COX-2 Is Downregulated in Human Stenotic Aortic Valves and Its Inhibition Promotes Dystrophic Calcification

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. COX-2 Expression in AVICs Is Decreased in Calcific Human Aortic Valves
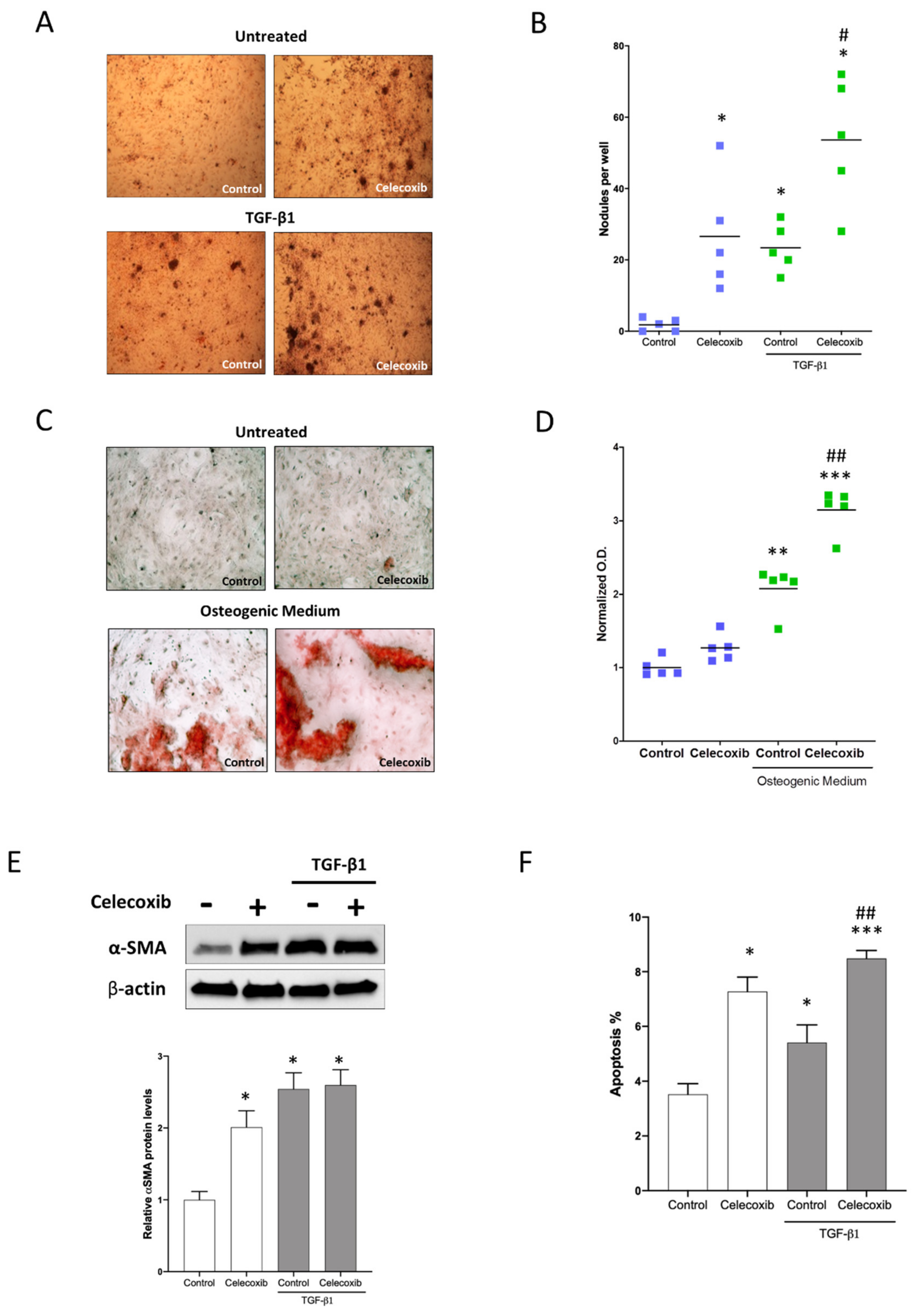
2.2. Effect of Celecoxib in Nodule Formation of Human Aortic Interstitial Cells
3. Discussion
4. Materials and Methods
4.1. Clinical Data
4.2. Gene Expression
4.3. AVIC Isolation and Cell Culture
4.4. Histology and Immunohistochemistry
4.5. Western Blots
4.6. Nodule Formation Assay
4.7. Apoptosis
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AS | aortic stenosis |
| AVIC | aortic valve interstitial cell |
| CAVD | calcific aortic valve disease |
| CN | calcific nodule |
| COX-2 | cyclooxygenase-2 |
| SMA | smooth muscle actin |
| TGF | transforming growth factor |
References
- Ngo, D.T.; Sverdlov, A.L.; Horowitz, J.D. Prevention of aortic valve stenosis: A realistic therapeutic target? Pharmacol. Ther. 2012, 135, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Nadir, M.A.; Wei, L.; Elder, D.H.; Libianto, R.; Lim, T.K.; Pauriah, M.; Pringle, S.D.; Doney, A.D.; Choy, A.M.; Struthers, A.D.; et al. Impact of renin-angiotensin system blockade therapy on outcome in aortic stenosis. J. Am. Coll. Cardiol. 2011, 58, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Thiago, L.; Tsuji, S.R.; Nyong, J.; Puga, M.E.; Gois, A.F.; Macedo, C.R.; Valente, O.; Atallah, Á. Statins for aortic valve stenosis. Cochrane Database Syst. Rev. 2016, 9, CD009571. [Google Scholar]
- Goody, P.R.; Hosen, M.R.; Christmann, D.; Niepmann, S.T.; Zietzer, A.; Adam, M.; Bönner, F.; Zimmer, S.; Nickenig, G.; Jansen, F. Aortic Valve Stenosis: From Basic Mechanisms to Novel Therapeutic Targets. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 885–900. [Google Scholar] [CrossRef]
- Cho, K.I.; Sakuma, I.; Sohn, I.S.; Jo, S.H.; Koh, K.K. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis 2018, 277, 60–65. [Google Scholar] [CrossRef]
- Sega, F.V.D.; Aquila, G.; Fortini, F.; Vaccarezza, M.; Secchiero, P.; Rizzo, P.; Campo, G. Context-dependent function of ROS in the vascular endothelium: The role of the Notch pathway and shear stress. Biofactors 2017, 43, 475–485. [Google Scholar] [CrossRef]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Peña-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, D.T.; Stafford, I.; Kelly, D.J.; Sverdlov, A.L.; Wuttke, R.D.; Weedon, H.; Nightingale, A.K.; Rosenkranz, A.C.; Smith, M.D.; Chirkov, Y.Y.; et al. Vitamin D(2) supplementation induces the development of aortic stenosis in rabbits: Interactions with endothelial function and thioredoxin-interacting protein. Eur. J. Pharmacol. 2008, 590, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Kopytek, M.; Kolasa-Trela, R.; Ząbczyk, M.; Undas, A.; Natorska, J. NETosis is associated with the severity of aortic stenosis: Links with inflammation. Int. J. Cardiol. 2019, 286, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Chirkov, Y.Y.; Holmes, A.S.; Willoughby, S.R.; Stewart, S.; Horowitz, J.D. Association of aortic stenosis with platelet hyperaggregability and impaired responsiveness to nitric oxide. Am. J. Cardiol. 2002, 90, 551–554. [Google Scholar] [CrossRef]
- Ngo, D.T.; Heresztyn, T.; Mishra, K.; Marwick, T.H.; Horowitz, J.D. Aortic stenosis is associated with elevated plasma levels of asymmetric dimethylarginine (ADMA). Nitric Oxide 2007, 16, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.A.; Hua, X.; Mishra, K.; Murphy, G.A.; Rosenkranz, A.C.; Horowitz, J.D. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur. J. Pharmacol. 2009, 602, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Andersson, D.C.; Caidahl, K.; Eriksson, M.J.; Eriksson, P.; Franco-Cereceda, A.; Hansson, G.K.; Bäck, M. Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation 2011, 123, 1316–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirrig, E.E.; Gomez, M.V.; Hinton, R.B.; Yutzey, K.E. COX2 inhibition reduces aortic valve calcification in vivo. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 938–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowler, M.A.; Raddatz, M.A.; Johnson, C.L.; Lindman, B.R.; Merryman, W.D. Celecoxib Is Associated With Dystrophic Calcification and Aortic Valve Stenosis. JACC Basic Transl. Sci. 2019, 4, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Rabkin-Aikawa, E.; Farber, M.; Aikawa, M.; Schoen, F.J. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J. Heart. Valve. Dis. 2004, 13, 841–847. [Google Scholar]
- Li, F.; Song, R.; Ao, L.; Reece, T.B.; Cleveland, J.C.; Dong, N.; Fullerton, D.A.; Meng, X. ADAMTS5 Deficiency in Calcified Aortic Valves Is Associated With Elevated Pro-Osteogenic Activity in Valvular Interstitial Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1339–1351. [Google Scholar] [CrossRef] [Green Version]
- Benton, J.A.; Kern, H.B.; Leinwand, L.A.; Mariner, P.D.; Anseth, K.S. Statins block calcific nodule formation of valvular interstitial cells by inhibiting alpha-smooth muscle actin expression. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1950–1957. [Google Scholar] [CrossRef] [Green Version]
- Mohler, E.R.; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone formation and inflammation in cardiac valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Sorli, C.H.; Zhang, H.J.; Armstrong, M.B.; Rajotte, R.V.; Maclouf, J.; Robertson, R.P. Basal expression of cyclooxygenase-2 and nuclear factor-interleukin 6 are dominant and coordinately regulated by interleukin 1 in the pancreatic islet. Proc. Natl. Acad. Sci. USA 1998, 95, 1788–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadji, F.; Boulanger, M.C.; Guay, S.P.; Gaudreault, N.; Amellah, S.; Mkannez, G.; Bouchareb, R.; Marchand, J.T.; Nsaibia, M.J.; Guauque-Olarte, S.; et al. Altered DNA Methylation of Long Noncoding RNA H19 in Calcific Aortic Valve Disease Promotes Mineralization by Silencing NOTCH1. Circulation 2016, 134, 1848–1862. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.; Murphy, B.; Woolington, S.; Ghafoory, S.; Chen, S.; Robison, T.; Ahamed, J. Inactivation of platelet-derived TGF-β1 attenuates aortic stenosis progression in a robust murine model. Blood Adv. 2019, 3, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, A.; della Mora, A.; Contin, M.; Gregoraci, G.; Tubaro, F.; Marchini, M.; Ortolani, F. Survival-Related Autophagic Activity Versus Procalcific Death in Cultured Aortic Valve Interstitial Cells Treated With Critical Normophosphatemic-Like Phosphate Concentrations. J. Histochem. Cytochem. 2017, 65, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, A.; Allegri, L.; Baldan, F.; Contin, M.; Battistella, C.; Damante, G.; Marchini, M.; Ortolani, F. Critical Involvement of Calcium-Dependent Cytosolic Phospholipase A2α in Aortic Valve Interstitial Cell Calcification. Int. J. Mol. Sci. 2020, 21, 6398. [Google Scholar] [CrossRef]
- Jian, B.; Narula, N.; Li, Q.Y.; Mohler, E.R.; Levy, R.J. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann. Thorac. Surg. 2003, 75, 457–465. [Google Scholar] [CrossRef]
- Wettlaufer, S.H.; Scott, J.P.; McEachin, R.C.; Peters-Golden, M.; Huang, S.K. Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am. J. Respir. Cell Mol. Biol. 2016, 54, 114–127. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, K.A.; Donnelly, M.P.; Gee, T.W.; Aibo, M.A.I.; Byers, S.; Butcher, J.T. Induction of aortic valve calcification by celecoxib and its COX-2 independent derivatives is glucocorticoid-dependent. Cardiovasc. Pathol. 2020, 46, 107194. [Google Scholar] [CrossRef]
- Lewis, G.D.; Campbell, W.B.; Johnson, A.R. Inhibition of prostaglandin synthesis by glucocorticoids in human endothelial cells. Endocrinology 1986, 119, 62–69. [Google Scholar] [CrossRef]
- Delaney, J.A.; Lehmann, N.; Jöckel, K.H.; Elmariah, S.; Psaty, B.M.; Mahabadi, A.A.; Budoff, M.; Kronmal, R.A.; Nasir, K.; O’Brien, K.D.; et al. Associations between aspirin and other non-steroidal anti-inflammatory drugs and aortic valve or coronary artery calcification: The Multi-Ethnic Study of Atherosclerosis and the Heinz Nixdorf Recall Study. Atherosclerosis 2013, 229, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurgers, L.J.; Cranenburg, E.C.; Vermeer, C. Matrix Gla-protein: The calcification inhibitor in need of vitamin K. Thromb. Haemost. 2008, 100, 593–603. [Google Scholar] [PubMed]
- Davin, L.; Dulgheru, R.; Lancellotti, P. ACE inhibitors in aortic stenosis: No fear just hope. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 828–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Lüscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef] [Green Version]
- Ali, O.A.; Chapman, M.; Nguyen, T.H.; Chirkov, Y.Y.; Heresztyn, T.; Mundisugih, J.; Horowitz, J.D. Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart 2014, 100, 800–805. [Google Scholar] [CrossRef]
- Hudzik, B.; Wilczek, K.; Gasior, M. Heyde syndrome: Gastrointestinal bleeding and aortic stenosis. CMAJ 2016, 188, 135–138. [Google Scholar] [CrossRef] [Green Version]
- Gould, R.A.; Butcher, J.T. Isolation of valvular endothelial cells. J. Vis. Exp. 2010, 46, e2158. [Google Scholar] [CrossRef] [Green Version]
- Kostina, A.; Shishkova, A.; Ignatieva, E.; Irtyuga, O.; Bogdanova, M.; Levchuk, K.; Golovkin, A.; Zhiduleva, E.; Uspenskiy, V.; Moiseeva, O.; et al. Different Notch signaling in cells from calcified bicuspid and tricuspid aortic valves. J. Mol. Cell. Cardiol. 2018, 114, 211–219. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Mastrocola, R.; Aquila, G.; Fortini, F.; Fornelli, C.; Zotta, A.; Cento, A.S.; Perrelli, A.; Boda, E.; Pannuti, A.; et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 4930. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| CAVD (n = 61) | Control (n = 22) | p | |
|---|---|---|---|
| Age (years) | 78 (73–81) | 71 (64–78) | 0.002 |
| Male sex | 33 (54) | 15 (68) | 0.251 |
| BMI (kg/m2) | 27 (24–29) | 28 (25–30) | 0.274 |
| Bicuspid | 4 (6.5) | 2 (11.0) | 0.653 |
| Medical history | |||
| Hypertension | 54 (89) | 22 (100) | 0.181 |
| Dyslipidemia | 41 (67) | 14 (64) | 0.761 |
| Diabetes | 13 (21) | 1 (5) | 0.099 |
Smoke
| 32 (53) 24 (39) 5 (8) | 7 (32) 9 (41) 6 (27) | 0.052 |
| Severe CAD | 9 (14) | 2 (9) | 0.719 |
| Prior PCI | 6 (10) | 2 (9) | 1.000 |
| Prior stroke | 2 (3) | 1 (5) | 1.000 |
| PAD | 11 (18) | 4 (18) | 1.000 |
| AF | 5 (8) | 8 (36) | 0.004 |
| COPD | 9 (15) | 3 (14) | 1.000 |
| CDK | 32 (63) | 9 (43) | 0.694 |
| Drug therapy | |||
| Warfarin | 6 (10) | 10 (45) | <0.01 |
| ASA | 30 (49) | 12 (54) | 0.804 |
| Laboratory data | |||
| Hemoglobin (g/dL) | 13 (12–14) | 14 (12–15) | 0.537 |
| Platelets (×103/mm3) | 207 (177–246) | 176 (155–191) | 0.069 |
| Glucose (mg/dL) | 106 (93–120) | 97 (90–113) | 0.407 |
| eGFR (mL/min) | 59 (49–72) | 61 (44–74) | 0.771 |
| LDL (mg/dL) | 94 (70–119) | 99 (77–119) | 0.513 |
| Albumin (g/dL) | 4.3 (4.1–4.5) | 4.3 (4.1–4.5) | 0.707 |
| Echocardiography data | |||
| LVEDVi (mL/m2) | 51 (42–62) | 76 (50–98) | 0.004 |
| LV ESVi (mL/m2) | 19 (16–26) | 28 (15–43) | 0.048 |
| LV EF (%) | 61 (51–68) | 65 (50–69) | 0.613 |
| LVMI (g/m2) | 118 (102–139) | 135 (91–187) | 0.410 |
| AV MPG (mmHg) | 47 (41–57) | 8 (7–25) | <0.001 |
| AV peak velocity (m/s) | 4.4 (4.1–4.8) | 2.0 (1.7–3.2) | <0.001 |
| Significant AR | 6 (10) | 17 (77) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieceli Dalla Sega, F.; Fortini, F.; Cimaglia, P.; Marracino, L.; Tonet, E.; Antonucci, A.; Moscarelli, M.; Campo, G.; Rizzo, P.; Ferrari, R. COX-2 Is Downregulated in Human Stenotic Aortic Valves and Its Inhibition Promotes Dystrophic Calcification. Int. J. Mol. Sci. 2020, 21, 8917. https://doi.org/10.3390/ijms21238917
Vieceli Dalla Sega F, Fortini F, Cimaglia P, Marracino L, Tonet E, Antonucci A, Moscarelli M, Campo G, Rizzo P, Ferrari R. COX-2 Is Downregulated in Human Stenotic Aortic Valves and Its Inhibition Promotes Dystrophic Calcification. International Journal of Molecular Sciences. 2020; 21(23):8917. https://doi.org/10.3390/ijms21238917
Chicago/Turabian StyleVieceli Dalla Sega, Francesco, Francesca Fortini, Paolo Cimaglia, Luisa Marracino, Elisabetta Tonet, Antonio Antonucci, Marco Moscarelli, Gianluca Campo, Paola Rizzo, and Roberto Ferrari. 2020. "COX-2 Is Downregulated in Human Stenotic Aortic Valves and Its Inhibition Promotes Dystrophic Calcification" International Journal of Molecular Sciences 21, no. 23: 8917. https://doi.org/10.3390/ijms21238917