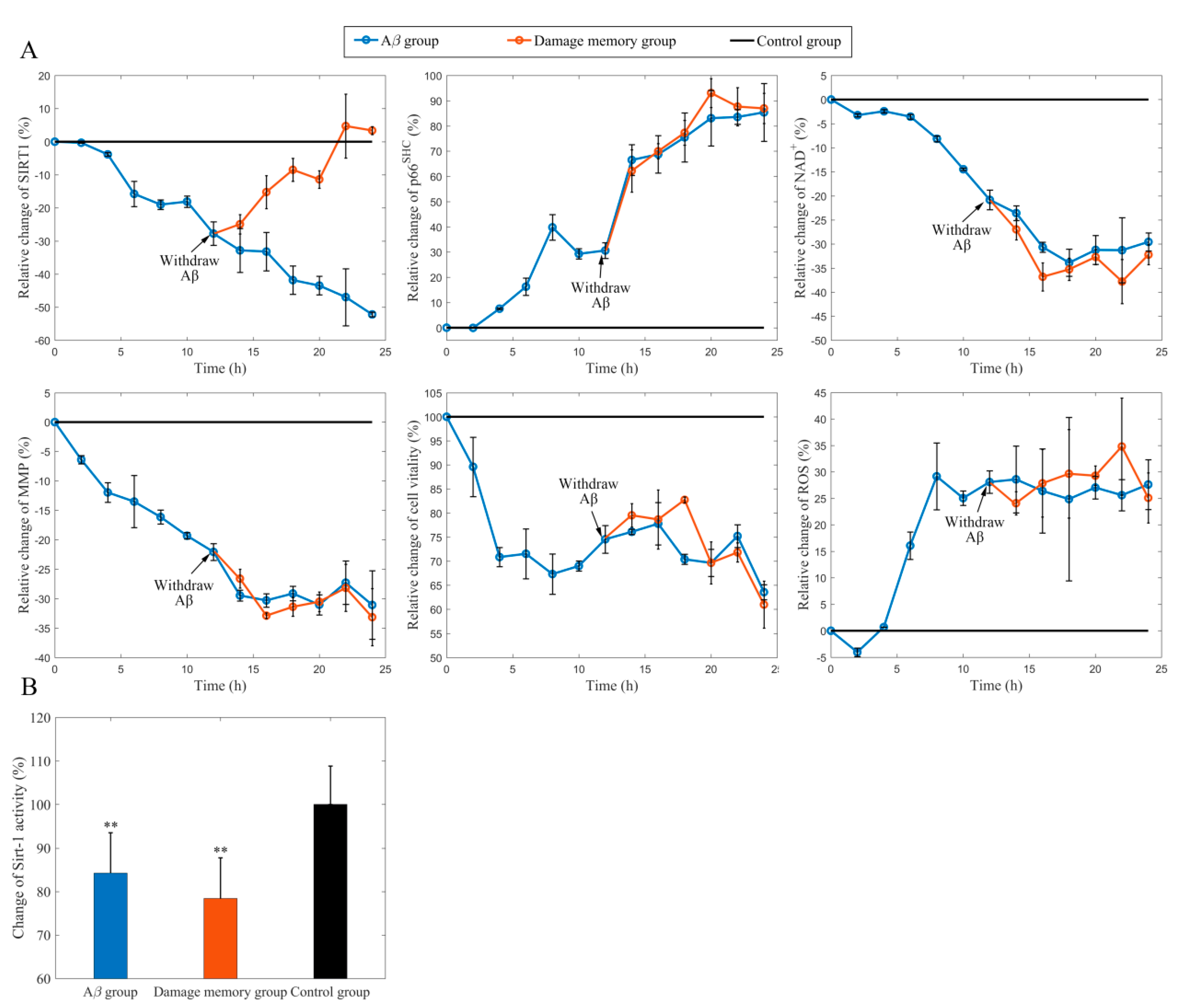
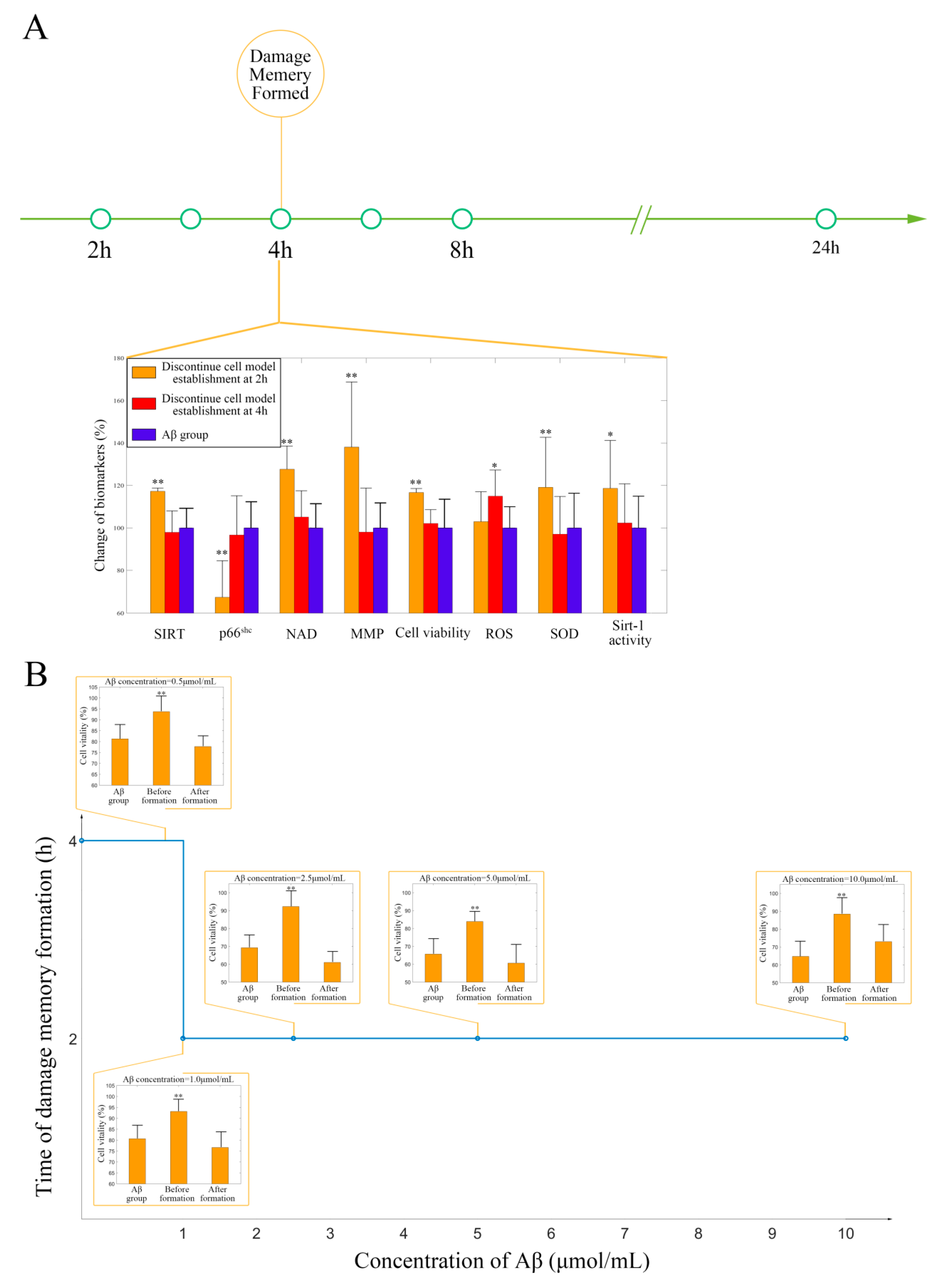
2.1. Withdrawing A Does Not Improve hCMEC/D3 Cell Vitality
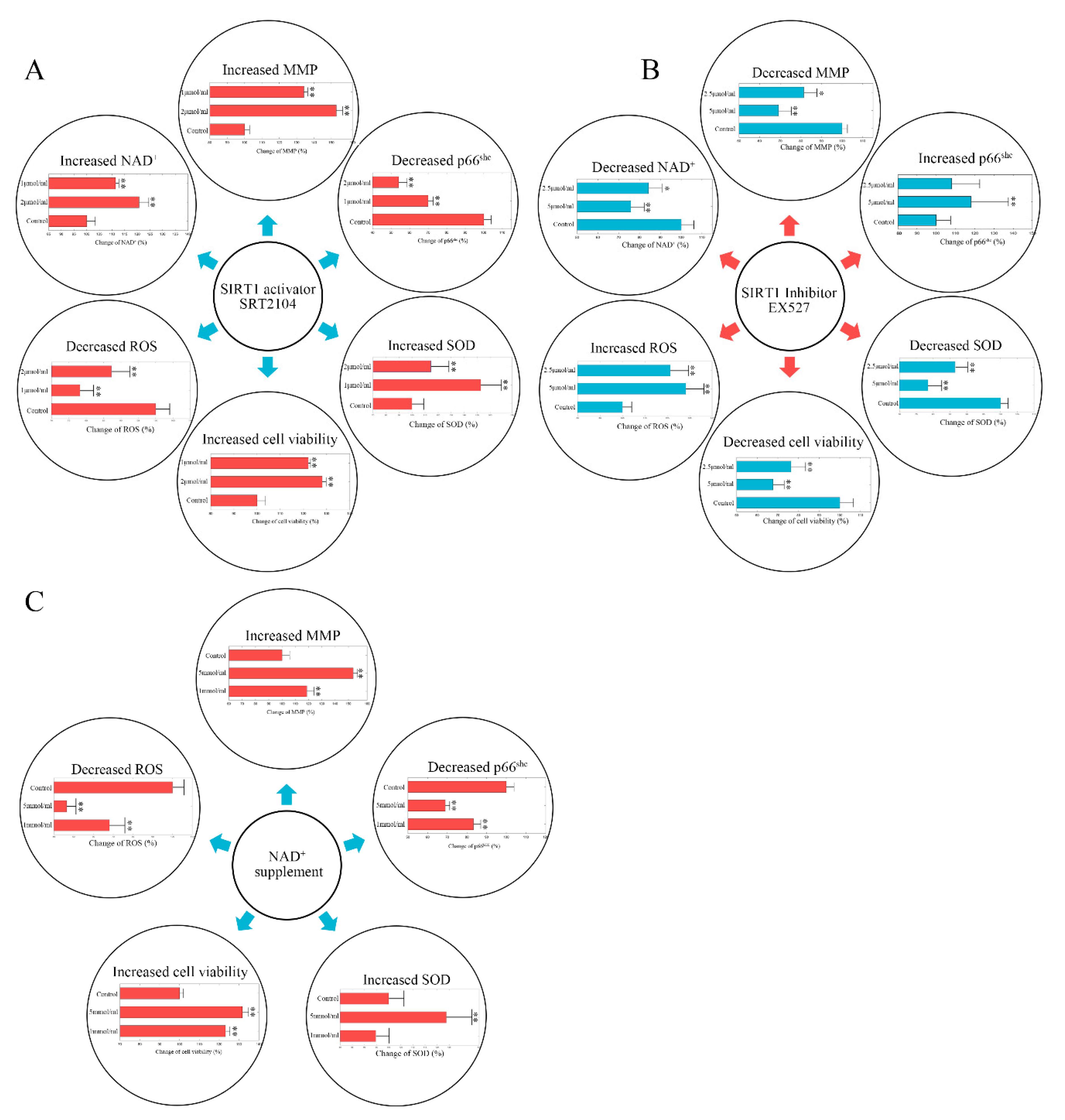
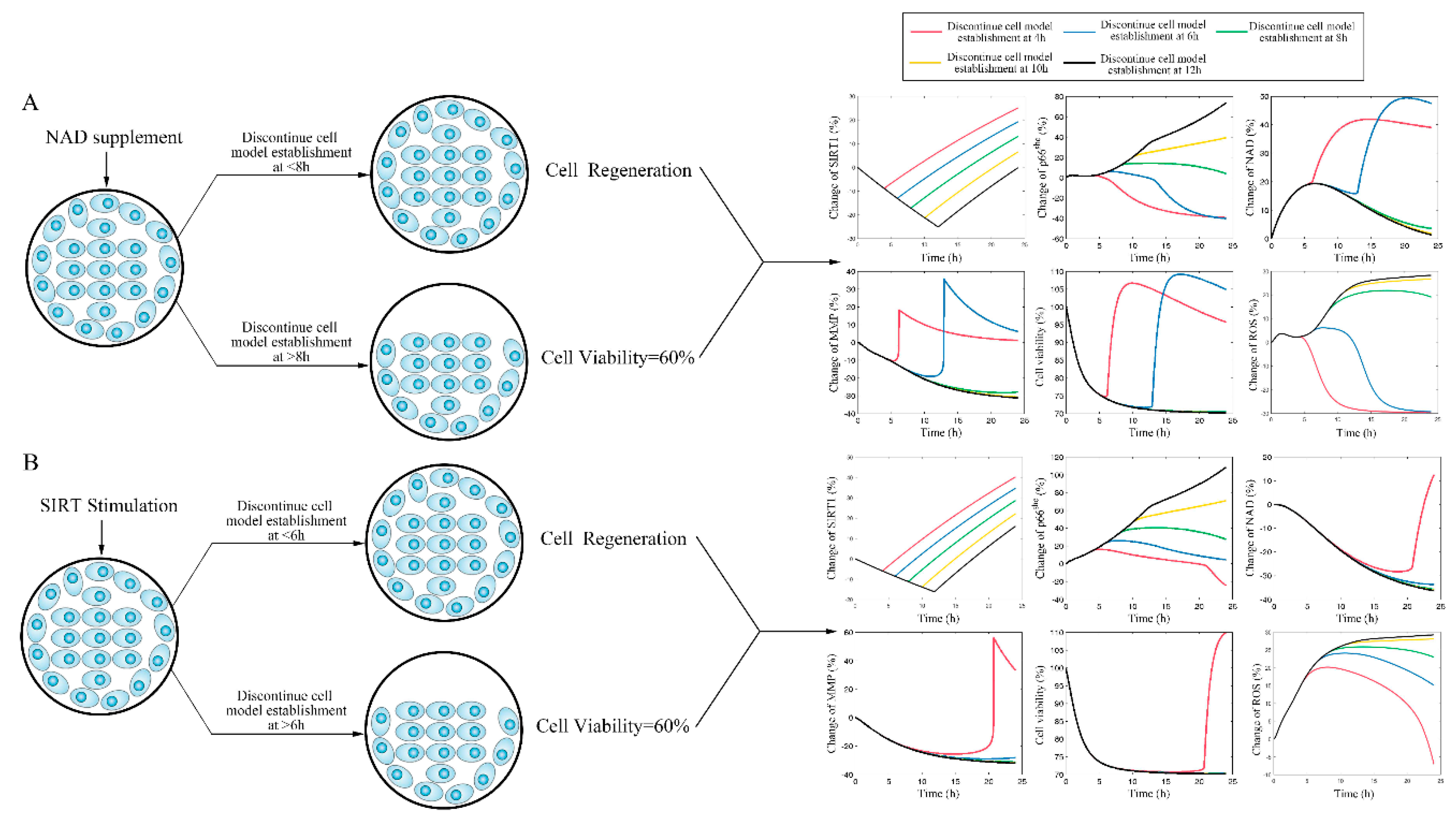
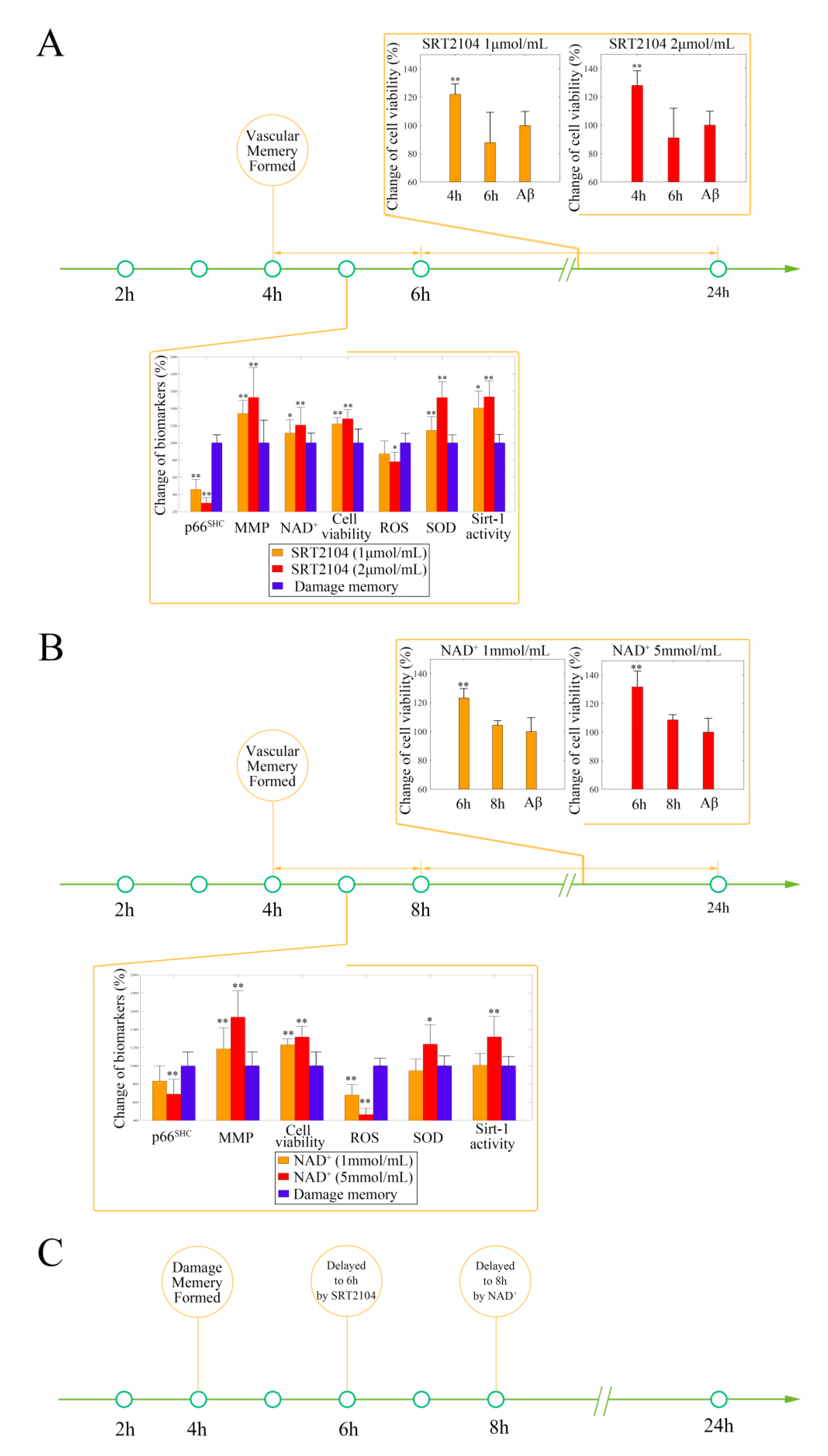
2.2. Stimulating Sirt-1 Relieves the Endothelial Damage Memory
2.3. Inhibiting Sirt-1 Exacerbates the Endothelial Damage Memory
2.4. NAD+ and Sirt-1 Play Different Roles in the Dynamic Process of Endothelial Damage Memory
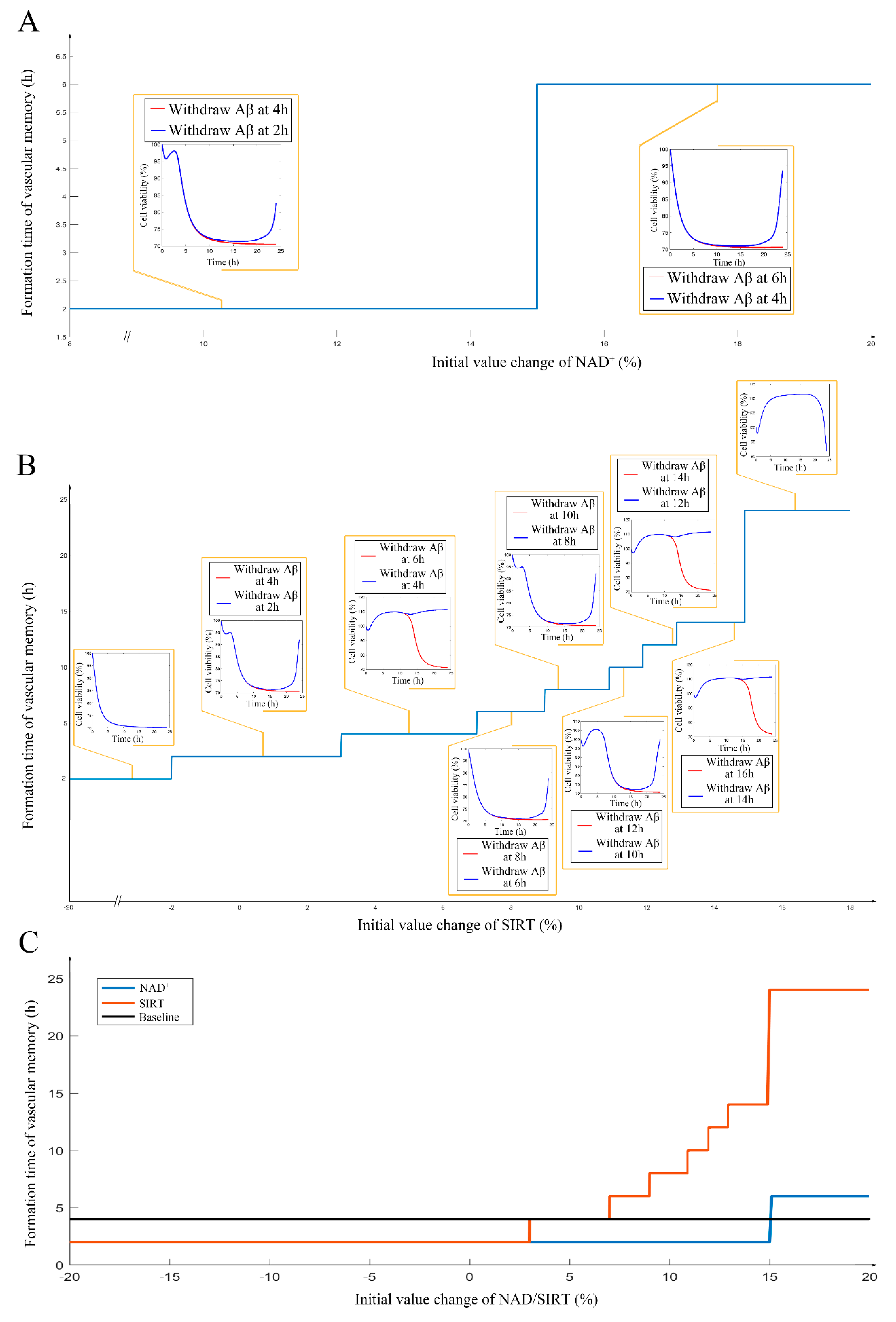
2.5. Different Roles of NAD+, and Sirt-1 in Delaying the Formation of Endothelial Damage Memory
3. Discussion
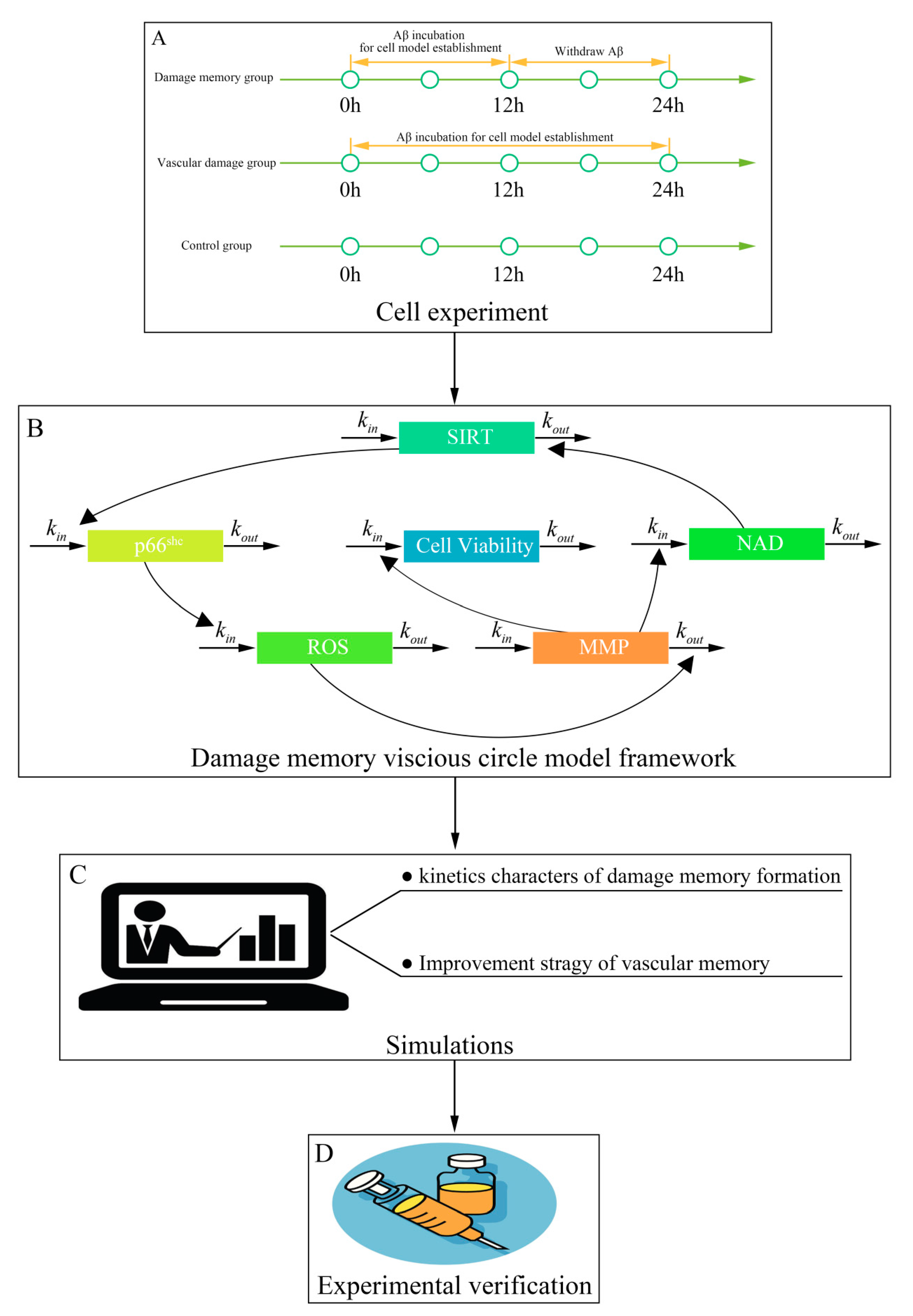
4. Method
4.1. Cell Culture
4.2. Cell Treatment
4.3. ELISA Kit
4.4. Immunoblot
4.5. Cell Vitality Assay
4.6. MMP Assay
4.7. ROS Measurement
4.8. NAD+ Sample Preparation and HPLC Condition
4.9. Sirt-1 Activity Assay
4.10. Mechanism-Based Kinetic Progression Model Development
4.11. Simulation
4.12. Simulation Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rózga, M.; Bittner, T.; Batrla, R.; Karl, J. Preanalytical sample handling recommendations for Alzheimer’s disease plasma biomarkers. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimers-disease–the amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Quaranta, V.; Glenner, G.G. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc. Natl. Acad. Sci. USA 1985, 82, 8729–8732. [Google Scholar] [CrossRef] [Green Version]
- Goure, W.F.; Krafft, G.A.; Jerecic, J.; Hefti, F. Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res. 2014, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.Y.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.M.; Ma, J.F. The role of amyloid beta clearance in cerebral amyloid angiopathy: More potential therapeutic targets. Transl. Neurodegener. 2017, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Burbach, G.J.; Vlachos, A.; Ghebremedhin, E.; Del Turco, D.; Coomaraswamy, J.; Staufenbiel, M.; Jucker, M.; Deller, T. Vessel ultrastructure in APP23 transgenic mice after passive anti-A beta immunotherapy and subsequent intracerebral hemorrhage. Neurobiol. Aging 2007, 28, 202–212. [Google Scholar] [CrossRef]
- Johnson, K.A.; Gregas, M.; Becker, J.A.; Kinnecom, C.; Salat, D.H.; Moran, E.K.; Smith, E.E.; Rosand, J.; Rentz, D.M.; Klunk, W.E.; et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann. Neurol. 2007, 62, 229–234. [Google Scholar] [CrossRef]
- Racke, M.M.; Boone, L.I.; Hepburn, D.L.; Parsadainian, M.; Bryan, M.T.; Ness, D.K.; Piroozi, K.S.; Jordan, W.H.; Brown, D.D.; Hoffman, W.P.; et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor proteintransgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J. Neurosci. 2005, 25, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, D.M.; Alamed, J.; Gottschall, P.E.; Grimm, J.; Rosenthal, A.; Pons, J.; Ronan, V.; Symmonds, K.; Gordon, M.N.; Morgan, D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5340–5346. [Google Scholar] [CrossRef]
- Berezin, A. Metabolic memory phenomenon in diabetes mellitus: Achieving and perspectives. Diabetes Metab. Syndr. Clin. Res. Rev. 2016, 10, S176–S183. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose Control and Vascular Complications in Veterans with Type 2 Diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiao, B.; Shen, L. The Epigenetics of Alzheimer’s Disease: Factors and Therapeutic Implications. Front. Genet. 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Norman, S.; Bennett, D.A.; Calon, F. Sirtuin 1 Reduction Parallels the Accumulation of Tau in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.D.; Margariti, A.; Zeng, L.F.; Xu, Q.B. Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc. Res. 2011, 90, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.; Kamermans, A.; van het Hof, B.; Castricum, K.; Aanhane, E.; van Horssen, J.; Thijssen, V.L.; Scheltens, P.; Teunissen, C.E.; Fontijn, R.D.; et al. Angiopoietin like-4 as a novel vascular mediator in capillary cerebral amyloid angiopathy. Brain 2018, 141, 3377–3388. [Google Scholar] [CrossRef]
- Hirschey, M.D. Old Enzymes, New Tricks: Sirtuins Are NAD(+)-Dependent De-acylases. Cell Metab. 2011, 14, 718–719. [Google Scholar] [CrossRef] [Green Version]
- Paneni, F.; Volpe, M.; Luscher, T.F.; Cosentino, F. SIRT1, p66(Shc), and Set7/9 in Vascular Hyperglycemic Memory. Diabetes 2013, 62, 1800–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.A.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc Expression by SIRT1 Contributes to the Prevention of Hyperglycemia-Induced Endothelial Dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Bourdette, D.; Forte, M. Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front. Physiol. 2013, 4, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, J.F.; Argaud, L.; Cottet-Rousselle, C.; Vial, G.; Gonzalez, C.; Detaille, D.; Leverve, X.; Fontaine, E. Effect of Transient and Permanent Permeability Transition Pore Opening on NAD(P)H Localization in Intact Cells. J. Biol. Chem. 2009, 284, 15117–15125. [Google Scholar] [CrossRef] [Green Version]
- Pallàs, M.; Pizarro, J.G.; Gutierrez-Cuesta, J.; Crespo-Biel, N.; Alvira, D.; Tajes, M.; Yeste-Velasco, M.; Folch, J.; Canudas, A.M.; Sureda, F.X.; et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience 2008, 154, 1388–1397. [Google Scholar] [CrossRef]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s Amyloid β-Peptide-Associated Free Radical Oxidative Stress and Neurotoxicity. J. Struct. Biol. 2000, 130, 184–208. [Google Scholar] [CrossRef]
- Li, N.C.; Lee, A.; Whitmer, R.A.; Kivipelto, M.; Lawler, E.; Kazis, L.E.; Wolozin, B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: Prospective cohort analysis. BMJ Br. Med. J. 2010, 340, b5465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhou, Y.G.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.L.; Mucke, L.; Li, G. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappa B signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donmez, G.; Guarente, L. Aging and disease: Connections to sirtuins. Aging Cell 2010, 9, 285–290. [Google Scholar] [CrossRef]
- Granchi, C.; Minutolo, F. Activators of Sirtuin-1 and their Involvement in Cardioprotection. Curr. Med. Chem. 2018, 25, 4432–4456. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Tekirian, T.L.; Yang, A.Y.; Glabe, C.; Geddes, J.W. Toxicity of Pyroglutaminated Amyloid β-Peptides 3(pE)-40 and -42 Is Similar to That of Aβ1-40 and -42. J. Neurochem. 1999, 73, 1584–1589. [Google Scholar] [CrossRef]
- Peck, B.; Chen, C.Y.; Ho, K.K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.D.; Lam, E.W.F. SIRT Inhibitors Induce Cell Death and p53 Acetylation through Targeting Both SIRT1 and SIRT2. Mol. Cancer 2010, 9, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Venkatasubramanian, S.; Noh, R.M.; Daga, S.; Langrish, J.P.; Joshi, N.V.; Mills, N.L.; Hoffmann, E.; Jacobson, E.W.; Vlasuk, G.P.; Waterhouse, B.R.; et al. Cardiovascular Effects of a Novel SIRT1 Activator, SRT2104, in Otherwise Healthy Cigarette Smokers. J. Am. Heart Assoc. 2013, 2, e000042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrow, N.J.; Shabir, G.A. Development and Validation of a HPLC Method for NAD: Application to Stability Studies in Buffered Solutions and Dry Test Strips. J. Liq. Chromatogr. Relat. Technol. 2009, 32, 2805–2821. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Zhang, Y.; Zhang, H.; Xu, S.; Zhao, H.; Liu, X. A?-Induced Damage Memory in hCMEC/D3 Cells Mediated by Sirtuin-1. Int. J. Mol. Sci. 2020, 21, 8226. https://doi.org/10.3390/ijms21218226
Liu H, Zhang Y, Zhang H, Xu S, Zhao H, Liu X. A?-Induced Damage Memory in hCMEC/D3 Cells Mediated by Sirtuin-1. International Journal of Molecular Sciences. 2020; 21(21):8226. https://doi.org/10.3390/ijms21218226
Chicago/Turabian StyleLiu, Haochen, Yixuan Zhang, Hong Zhang, Sheng Xu, Huimin Zhao, and Xiaoquan Liu. 2020. "A?-Induced Damage Memory in hCMEC/D3 Cells Mediated by Sirtuin-1" International Journal of Molecular Sciences 21, no. 21: 8226. https://doi.org/10.3390/ijms21218226
APA StyleLiu, H., Zhang, Y., Zhang, H., Xu, S., Zhao, H., & Liu, X. (2020). A?-Induced Damage Memory in hCMEC/D3 Cells Mediated by Sirtuin-1. International Journal of Molecular Sciences, 21(21), 8226. https://doi.org/10.3390/ijms21218226