Depleting RhoA/Stress Fiber-Organized Fibronectin Matrices on Tumor Cells Non-Autonomously Aggravates Fibroblast-Driven Tumor Cell Growth

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
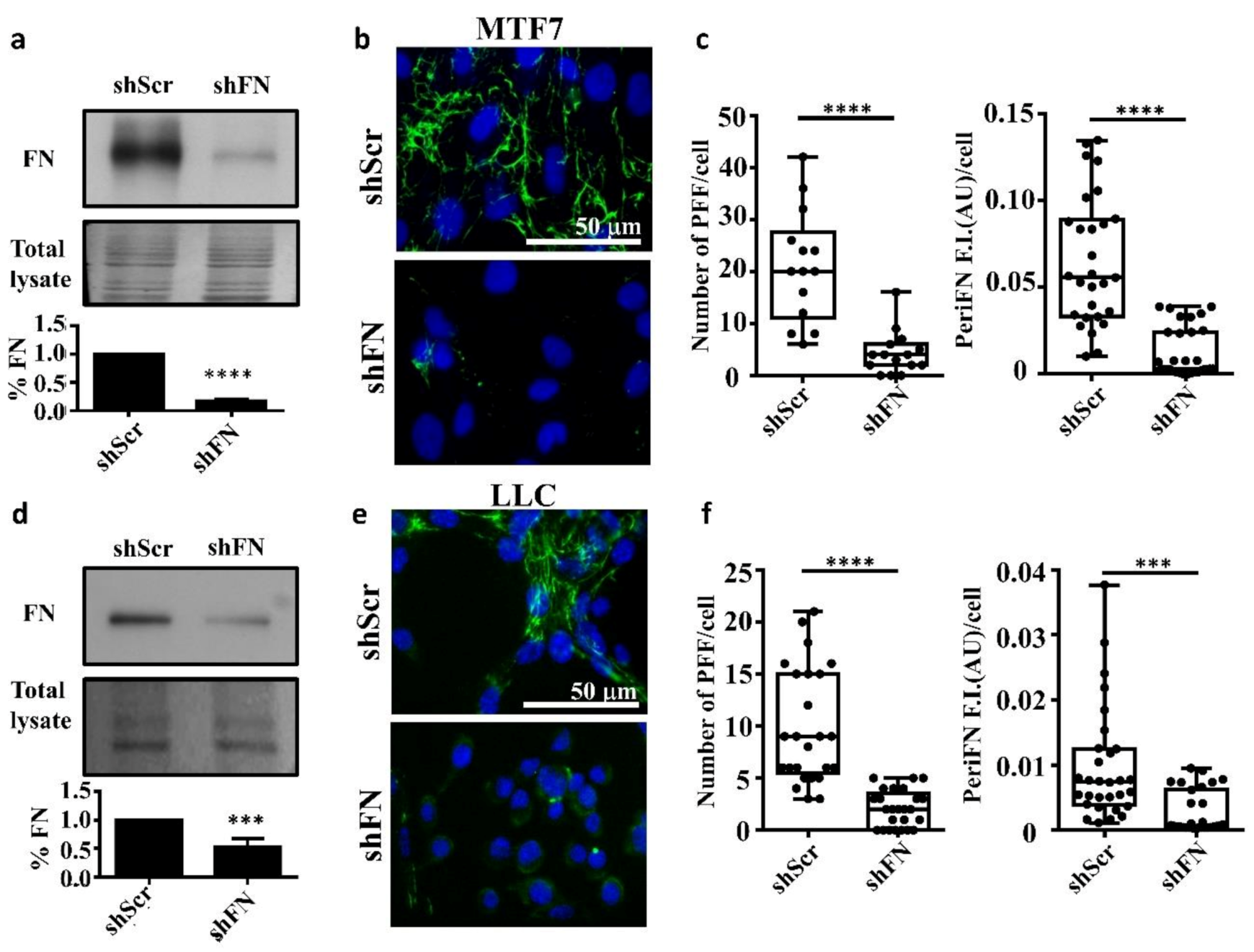
2.1. The Endogenous FN-Dependent periFN Assembly on Tumor Cells Is Regulated by SF Actin Cytoskeleton
2.2. The Stress Fiber-Organized Tumor periFN Assembly Is Regulated by RhoA Activity
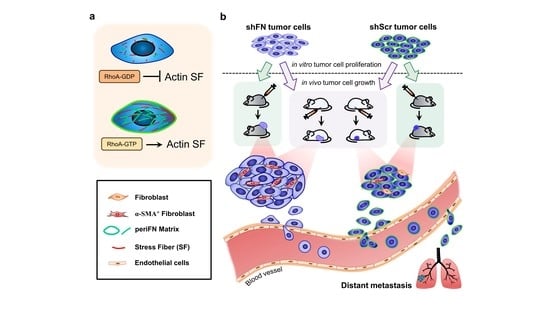
2.3. PeriFN Assembled on Tumor Cells Displays A Suppressive Role in In Vivo Tumor Growth
2.4. Demolishing periFN Does not Promote Tumor Cell Proliferative Activity
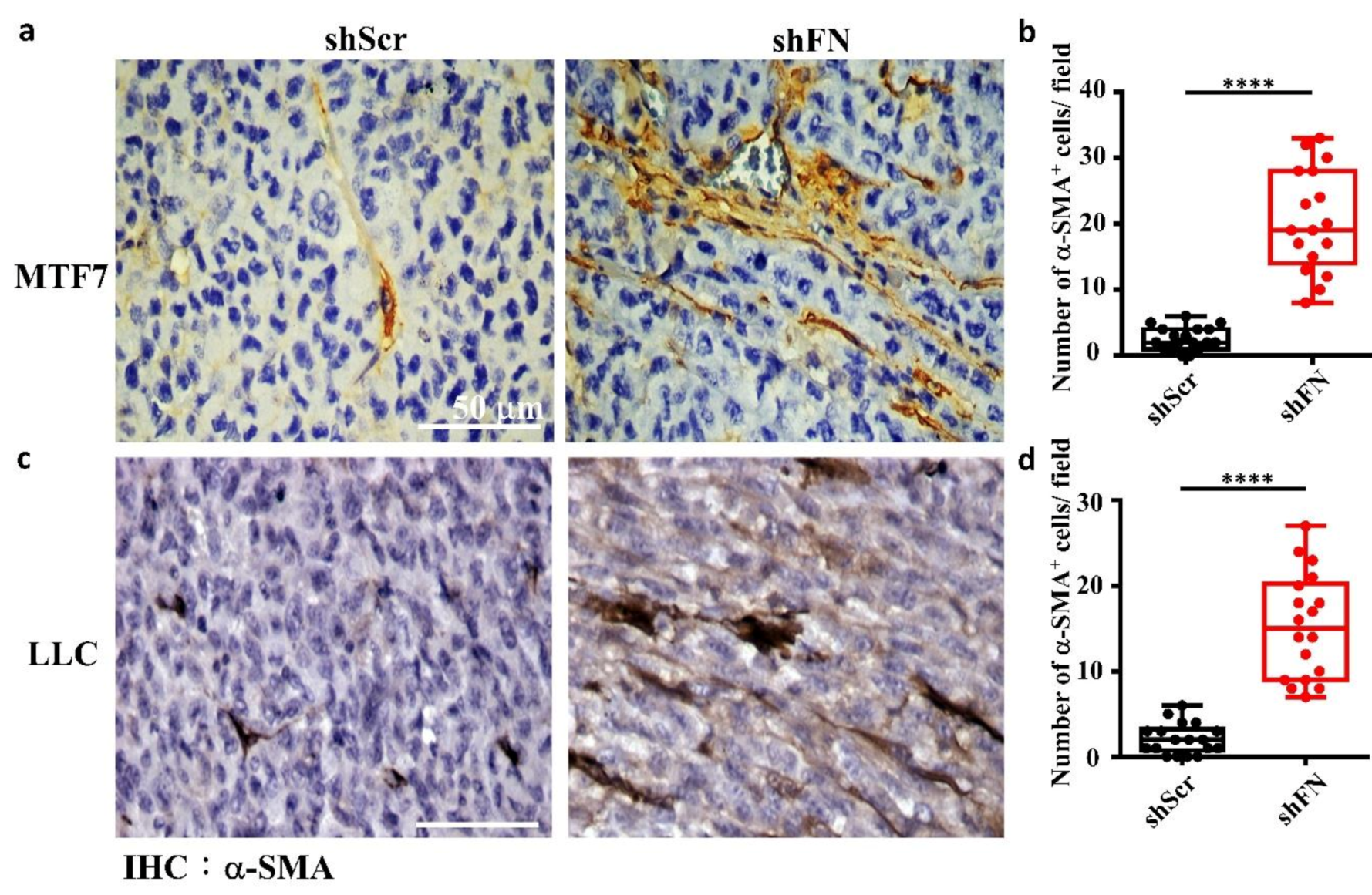
2.5. Depleting Cancerous PeriFN with shFN Non-autonomously Promotes Fibroblast-Mediated Tumor Growth
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Materials
4.3. Silencing Endogenous FN Expression in Tumor Cells with Short Hairpin RNAs Complementary to FN mRNA
4.4. Immunoblotting
4.5. Immunofluorescence Staining for periFN Matrices on Adherent Tumor Cell Surfaces
4.6. Fluorescence Staining for Actin Cytoskeleton
4.7. Double Fluorescence Staining for periFN and Actin Stress Fiber in Tumor Cells
4.8. Plasmid Transfection of Tumor Cells by Electroporation
4.9. Real-Time Cell Proliferation and Co-Culture Assays
4.10. Apoptosis Assay
4.11. In Vivo Tumor Growth Animal Models
4.12. Immunohistochemistry Staining
4.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FN | fibronectin |
| periFN | pericellular FN |
| SF | stress fiber |
| TMEs | tumor microenvironments |
| VHL | von Hippel–Lindau |
| RhoA-DN | dominant-negative Thr19Asn RhoA |
| GEF | guanidine nucleotide exchanging factor |
| RhoA-CA | constitutively active Gln63Leu RhoA |
| CAFs | cancer associated fibroblasts |
| α-SMA | α-smooth muscle actin |
| ECM | extracellular matrix |
| IL-1 | interleukin-1 |
| IL-6 | interleukin-6 |
| hHGF | human hepatocyte growth factor |
| HIF-1α | hypoxia-induced factor-1α |
| ROPN-1 | Rhophilin-associated tail protein 1 |
| FAP | fibroblast activation protein |
References
- Lin, T.C.; Yang, C.H.; Chang, W.T.; Lin, Y.R. Fibronectin in Cancer: Friend or Foe. Cells 2020, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, P.C. Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin. Cancer Biol. 2017, 47, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Renna, M.; Park, S.J.; Menzies, F.M.; Ricketts, T.; Füllgrabe, J.; Ashkenazi, A.; Frake, R.A.; Lombarte, A.C.; Bento, C.F.; et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat. Commun. 2018, 9, 2961. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cui, L.; Sun, P.; Shi, L.; Yue, C.; Li, F. Reusable N-Heterocyclic Carbene Complex Catalysts and Beyond: A Perspective on Recycling Strategies. Chem. Rev. 2018, 118, 9843–9929. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.E.; Neu, C.P.; Seelbinder, B.; McCreery, K.P. The Role of Mechanobiology in Cancer Metastasis. In Mechanobiology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 65–78. [Google Scholar]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharm. 2014, 171, 5507–5523. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Cheng, H.C.; Abdel-Ghany, M.; Elble, R.C.; Pauli, B.U. Lung endothelial dipeptidyl peptidase IV promotes adhesion and metastasis of rat breast cancer cells via tumor cell surface-associated fibronectin. J. Biol. Chem. 1998, 273, 24207–24215. [Google Scholar] [CrossRef]
- Cheng, H.C.; Abdel-Ghany, M.; Pauli, B.U. A novel consensus motif in fibronectin mediates dipeptidyl peptidase IV adhesion and metastasis. J. Biol. Chem. 2003, 278, 24600–24607. [Google Scholar] [CrossRef]
- Chang, Y.H.; Lee, S.H.; Liao, I.C.; Huang, S.H.; Cheng, H.C.; Liao, P.C. Secretomic analysis identifies alpha-1 antitrypsin (A1AT) as a required protein in cancer cell migration, invasion, and pericellular fibronectin assembly for facilitating lung colonization of lung adenocarcinoma cells. Mol. Cell. Proteom. 2012, 11, 1320–1339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Lin, J.F.; Cheng, L.H.; Chang, W.T.; Kao, Y.H.; Chang, M.M.; Wang, B.J.; Cheng, H.C. Pterostilbene prevents AKT-ERK axis-mediated polymerization of surface fibronectin on suspended lung cancer cells independently of apoptosis and suppresses metastasis. J. Hematol. Oncol. 2017, 10, 72. [Google Scholar] [CrossRef]
- Huang, L.; Cheng, H.C.; Isom, R.; Chen, C.S.; Levine, R.A.; Pauli, B.U. Protein kinase Cepsilon mediates polymeric fibronectin assembly on the surface of blood-borne rat breast cancer cells to promote pulmonary metastasis. J. Biol. Chem. 2008, 283, 7616–7627. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000, 406, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Niknami, Z.; Eslamifar, A.; Emamirazavi, A.; Ebrahimi, A.; Shirkoohi, R. The association of vimentin and fibronectin gene expression with epithelial-mesenchymal transition and tumor malignancy in colorectal carcinoma. EXCLI J. 2017, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Miao, L.; Yu, M.; Shi, M.; Wang, Y.; Yang, J.; Xiao, Y.; Cai, H. α1-antitrypsin promotes lung adenocarcinoma metastasis through upregulating fibronectin expression. Int. J. Oncol. 2017, 50, 1955–1964. [Google Scholar] [CrossRef]
- Wong, F.H.; Huang, C.Y.; Su, L.J.; Wu, Y.C.; Lin, Y.S.; Hsia, J.Y.; Tsai, H.T.; Lee, S.A.; Lin, C.H.; Tzeng, C.H.; et al. Combination of microarray profiling and protein-protein interaction databases delineates the minimal discriminators as a metastasis network for esophageal squamous cell carcinoma. Int. J. Oncol. 2009, 34, 117–128. [Google Scholar] [PubMed]
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, X.; Lu, W.; Chen, Y.; Wu, X.; Li, M.; Wang, X.-A.; Zhang, F.; Jiang, L.; Zhang, Y.; et al. Fibronectin promotes cell proliferation and invasion through mTOR signaling pathway activation in gallbladder cancer. Cancer Lett. 2015, 360, 141–150. [Google Scholar] [CrossRef]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Ruoslahti, E. Fibronectin in cell adhesion and invasion. Cancer Metastasis Rev. 1984, 3, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R. Molecular biology of fibronectin. Annu. Rev. Cell Biol. 1985, 1, 67–90. [Google Scholar] [CrossRef]
- Akiyama, S.K.; Olden, K.; Yamada, K.M. Fibronectin and integrins in invasion and metastasis. Cancer Metastasis Rev. 1995, 14, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cheng, S.; Asa, S.L.; Ezzat, S. The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res. 2008, 68, 8104–8112. [Google Scholar] [CrossRef]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. NKp46 Receptor-Mediated Interferon-γ Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis. Immunity 2018, 48, 107–119. [Google Scholar] [CrossRef]
- Brennan, J.R.; Hocking, D.C. Cooperative effects of fibronectin matrix assembly and initial cell-substrate adhesion strength in cellular self-assembly. Acta Biomater. 2016, 32, 198–209. [Google Scholar] [CrossRef]
- Gagné, D.; Benoit, Y.D.; Groulx, J.-F.; Vachon, P.H.; Beaulieu, J.-F. ILK supports RhoA/ROCK-mediated contractility of human intestinal epithelial crypt cells by inducing the fibrillogenesis of endogenous soluble fibronectin during the spreading process. BMC Mol. Biol. 2020, 21, 14. [Google Scholar] [CrossRef]
- Cali, G.; Mazzarella, C.; Chiacchio, M.; Negri, R.; Retta, S.F.; Zannini, M.; Gentile, F.; Tarone, G.; Nitsch, L.; Garbi, C. RhoA activity is required for fibronectin assembly and counteracts beta1B integrin inhibitory effect in FRT epithelial cells. J. Cell Sci. 1999, 112, 957. [Google Scholar]
- Menager, C.; Vassy, J.; Doliger, C.; Legrand, Y.; Karniguian, A. Subcellular localization of RhoA and ezrin at membrane ruffles of human endothelial cells: Differential role of collagen and fibronectin. Exp. Cell Res. 1999, 249, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Chrzanowska-Wodnicka, M.; Brown, J.; Shaub, A.; Belkin, A.M.; Burridge, K. Rho-mediated Contractility Exposes a Cryptic Site in Fibronectin and Induces Fibronectin Matrix Assembly. Int. J. Cell Biol. 1998, 141, 539–551. [Google Scholar] [CrossRef]
- Danen, E.H.J.; Sonneveld, P.; Brakebusch, C.; Fässler, R.; Sonnenberg, A. The fibronectin-binding integrins α5β1 and αvβ3 differentially modulate RhoA–GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. Int. J. Cell Biol. 2002, 159, 1071–1086. [Google Scholar] [CrossRef]
- Ohh, M.; Yauch, R.L.; Lonergan, K.M.; Whaley, J.M.; Stemmer-Rachamimov, A.O.; Louis, D.N.; Gavin, B.J.; Kley, N.; Kaelin, W.G., Jr.; Iliopoulos, O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol. Cell 1998, 1, 959–968. [Google Scholar] [CrossRef]
- Stickle, N.H.; Chung, J.; Klco, J.M.; Hill, R.P.; Kaelin, W.G., Jr.; Ohh, M. pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol. Cell. Biol. 2004, 24, 3251–3261. [Google Scholar] [CrossRef]
- Vikkath, N.; Ariyannur, P.; Menon, K.N.; Mr, B.; Pillai, A. Exploring the role of defective fibronectin matrix assembly in the VHL-associated CNS hemangioblastoma. Drug Metab. Pers. Ther. 2018, 33, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Feijóo-Cuaresma, M.; Méndez, F.; Maqueda, A.; Esteban, M.A.; Naranjo-Suarez, S.; Castellanos, M.C.; del Cerro, M.H.; Vazquez, S.N.; García-Pardo, A.; Landázuri, M.O.; et al. Inadequate activation of the GTPase RhoA contributes to the lack of fibronectin matrix assembly in von Hippel-Lindau protein-defective renal cancer cells. J. Biol. Chem. 2008, 283, 24982–24990. [Google Scholar] [CrossRef]
- Kamada, M.; Suzuki, K.; Kato, Y.; Okuda, H.; Shuin, T. von Hippel-Lindau Protein Promotes the Assembly of Actin and Vinculin and inhibits cell motility. Cancer Res. 2001, 61, 4184–4189. [Google Scholar]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar] [CrossRef]
- Kranenburg, O.; Poland, M.; Gebbink, M.; Oomen, L.; Moolenaar, W.H. Dissociation of LPA-induced cytoskeletal contraction from stress fiber formation by differential localization of RhoA. J. Cell Sci. 1997, 110, 2417. [Google Scholar]
- Ghosh, P.M.; Ghosh-Choudhury, N.; Moyer, M.L.; Mott, G.E.; Thomas, C.A.; Foster, B.A.; Greenberg, N.M.; Kreisberg, J.I. Role of RhoA activation in the growth and morphology of a murine prostate tumor cell line. Oncogene 1999, 18, 4120–4130. [Google Scholar] [CrossRef]
- Qiu, R.G.; Chen, J.; McCormick, F.; Symons, M. A role for Rho in Ras transformation. Proc. Natl. Acad. Sci. USA 1995, 92, 11781. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Barker, T.H.; Lindsey, M.L. Fibroblasts: The arbiters of extracellular matrix remodeling. Matrix Biol. 2020, 91–92, 1–7. [Google Scholar] [CrossRef]
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. 2018, 74, 52–61. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO Rep. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zai-Chun, X.; Wun-Lun, H.; Yun-Yun, Z. BMP-7 Attenuates TGF-β1-Induced Fibronectin Secretion and Apoptosis of NRK-52E Cells by the Suppression of miRNA-21. Oncol. Res. 2016, 23, 147–154. [Google Scholar] [CrossRef]
- Sridurongrit, S. Tumor-suppressive and tumor-promoting role of Tgf-Beta in Hepatocellular Carcinoma. Int. J. Biol. 2016, 9, 41–49. [Google Scholar] [CrossRef]
- Moses, H.L.; Roberts, A.B.; Derynck, R. The Discovery and Early Days of TGF-β: A Historical Perspective. Cold Spring Harb. Perspect. Biol. 2016, 8, a021865. [Google Scholar] [CrossRef]
- Gohda, E.; Matsunaga, T.; Kataoka, H.; Yamamoto, I. TGF-beta is a potent inhibitor of hepatocyte growth factor secretion by human fibroblasts. Cell Biol. Int. Rep. 1992, 16, 917–926. [Google Scholar] [CrossRef]
- Modica, C.; Tortarolo, D.; Comoglio, P.M.; Basilico, C.; Vigna, E. MET/HGF Co-Targeting in Pancreatic Cancer: A Tool to Provide Insight into the Tumor/Stroma Crosstalk. Int. J. Mol. Sci. 2018, 19, 3920. [Google Scholar] [CrossRef]
- Kamijo, R.; Takeda, K.; Nagumo, M.; Konno, K. Suppression of TNF-stimulated proliferation of diploid fibroblasts and TNF-induced cytotoxicity against transformed fibroblasts by TGF-beta. Biochem. Biophys. Res. Commun. 1989, 158, 155–162. [Google Scholar] [CrossRef]
- Errarte, P.; Larrinaga, G.; López, J.I. The role of cancer-associated fibroblasts in renal cell carcinoma. An example of tumor modulation through tumor/non-tumor cell interactions. J. Adv. Res. 2020, 21, 103–108. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Semenza, G.L. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol. Med. 2012, 18, 534–543. [Google Scholar] [CrossRef]
- Ryu, M.H.; Park, H.M.; Chung, J.; Lee, C.H.; Park, H.R. Hypoxia-inducible factor-1alpha mediates oral squamous cell carcinoma invasion via upregulation of alpha5 integrin and fibronectin. Biochem. Biophys. Res. Commun. 2010, 393, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, Y.J.; Han, H.J. Role of hypoxia-induced fibronectin-integrin β1 expression in embryonic stem cell proliferation and migration: Involvement of PI3K/Akt and FAK. J. Cell. Physiol. 2011, 226, 484–493. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Fischer, A. Notch1 induces endothelial senescence and promotes tumor progression. Cell Cycle 2017, 16, 911–912. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Zhan, Y.; Zeng, T.; Zhang, X.; Guan, X.Y.; Li, Y. Membrane Metalloendopeptidase (MME) Suppresses Metastasis of Esophageal Squamous Cell Carcinoma (ESCC) by Inhibiting FAK-RhoA Signaling Axis. Am. J. Pathol. 2019, 189, 1462–1472. [Google Scholar] [CrossRef]
- Fife, C.M.; Sagnella, S.M.; Teo, W.S.; Po’uha, S.T.; Byrne, F.L.; Yeap, Y.Y.; Ng, D.C.; Davis, T.P.; McCarroll, J.A.; Kavallaris, M. Stathmin mediates neuroblastoma metastasis in a tubulin-independent manner via RhoA/ROCK signaling and enhanced transendothelial migration. Oncogene 2017, 36, 501–511. [Google Scholar] [CrossRef]
- García-Mariscal, A.; Li, H.; Pedersen, E.; Peyrollier, K.; Ryan, K.M.; Stanley, A.; Quondamatteo, F.; Brakebusch, C. Loss of RhoA promotes skin tumor formation and invasion by upregulation of RhoB. Oncogene 2018, 37, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, T.; Malik, R.; Thomas, S.; Sage, J.; Johnson, P.F. C/EBPβ cooperates with RB:E2F to implement RasV12-induced cellular senescence. EMBO Rep. 2005, 24, 3301–3312. [Google Scholar] [CrossRef]
- Chen, Q.M.; Tu, V.C.; Catania, J.; Burton, M.; Toussaint, O.; Dilley, T. Involvement of Rb family proteins, focal adhesion proteins and protein synthesis in senescent morphogenesis induced by hydrogen peroxide. J. Cell Sci. 2000, 113, 4087. [Google Scholar]
- Maria-Engler, S.S.; Soengas, M.S.; Campa, A.; Reiter, R.J.; Souza, P.d.C.; Pegoraro, R.; Pagni, R.L.; Massaro, R.R.; Tiago, M.; Saito, R.d.F.; et al. Melatonin inhibits human melanoma cells proliferation and invasion via cell cycle arrest and cytoskeleton remodeling. Melatonin Res. 2020, 3, 194–209. [Google Scholar] [CrossRef]
- Natarajan, S.; Williamson, D.; Stiltz, A.J.; Harding, K. Advances in wound care and healing technology. Am. J. Clin. Dermatol. 2000, 1, 269–275. [Google Scholar] [CrossRef]
- Raz, Y.; van den Akker, E.B.; Roest, T.; Riaz, M.; van de Rest, O.; Suchiman, H.E.D.; Lakenberg, N.; Stassen, S.A.; van Putten, M.; Feskens, E.J.M.; et al. A data-driven methodology reveals novel myofiber clusters in older human muscles. FASEB J. 2020, 34, 5525–5537. [Google Scholar] [CrossRef]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. ∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nat. Commun. 2018, 9, 254. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Park, J.E.; Schubert, R.D.; Rettig, W.J.; Peter, R.U.; Garin-Chesa, P. Fibroblast activation protein: Differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J. Investig. Dermatol. 2003, 120, 182–188. [Google Scholar] [CrossRef]
- Santos, A.M.; Jung, J.; Aziz, N.; Kissil, J.L.; Puré, E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Investig. 2009, 119, 3613–3625. [Google Scholar] [CrossRef]
- Loeffler, M.; Krüger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Investig. 2006, 116, 1955–1962. [Google Scholar] [CrossRef]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-β-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005, 65, 3772–3780. [Google Scholar] [CrossRef]
- Zhang, X.H.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massagué, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 154, 1060–1073. [Google Scholar] [CrossRef]
- Wu, M.-H.; Hong, H.-C.; Hong, T.-M.; Chiang, W.-F.; Jin, Y.-T.; Chen, Y.-L. Targeting Galectin-1 in Carcinoma-Associated Fibroblasts Inhibits Oral Squamous Cell Carcinoma Metastasis by Downregulating MCP-1/CCL2 Expression. Clin. Cancer Res. 2011, 17, 1306. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999, 144, 1235–1244. [Google Scholar] [CrossRef]
- Nieradka, A.; Ufer, C.; Thiadens, K.; Grech, G.; Horos, R.; van Coevorden-Hameete, M.; van den Akker, E.; Sofi, S.; Kuhn, H.; von Lindern, M. Grsf1-induced translation of the SNARE protein Use1 is required for expansion of the erythroid compartment. PLoS ONE 2014, 9, e104631. [Google Scholar] [CrossRef] [PubMed]
- Pasztorek, M.; Rossmanith, E.; Mayr, C.; Ebner, A. Influence of Platelet Lysate on 2D and 3D Amniotic Mesenchymal Stem Cell Cultures. Front. Bioeng. Biotechnol. 2019, 7, 338. [Google Scholar] [CrossRef]
- Olumi, A.F.; Dazin, P.; Tlsty, T.D. A Novel Coculture Technique Demonstrates That Normal Human Prostatic. Cancer Res. 1998, 58, 4525–4530. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.-T.; Tsai, C.-L.; Huang, S.-H.; Chang, M.-M.; Chang, W.-T.; Cheng, L.-H.; Cheng, H.-C. Depleting RhoA/Stress Fiber-Organized Fibronectin Matrices on Tumor Cells Non-Autonomously Aggravates Fibroblast-Driven Tumor Cell Growth. Int. J. Mol. Sci. 2020, 21, 8272. https://doi.org/10.3390/ijms21218272
Huang L-T, Tsai C-L, Huang S-H, Chang M-M, Chang W-T, Cheng L-H, Cheng H-C. Depleting RhoA/Stress Fiber-Organized Fibronectin Matrices on Tumor Cells Non-Autonomously Aggravates Fibroblast-Driven Tumor Cell Growth. International Journal of Molecular Sciences. 2020; 21(21):8272. https://doi.org/10.3390/ijms21218272
Chicago/Turabian StyleHuang, Li-Tzu, Chen-Lung Tsai, Shin-Huei Huang, Ming-Min Chang, Wen-Tsan Chang, Li-Hsin Cheng, and Hung-Chi Cheng. 2020. "Depleting RhoA/Stress Fiber-Organized Fibronectin Matrices on Tumor Cells Non-Autonomously Aggravates Fibroblast-Driven Tumor Cell Growth" International Journal of Molecular Sciences 21, no. 21: 8272. https://doi.org/10.3390/ijms21218272
APA StyleHuang, L.-T., Tsai, C.-L., Huang, S.-H., Chang, M.-M., Chang, W.-T., Cheng, L.-H., & Cheng, H.-C. (2020). Depleting RhoA/Stress Fiber-Organized Fibronectin Matrices on Tumor Cells Non-Autonomously Aggravates Fibroblast-Driven Tumor Cell Growth. International Journal of Molecular Sciences, 21(21), 8272. https://doi.org/10.3390/ijms21218272