Mechanisms of Colorectal Cancer Prevention by Aspirin—A Literature Review and Perspective on the Role of COX-Dependent and -Independent Pathways

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pharmacological Effects of Aspirin
3. Mechanisms Proposed for Aspirin’s Chemopreventive Effects
3.1. Inhibition of COX Enzymes: The Platelet Hypothesis
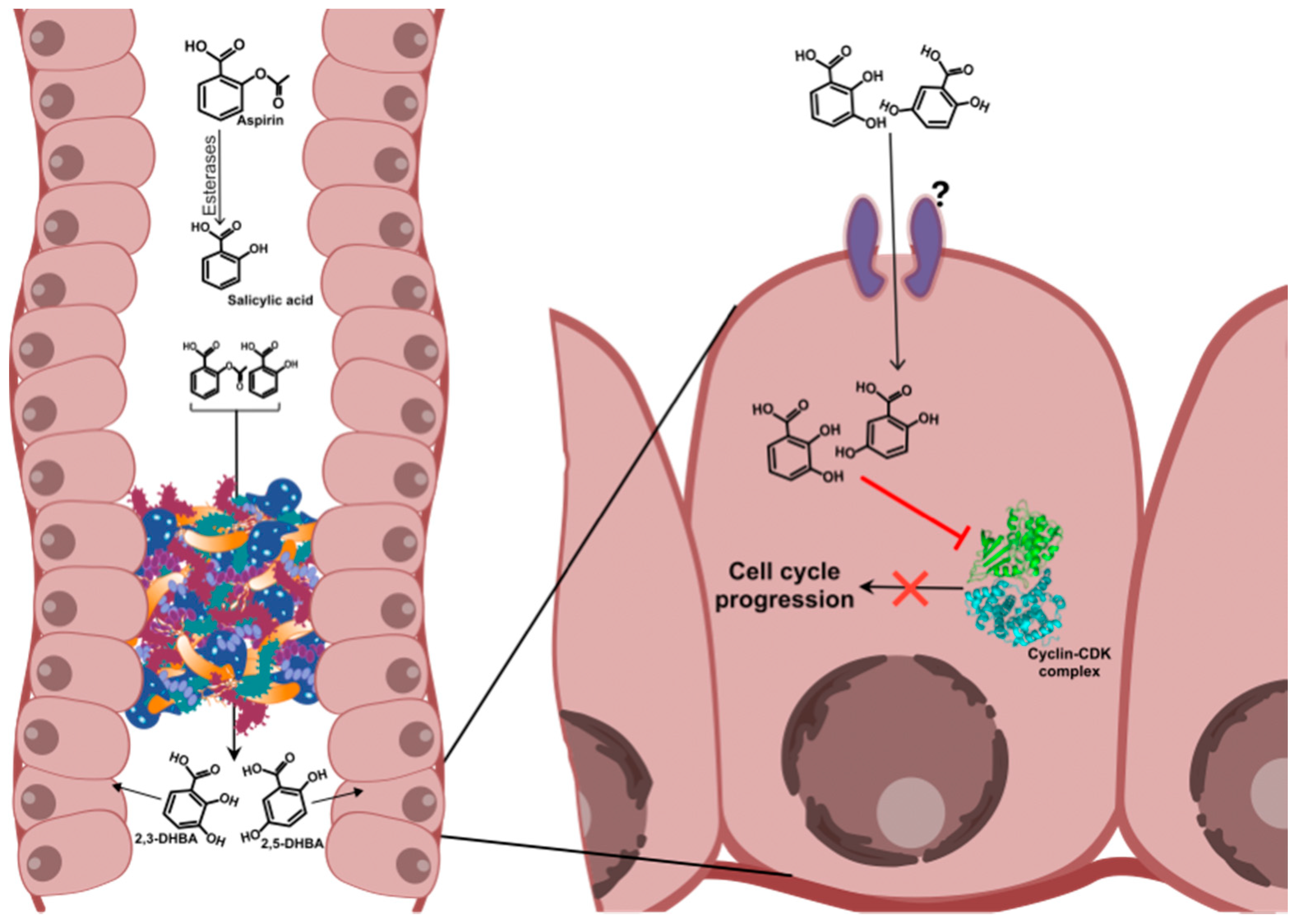
3.2. Inhibition of Cyclin Dependent Kinases (CDKs) by Aspirin Metabolites: The Metabolite Hypothesis
3.3. Inhibition of Nuclear Factor (NF)-κB Signalling
3.4. Activation of AMP-Kinase and Inhibition of mTOR Signaling
3.5. Inhibition of Wnt Signalling and β-Catenin Phosphorylation
3.6. Downregulation of c-Myc, Cyclin A2 and CDK2
3.7. Induction of Polyamine Catabolism
3.8. Induction of DNA Mismatch Repair Proteins
3.9. Acetylation of p53, Glucose-6-Phosphate Dehydrogenase and Other Proteins
3.10. Other Mechanisms
4. Potential Role of the Gut Microbiota in Aspirin’s Effect against CRC
5. Perspective and Future Studies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2,3-DHBA | 2,3-dihydroxybenzoic acid |
| 2,5-DHBA | 2,5-dihydroxybenzoic acid |
| 5-ASA | 5-aminosalicylic acid |
| AMPK | Adenosine monophosphate activated protein kinase |
| AMP | Adenosine MonoPhosphate |
| APC | Adenomatous polyposis coli |
| ATP | Adenosine TriPhosphate |
| CDK | Cyclin Dependent Kinase |
| COX | Cyclooxygenase |
| CYP450 | Cytochrome P450 |
| CRC | Colorectal cancer |
| EMT | Epithelial-mesenchymal transition |
| FGF | Fibroblast growth factor |
| G6PD | glucose-6-phosphate dehydrogenase |
| GI | Gastrointestinal |
| HBA | Hydroxybenzoic acid |
| IKK | IκB Kinase |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor -κB |
| NSAID | Non-steroidal anti-inflammatory drug |
| ODC | Ornithine decarboxylase |
| PDGF | Platelet-derived growth factor |
| PG | Prostaglandin |
| PGI2 | Prostacyclin |
| SSAT | Spermidine/spermine N1-acetyltransferase |
| TXA-2 | Thromboxane A2 |
| USPSTF | United States Preventive Services Task Force |
References
- Bardhan, K.; Liu, K. Epigenetics and colorectal cancer pathogenesis. Cancers 2013, 5, 676–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M. Colon Cancer: A Clinician’s Perspective in 2019. Gastroenterol. Res. 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.T.; Giovannucci, E.L.; Meyerhardt, J.A.; Schernhammer, E.S.; Curhan, G.C.; Fuchs, C.S. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. JAMA 2005, 294, 914–923. [Google Scholar] [CrossRef]
- Thun, M.J.; Jacobs, E.J.; Patrono, C. The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol. 2012, 9, 259–267. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Kune, G.A.; Kune, S.; Watson, L.F. Colorectal cancer risk, chronic illnesses, operations, and medications: Case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar] [CrossRef] [Green Version]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Cole, B.F.; Logan, R.F.; Halabi, S.; Benamouzig, R.; Sandler, R.S.; Grainge, M.J.; Chaussade, S.; Baron, J.A. Aspirin for the chemoprevention of colorectal adenomas: Meta-analysis of the randomized trials. J. Natl. Cancer Inst. 2009, 101, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burn, J.; Bishop, D.T.; Mecklin, J.P.; Macrae, F.; Moslein, G.; Olschwang, S.; Bisgaard, M.L.; Ramesar, R.; Eccles, D.; Maher, E.R.; et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N. Engl. J. Med. 2008, 359, 2567–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Cuzick, J.; Thorat, M.A.; Bosetti, C.; Brown, P.H.; Burn, J.; Cook, N.R.; Ford, L.G.; Jacobs, E.J.; Jankowski, J.A.; La Vecchia, C.; et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann. Oncol. 2015, 26, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Tacconelli, S.; Sostres, C.; Ricciotti, E.; Patrignani, P. Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals 2012, 5, 1346–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpitz, B.; Bomstein, Y.; Kariv, N.; Shalev, M.; Buklan, G.; Bernheim, J. Chemopreventive effect of aspirin on growth of aberrant crypt foci in rats. Int. J. Colorectal Dis. 1998, 13, 169–172. [Google Scholar] [CrossRef]
- Rohwer, N.; Kuhl, A.A.; Ostermann, A.I.; Hartung, N.M.; Schebb, N.H.; Zopf, D.; McDonald, F.M.; Weylandt, K.H. Effects of chronic low-dose aspirin treatment on tumor prevention in three mouse models of intestinal tumorigenesis. Cancer Med. 2020, 9, 2535–2550. [Google Scholar] [CrossRef]
- Tian, Y.; Ye, Y.; Gao, W.; Chen, H.; Song, T.; Wang, D.; Mao, X.; Ren, C. Aspirin promotes apoptosis in a murine model of colorectal cancer by mechanisms involving downregulation of IL-6-STAT3 signaling pathway. Int. J. Colorectal Dis. 2011, 26, 13–22. [Google Scholar] [CrossRef]
- Reddy, B.S.; Rao, C.V.; Rivenson, A.; Kelloff, G. Inhibitory effect of aspirin on azoxymethane-induced colon carcinogenesis in F344 rats. Carcinogenesis 1993, 14, 1493–1497. [Google Scholar] [CrossRef]
- Garcia-Albeniz, X.; Chan, A.T. Aspirin for the prevention of colorectal cancer. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia Rodriguez, L.A.; Martin-Perez, M.; Hennekens, C.H.; Rothwell, P.M.; Lanas, A. Bleeding Risk with Long-Term Low-Dose Aspirin: A Systematic Review of Observational Studies. PLoS ONE 2016, 11, e0160046. [Google Scholar] [CrossRef] [PubMed]
- Cea Soriano, L.; Lanas, A.; Soriano-Gabarro, M.; Garcia Rodriguez, L.A. Incidence of Upper and Lower Gastrointestinal Bleeding in New Users of Low-Dose Aspirin. Clin. Gastroenterol. Hepatol. 2019, 17, 887–895 e886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibbins-Domingo, K.; U.S. Preventive Services Task Force. Aspirin Use for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: U.S. Preventive Services Task Force Recommendation StatementAspirin Use for the Primary Prevention of CVD and CRC. Ann. Intern. Med. 2016, 164, 836–845. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, C.; Santucci, C.; Gallus, S.; Martinetti, M.; La Vecchia, C. Aspirin and the risk of colorectal and other digestive tract cancers: An updated meta-analysis through 2019. Ann. Oncol. 2020, 31, 558–568. [Google Scholar] [CrossRef]
- Drew, D.A.; Chin, S.M.; Gilpin, K.K.; Parziale, M.; Pond, E.; Schuck, M.M.; Stewart, K.; Flagg, M.; Rawlings, C.A.; Backman, V.; et al. ASPirin Intervention for the REDuction of colorectal cancer risk (ASPIRED): A study protocol for a randomized controlled trial. Trials 2017, 18, 50. [Google Scholar] [CrossRef] [Green Version]
- Roy, H.K.; Turzhitsky, V.; Wali, R.; Radosevich, A.J.; Jovanovic, B.; Della’Zanna, G.; Umar, A.; Rubin, D.T.; Goldberg, M.J.; Bianchi, L.; et al. Spectral biomarkers for chemoprevention of colonic neoplasia: A placebo-controlled double-blinded trial with aspirin. Gut 2017, 66, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Nishihara, R.; Wu, K.; Wang, M.; Ogino, S.; Willett, W.C.; Spiegelman, D.; Fuchs, C.S.; Giovannucci, E.L.; Chan, A.T. Population-wide Impact of Long-term Use of Aspirin and the Risk for Cancer. JAMA Oncol. 2016, 2, 762–769. [Google Scholar] [CrossRef] [Green Version]
- Patrignani, P.; Patrono, C. Aspirin and Cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976. [Google Scholar] [CrossRef]
- Needs, C.J.; Brooks, P.M. Clinical pharmacokinetics of the salicylates. Clin. Pharmacokinet. 1985, 10, 164–177. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Phan, T.; Fang, D.; Edler, S.; Philip, J.; Li-Geng, T.; Dial, E.J. Bioavailability of aspirin in rats comparing the drug’s uptake into gastrointestinal tissue and vascular and lymphatic systems: Implications on aspirin’s chemopreventive action. J. Physiol. Pharmacol. 2016, 67, 635–642. [Google Scholar] [PubMed]
- Rowland, M.; Riegelman, S.; Harris, P.A.; Sholkoff, S.D. Absorption kinetics of aspirin in man following oral administration of an aqueous solution. J. Pharm. Sci. 1972, 61, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Maree, A.O.; Dooley, M.; Conroy, R.; Byrne, M.F.; Fitzgerald, D.J. Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke 2006, 37, 2153–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovizio, M.; Alberti, S.; Guillem-Llobat, P.; Patrignani, P. Role of platelets in inflammation and cancer: Novel therapeutic strategies. Basic Clin. Pharmacol. Toxicol. 2014, 114, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, M.; Laneuville, O.; Ji, C.; DeWitt, D.L.; Smith, W.L. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J. Biol. Chem. 1994, 269, 13207–13215. [Google Scholar] [PubMed]
- Roth, G.J.; Machuga, E.T.; Ozols, J. Isolation and covalent structure of the aspirin-modified, active-site region of prostaglandin synthetase. Biochemistry 1983, 22, 4672–4675. [Google Scholar] [CrossRef]
- DeWitt, D.L.; el-Harith, E.A.; Kraemer, S.A.; Andrews, M.J.; Yao, E.F.; Armstrong, R.L.; Smith, W.L. The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J. Biol. Chem. 1990, 265, 5192–5198. [Google Scholar]
- Vane, J.R.; Botting, R.M. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: A full in vitro analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 7563–7568. [Google Scholar] [CrossRef] [Green Version]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Chulada, P.C.; Thompson, M.B.; Mahler, J.F.; Doyle, C.M.; Gaul, B.W.; Lee, C.; Tiano, H.F.; Morham, S.G.; Smithies, O.; Langenbach, R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60, 4705–4708. [Google Scholar] [PubMed]
- Menter, D.G.; Dubois, R.N. Prostaglandins in cancer cell adhesion, migration, and invasion. Int. J. Cell Biol. 2012, 2012, 723419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaranarayanan, R.; Kumar, D.R.; Patel, J.; Bhat, G.J. Do Aspirin and Flavonoids Prevent Cancer through a Common Mechanism Involving Hydroxybenzoic Acids? The Metabolite Hypothesis. Molecules 2020, 25, 2243. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Valiveti, C.K.; Dachineni, R.; Kumar, D.R.; Lick, T.; Bhat, G.J. Aspirin metabolites 2,3DHBA and 2,5DHBA inhibit cancer cell growth: Implications in colorectal cancer prevention. Mol. Med. Rep. 2020, 21, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Dachineni, R.; Kumar, D.R.; Callegari, E.; Kesharwani, S.S.; Sankaranarayanan, R.; Seefeldt, T.; Tummala, H.; Bhat, G.J. Salicylic acid metabolites and derivatives inhibit CDK activity: Novel insights into aspirin’s chemopreventive effects against colorectal cancer. Int. J. Oncol. 2017, 51, 1661–1673. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.S.; Yoo, D.H.; Jung, I.H.; Lim, S.; Jeong, J.J.; Kim, K.A.; Bae, O.N.; Yoo, H.H.; Kim, D.H. Reduced metabolic activity of gut microbiota by antibiotics can potentiate the antithrombotic effect of aspirin. Biochem. Pharmacol. 2016, 122, 72–79. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, Y.; Wang, R.; Zhang, J. Gut Microbiota-Mediated Drug-Drug Interaction between Amoxicillin and Aspirin. Sci. Rep. 2019, 9, 16194. [Google Scholar] [CrossRef] [Green Version]
- Hutt, A.J.; Caldwell, J.; Smith, R.L. The metabolism of aspirin in man: A population study. Xenobiotica 1986, 16, 239–249. [Google Scholar] [CrossRef]
- Schiller, C.; Frohlich, C.P.; Giessmann, T.; Siegmund, W.; Monnikes, H.; Hosten, N.; Weitschies, W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef]
- Grootveld, M.; Halliwell, B. Aromatic hydroxylation as a potential measure of hydroxyl-radical formation in vivo. Identification of hydroxylated derivatives of salicylate in human body fluids. Biochem. J. 1986, 237, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dull, B.J.; Salata, K.; Van Langenhove, A.; Goldman, P. 5-Aminosalicylate: Oxidation by activated leukocytes and protection of cultured cells from oxidative damage. Biochem. Pharmacol. 1987, 36, 2467–2472. [Google Scholar] [CrossRef]
- Lopez, A.; Pouillon, L.; Beaugerie, L.; Danese, S.; Peyrin-Biroulet, L. Colorectal cancer prevention in patients with ulcerative colitis. Best Pract. Res. Clin. Gastroenterol. 2018, 32–33, 103–109. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Elmaci, I.; Cengiz, S.; Emekli-Alturfan, E.; Ozpinar, A. From epidemiology to treatment: Aspirin’s prevention of brain and breast-cancer and cardioprotection may associate with its metabolite gentisic acid. Chem. Biol. Interact. 2018, 291, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.S.; Cuevas, P.; Angulo, J.; Lopez-Navajas, P.; Canales-Mayordomo, A.; Gonzalez-Corrochano, R.; Lozano, R.M.; Valverde, S.; Jimenez-Barbero, J.; Romero, A.; et al. Gentisic acid, a compound associated with plant defense and a metabolite of aspirin, heads a new class of in vivo fibroblast growth factor inhibitors. J. Biol. Chem. 2010, 285, 11714–11729. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Kraus, V.; Pahl, A.; Brune, K. Salicylate metabolites inhibit cyclooxygenase-2-dependent prostaglandin E(2) synthesis in murine macrophages. Biochem. Biophys. Res. Commun. 2000, 274, 197–202. [Google Scholar] [CrossRef]
- Borges, R.S.; Castle, S.L. The antioxidant properties of salicylate derivatives: A possible new mechanism of anti-inflammatory activity. Bioorg. Med. Chem. Lett. 2015, 25, 4808–4811. [Google Scholar] [CrossRef]
- Snigireva, A.V.; Morenkov, O.S.; Skarga, Y.Y.; Lisov, A.V.; Lisova, Z.A.; Leontievsky, A.A.; Zhmurina, M.A.; Petrenko, V.S.; Vrublevskaya, V.V. A 2,5-Dihydroxybenzoic Acid-Gelatin Conjugate Inhibits the Basal and Hsp90-Stimulated Migration and Invasion of Tumor Cells. J. Funct. Biomater. 2020, 11, 39. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Elmaci, I.; Ozpinar, A. Gentisic Acid, a Quinonoid Aspirin Metabolite in Cancer Prevention and Treatment. New Horizons in Management of Brain Tumors and Systemic Cancers. J. Cancer Res. Oncobiol. 2018, 1, 109. [Google Scholar] [CrossRef] [Green Version]
- Russell, W.R.; Scobbie, L.; Labat, A.; Duthie, G.G. Selective bio-availability of phenolic acids from Scottish strawberries. Mol. Nutr. Food Res. 2009, 53 (Suppl. S1), S85–S91. [Google Scholar] [CrossRef]
- Tomás-Barberán, F.A.; Clifford, M.N. Dietary hydroxybenzoic acid derivatives—Nature, occurrence and dietary burden. J. Sci. Food Agric. 2000, 80, 1024–1032. [Google Scholar] [CrossRef]
- Williamson, G.; Clifford, M.N. Colonic metabolites of berry polyphenols: The missing link to biological activity? Br. J. Nutr. 2010, 104 (Suppl. S3), S48–S66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Dandapani, M.; Hardie, D.G. AMPK: Mediating the metabolic effects of salicylate-based drugs? Trends Endocrinol. Metab. 2013, 24, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515.e1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene 2006, 25, 6447–6456. [Google Scholar] [CrossRef] [Green Version]
- Ai, G.; Dachineni, R.; Muley, P.; Tummala, H.; Bhat, G.J. Aspirin and salicylic acid decrease c-Myc expression in cancer cells: A potential role in chemoprevention. Tumour Biol. 2016, 37, 1727–1738. [Google Scholar] [CrossRef]
- Law, B.K.; Waltner-Law, M.E.; Entingh, A.J.; Chytil, A.; Aakre, M.E.; Norgaard, P.; Moses, H.L. Salicylate-induced growth arrest is associated with inhibition of p70s6k and down-regulation of c-myc, cyclin D1, cyclin A, and proliferating cell nuclear antigen. J. Biol. Chem. 2000, 275, 38261–38267. [Google Scholar] [CrossRef] [Green Version]
- Dachineni, R.; Ai, G.; Kumar, D.R.; Sadhu, S.S.; Tummala, H.; Bhat, G.J. Cyclin A2 and CDK2 as Novel Targets of Aspirin and Salicylic Acid: A Potential Role in Cancer Prevention. Mol. Cancer Res. 2016, 14, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babbar, N.; Gerner, E.W.; Casero, R.A., Jr. Induction of spermidine/spermine N1-acetyltransferase (SSAT) by aspirin in Caco-2 colon cancer cells. Biochem. J. 2006, 394, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.K.H.; Martin, A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol. Med. 2016, 22, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Chang, D.K.; Ricciardiello, L.; Gasche, C.; Boland, C.R. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin. Cancer Res. 2003, 9, 383–390. [Google Scholar]
- Ai, G.; Dachineni, R.; Kumar, D.R.; Marimuthu, S.; Alfonso, L.F.; Bhat, G.J. Aspirin acetylates wild type and mutant p53 in colon cancer cells: Identification of aspirin acetylated sites on recombinant p53. Tumour Biol. 2016, 37, 6007–6016. [Google Scholar] [CrossRef]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Alfonso, L.F.; Srivenugopal, K.S.; Arumugam, T.V.; Abbruscato, T.J.; Weidanz, J.A.; Bhat, G.J. Aspirin inhibits camptothecin-induced p21CIP1 levels and potentiates apoptosis in human breast cancer cells. Int. J. Oncol. 2009, 34, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Marimuthu, S.; Chivukula, R.S.; Alfonso, L.F.; Moridani, M.; Hagen, F.K.; Bhat, G.J. Aspirin acetylates multiple cellular proteins in HCT-116 colon cancer cells: Identification of novel targets. Int. J. Oncol. 2011, 39, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Ai, G.; Dachineni, R.; Kumar, D.R.; Alfonso, L.F.; Marimuthu, S.; Bhat, G.J. Aspirin inhibits glucose6phosphate dehydrogenase activity in HCT 116 cells through acetylation: Identification of aspirin-acetylated sites. Mol. Med. Rep. 2016, 14, 1726–1732. [Google Scholar] [CrossRef] [Green Version]
- Dore, M.P.; Davoli, A.; Longo, N.; Marras, G.; Pes, G.M. Glucose-6-phosphate dehydrogenase deficiency and risk of colorectal cancer in Northern Sardinia: A retrospective observational study. Medicine 2016, 95, e5254. [Google Scholar] [CrossRef] [PubMed]
- Bateman, L.A.; Zaro, B.W.; Miller, S.M.; Pratt, M.R. An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J. Am. Chem. Soc. 2013, 135, 14568–14573. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Cole, C.; Scullion, P.; Wilkie, R.; Westwood, N.J.; Stark, L.A.; Hay, R.T. A Proteomic Approach to Analyze the Aspirin-mediated Lysine Acetylome. Mol. Cell. Proteom. 2017, 16, 310–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhu, F.; Boardman, L.A.; Wang, L.; Oi, N.; Liu, K.; Li, X.; Fu, Y.; Limburg, P.J.; Bode, A.M.; et al. Aspirin Prevents Colorectal Cancer by Normalizing EGFR Expression. EBioMedicine 2015, 2, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Huang, C.; Brown, R.E.; Ma, W.Y. Inhibition of activator protein 1 activity and neoplastic transformation by aspirin. J. Biol. Chem. 1997, 272, 9962–9970. [Google Scholar] [CrossRef] [Green Version]
- Pathi, S.; Jutooru, I.; Chadalapaka, G.; Nair, V.; Lee, S.O.; Safe, S. Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (Sp) transcription factors. PLoS ONE 2012, 7, e48208. [Google Scholar] [CrossRef] [Green Version]
- Bellosillo, B.; Pique, M.; Barragan, M.; Castano, E.; Villamor, N.; Colomer, D.; Montserrat, E.; Pons, G.; Gil, J. Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells. Blood 1998, 92, 1406–1414. [Google Scholar] [CrossRef]
- Vad, N.M.; Yount, G.; Moridani, M.Y. Biochemical mechanism of acetylsalicylic acid (Aspirin) selective toxicity toward melanoma cell lines. Melanoma Res. 2008, 18, 386–399. [Google Scholar] [CrossRef]
- Patrignani, P.; Sacco, A.; Sostres, C.; Bruno, A.; Dovizio, M.; Piazuelo, E.; Di Francesco, L.; Contursi, A.; Zucchelli, M.; Schiavone, S.; et al. Low-Dose Aspirin Acetylates Cyclooxygenase-1 in Human Colorectal Mucosa: Implications for the Chemoprevention of Colorectal Cancer. Clin. Pharmacol. Ther. 2017, 102, 52–61. [Google Scholar] [CrossRef]
- Bigler, J.; Whitton, J.; Lampe, J.W.; Fosdick, L.; Bostick, R.M.; Potter, J.D. CYP2C9 and UGT1A6 genotypes modulate the protective effect of aspirin on colon adenoma risk. Cancer Res. 2001, 61, 3566–3569. [Google Scholar]
- Ozdal, T.; Sela, D.A.; Xiao, J.; Boyacioglu, D.; Chen, F.; Capanoglu, E. The Reciprocal Interactions between Polyphenols and Gut Microbiota and Effects on Bioaccessibility. Nutrients 2016, 8, 78. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef] [Green Version]
- Prizment, A.E.; Staley, C.; Onyeaghala, G.C.; Vivek, S.; Thyagarajan, B.; Straka, R.J.; Demmer, R.T.; Knights, D.; Meyer, K.A.; Shaukat, A.; et al. Randomised clinical study: Oral aspirin 325 mg daily vs. placebo alters gut microbial composition and bacterial taxa associated with colorectal cancer risk. Aliment. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Zhao, R.; Coker, O.O.; Wu, J.; Zhou, Y.; Zhao, L.; Nakatsu, G.; Bian, X.; Wei, H.; Chan, A.W.H.; Sung, J.J.Y.; et al. Aspirin Reduces Colorectal Tumor Development in Mice and Gut Microbes Reduce its Bioavailability and Chemopreventive Effects. Gastroenterology 2020, 159, 969–983 e964. [Google Scholar] [CrossRef]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [Green Version]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996, 52, 237–245. [Google Scholar] [CrossRef]
- Hsi, L.C.; Baek, S.J.; Eling, T.E. Lack of cyclooxygenase-2 activity in HT-29 human colorectal carcinoma cells. Exp. Cell. Res. 2000, 256, 563–570. [Google Scholar] [CrossRef]
- Birkenkamp-Demtroder, K.; Olesen, S.H.; Sorensen, F.B.; Laurberg, S.; Laiho, P.; Aaltonen, L.A.; Orntoft, T.F. Differential gene expression in colon cancer of the caecum versus the sigmoid and rectosigmoid. Gut 2005, 54, 374–384. [Google Scholar] [CrossRef] [Green Version]
- Ornelas, A.; Zacharias-Millward, N.; Menter, D.G.; Davis, J.S.; Lichtenberger, L.; Hawke, D.; Hawk, E.; Vilar, E.; Bhattacharya, P.; Millward, S. Beyond COX-1: The effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metastasis Rev. 2017, 36, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Vijayan, K.V. Are Platelets the Primary Target of Aspirin’s Remarkable Anticancer Activity? Cancer Res. 2019, 79, 3820–3823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenberger, L.M.; Fang, D.; Bick, R.J.; Poindexter, B.J.; Phan, T.; Bergeron, A.L.; Pradhan, S.; Dial, E.J.; Vijayan, K.V. Unlocking Aspirin’s Chemopreventive Activity: Role of Irreversibly Inhibiting Platelet Cyclooxygenase-1. Cancer Prev. Res. 2017, 10, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, A.K.; FitzGerald, G.A. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N. Engl. J. Med. 1984, 311, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Sagar, K.A.; Smyth, M.R. A comparative bioavailability study of different aspirin formulations using on-line multidimensional chromatography. J. Pharm. Biomed. Anal. 1999, 21, 383–392. [Google Scholar] [CrossRef]
- De Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The pharmacokinetics of anthocyanins and their metabolites in humans. Br. J. Pharmacol. 2014, 171, 3268–3282. [Google Scholar] [CrossRef] [Green Version]
- Hanske, L.; Engst, W.; Loh, G.; Sczesny, S.; Blaut, M.; Braune, A. Contribution of gut bacteria to the metabolism of cyanidin 3-glucoside in human microbiota-associated rats. Br. J. Nutr. 2013, 109, 1433–1441. [Google Scholar] [CrossRef]
- Thilakarathna, W.; Rupasinghe, H.P.V. Microbial metabolites of proanthocyanidins reduce chemical carcinogen-induced DNA damage in human lung epithelial and fetal hepatic cells in vitro. Food Chem. Toxicol. 2019, 125, 479–493. [Google Scholar] [CrossRef]
- Pace, E.; Jiang, Y.; Clemens, A.; Crossman, T.; Rupasinghe, H.P.V. Impact of Thermal Degradation of Cyanidin-3-O-Glucoside of Haskap Berry on Cytotoxicity of Hepatocellular Carcinoma HepG2 and Breast Cancer MDA-MB-231 Cells. Antioxidants 2018, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, R.; Valiveti, C.K.; Kumar, D.R.; Van Slambrouck, S.; Kesharwani, S.S.; Seefeldt, T.; Scaria, J.; Tummala, H.; Bhat, G.J. The Flavonoid Metabolite 2,4,6-Trihydroxybenzoic Acid Is a CDK Inhibitor and an Anti-Proliferative Agent: A Potential Role in Cancer Prevention. Cancers 2019, 11, 427. [Google Scholar] [CrossRef] [Green Version]
- Jenner, A.M.; Rafter, J.; Halliwell, B. Human fecal water content of phenolics: The extent of colonic exposure to aromatic compounds. Free Radic. Biol. Med. 2005, 38, 763–772. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sankaranarayanan, R.; Kumar, D.R.; Altinoz, M.A.; Bhat, G.J. Mechanisms of Colorectal Cancer Prevention by Aspirin—A Literature Review and Perspective on the Role of COX-Dependent and -Independent Pathways. Int. J. Mol. Sci. 2020, 21, 9018. https://doi.org/10.3390/ijms21239018
Sankaranarayanan R, Kumar DR, Altinoz MA, Bhat GJ. Mechanisms of Colorectal Cancer Prevention by Aspirin—A Literature Review and Perspective on the Role of COX-Dependent and -Independent Pathways. International Journal of Molecular Sciences. 2020; 21(23):9018. https://doi.org/10.3390/ijms21239018
Chicago/Turabian StyleSankaranarayanan, Ranjini, D. Ramesh Kumar, Meric A. Altinoz, and G. Jayarama Bhat. 2020. "Mechanisms of Colorectal Cancer Prevention by Aspirin—A Literature Review and Perspective on the Role of COX-Dependent and -Independent Pathways" International Journal of Molecular Sciences 21, no. 23: 9018. https://doi.org/10.3390/ijms21239018
APA StyleSankaranarayanan, R., Kumar, D. R., Altinoz, M. A., & Bhat, G. J. (2020). Mechanisms of Colorectal Cancer Prevention by Aspirin—A Literature Review and Perspective on the Role of COX-Dependent and -Independent Pathways. International Journal of Molecular Sciences, 21(23), 9018. https://doi.org/10.3390/ijms21239018