DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands



Abstract
1. Introduction

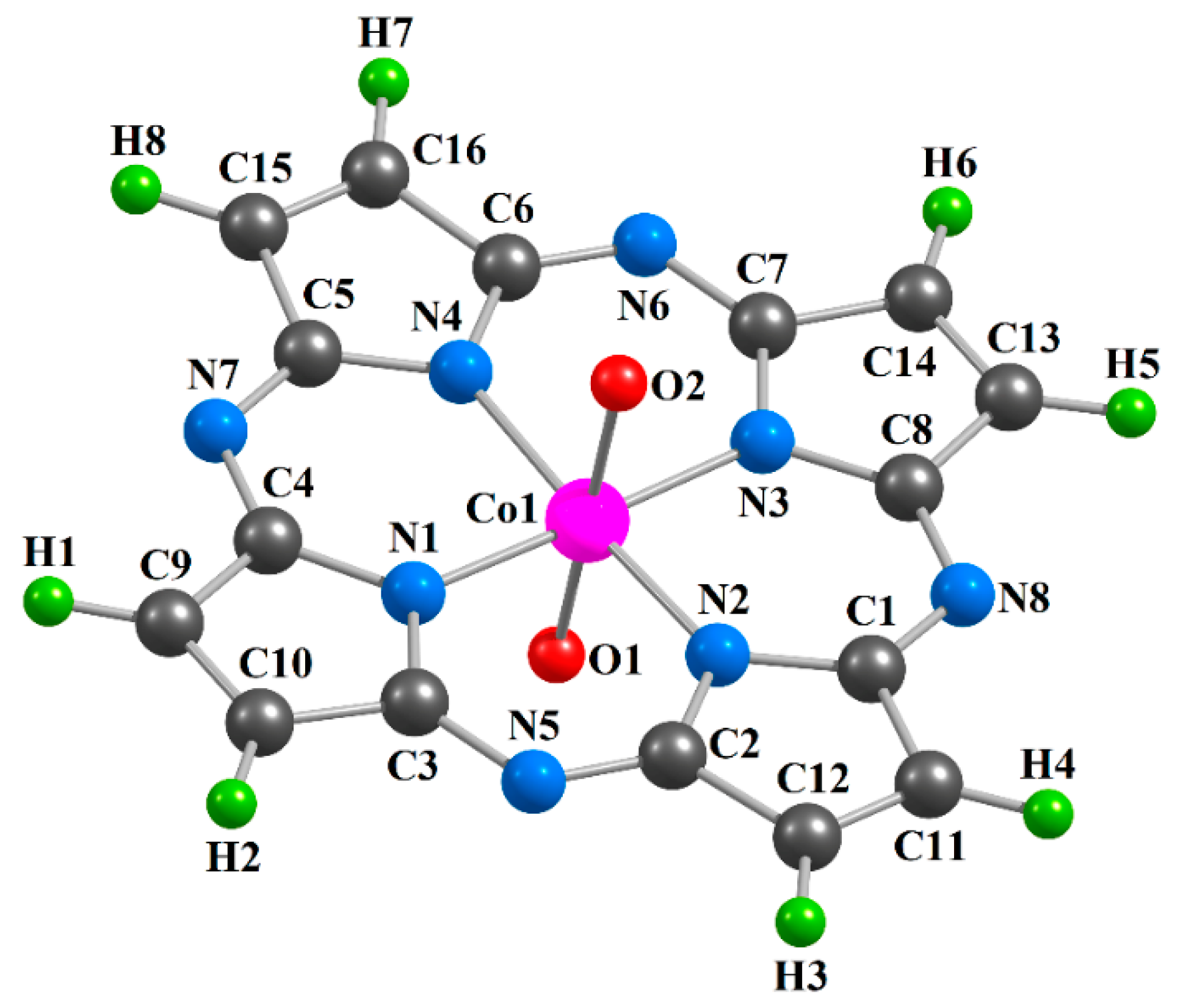
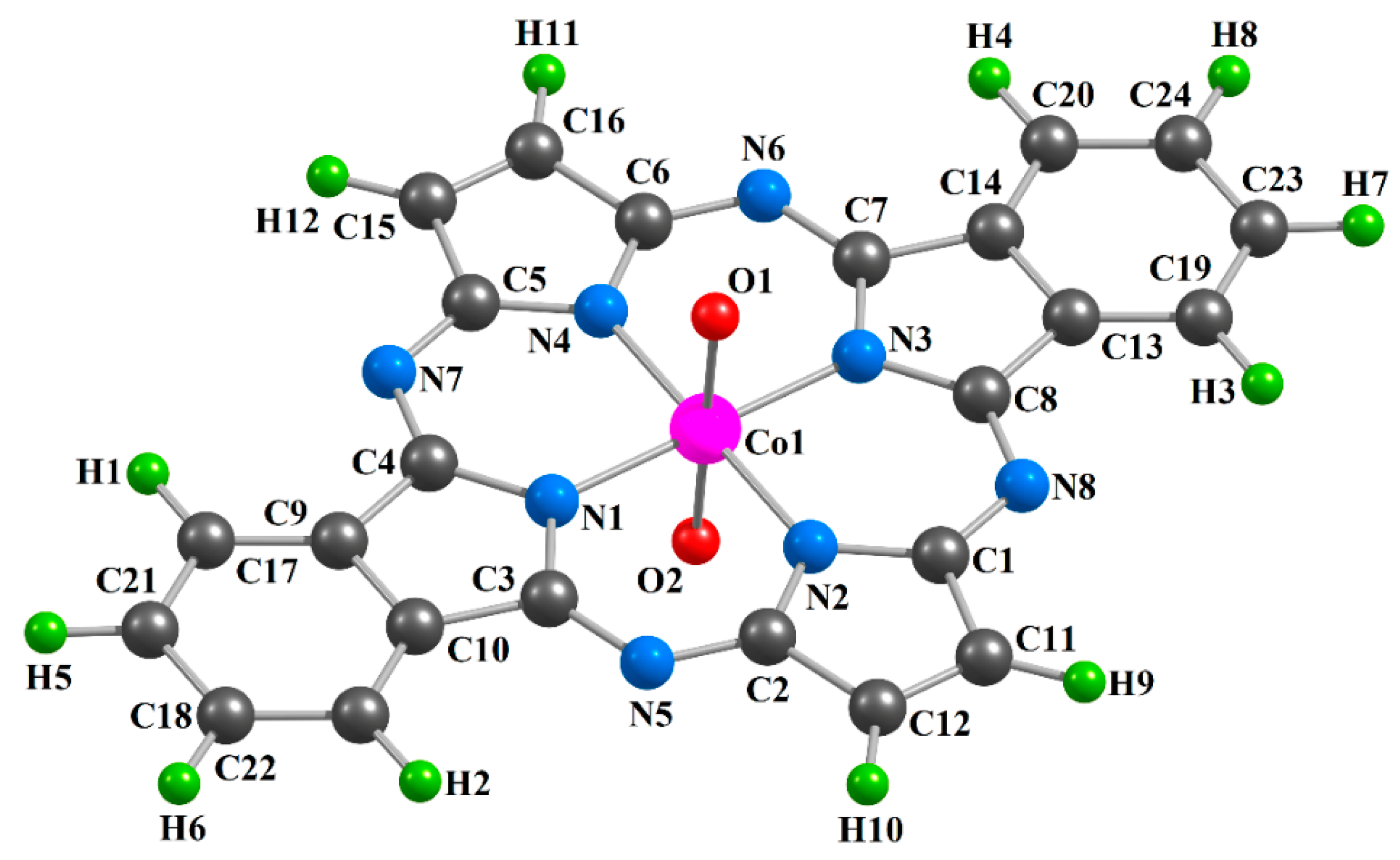
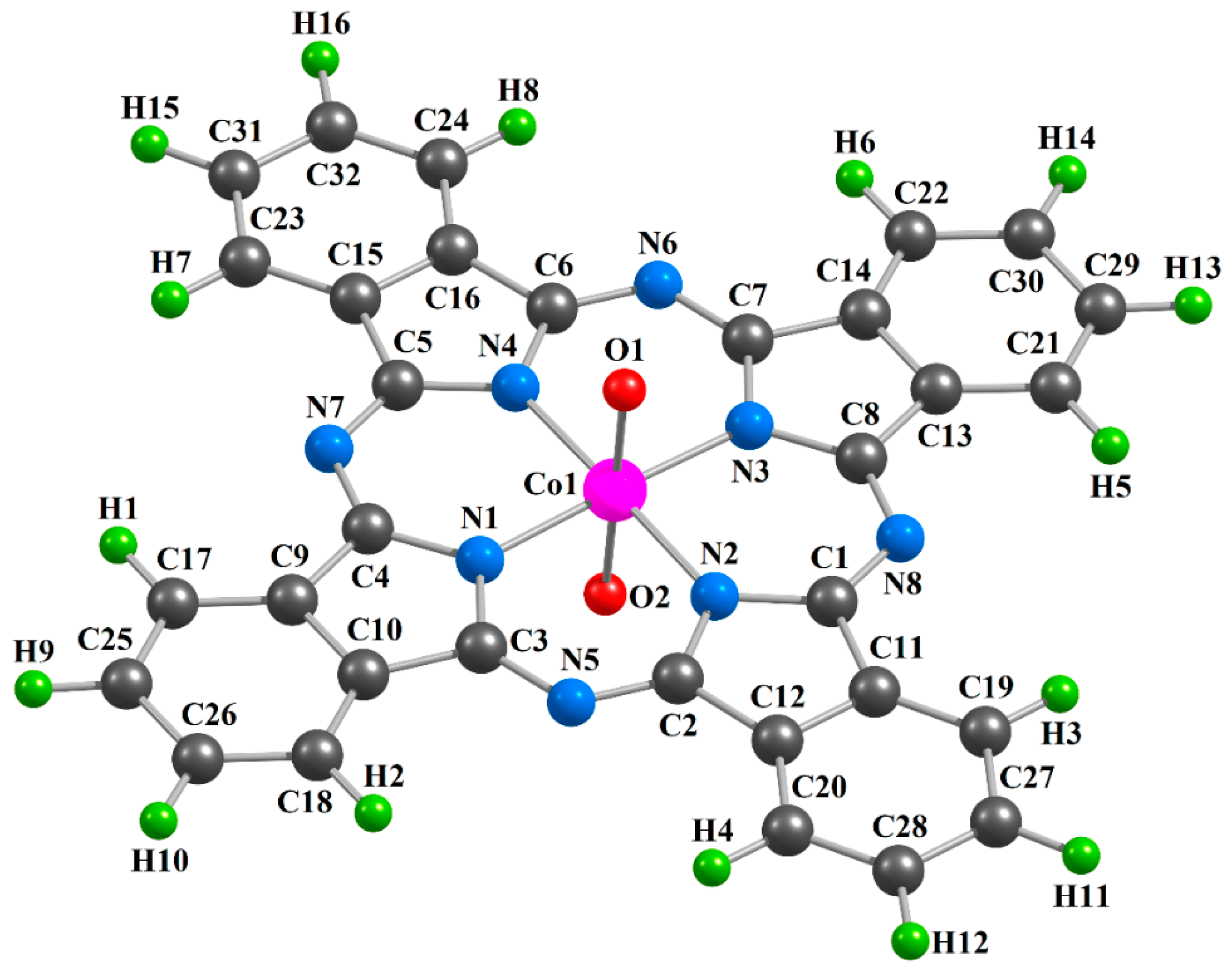
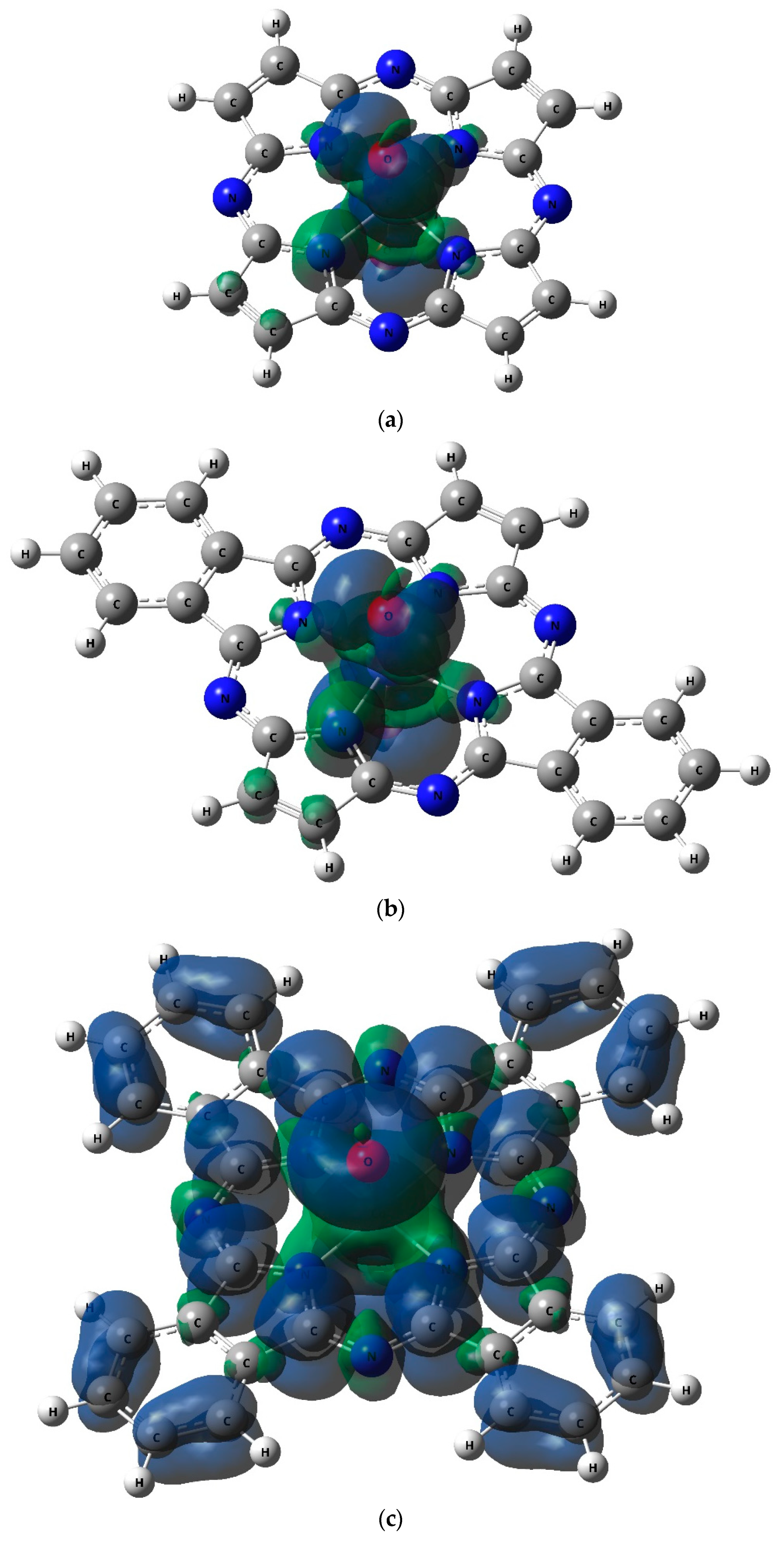
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weller, M.; Overton, T.; Rourke, J.; Armstrong, F. Inorganic Chemistry, 6th ed.; OUP: Oxford, UK, 2014. [Google Scholar]
- Kiselev, Y.M.; Tretiyakov, Y.D. The Problem of Oxidation State Stabilization and Some Regularities of a Periodic System of the Elements. Russ. Chem. Rev. 1999, 68, 365–379. [Google Scholar] [CrossRef]
- Riedel, S.; Kaupp, M. The highest oxidation states of the transition metal elements. Coord. Chem. Rev. 2009, 253, 606–624. [Google Scholar] [CrossRef]
- Quail, J.W.; Rivett, G.A. Complex Fluorides of Tetravalent Cobalt. Can. J. Chem. 1972, 50, 2447–2450. [Google Scholar] [CrossRef]
- Allen, G.C.; Warren, K.D. Electronic spectrum of the hexafluorocobaltate(IV) anion. Inorg. Chem. 1969, 8, 1902–1904. [Google Scholar] [CrossRef]
- Brendel, C.; Klemm, W. Weitere Versuche zur Darstellung von Kaliumcobaltat (V). Z. Anorg. Allgem. Chem. 1963, 320, 59–63. [Google Scholar] [CrossRef]
- Nicholls, D. The Chemistry of Iron, Cobalt and Nickel: Comprehensive Inorganic Chemistry; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Byrne, E.K.; Theopold, K.H. Redox chemistry of tetrakis(1-norbornyl)cobalt. Synthesis and characterization of a cobalt(V) alkyl and self-exchange rate of a Co(III)/Co(IV) couple. J. Am. Chem. Soc. 1987, 109, 1282–1283. [Google Scholar] [CrossRef]
- Brookhart, M.; Grant, B.E.; Lenges, C.P.; Prosenc, M.H.; White, P.S. High Oxidation State Organocobalt Complexes: Synthesis and Characterization of Dihydridodisilyl Cobalt(V) Species. Angew. Chem. Int. Ed. 2000, 39, 1676–1679. [Google Scholar] [CrossRef]
- Ingleson, M.; Fan, H.; Pink, M.; Tomaszewski, J.; Caulton, K.G. Three-Coordinate Co(I) Provides Access to Unsaturated Dihydrido-Co(III) and Seven-Coordinate Co(V). J. Am. Chem. Soc. 2006, 128, 1804–1805. [Google Scholar] [CrossRef]
- Kopelev, N.; Kulikov, L.; Peréliev, Y. Mössbauer effect study of Cs2Fe(57Co)O4. Hyperfine Interact. 1994, 90, 377–381. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. About of Possibility of Existence of Zn(IV) Oxidation State in Heteroligand Complexes with Porphyrazine, trans-Di[benzo]porphyrazine, Phthalocyanine, and Oxo Ligands: Quantum-Chemical Review. Comm. Inorg. Chem. 2020, 40, 107–115. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Copper (IV) Stabilization in Macrocyclic Complexes with 3,7,11,15-Tetraazaporphine, Its Di[benzo]- or Tetra[benzo] Derivatives and Oxide Anion: Quantum-Chemical Research. Materials 2020, 13, 3162. [Google Scholar] [CrossRef] [PubMed]
- Kasuda, K.; Tsutsui, M. Some new developments in the chemistry of metallophthalocyanines. Coord. Chem. Rev. 1980, 32, 67–95. [Google Scholar] [CrossRef]
- Thomas, A.L. Phthalocyanines. Research & Applications; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Sliva, W.; Mianovska, B. Metalloporphyrin arrays. Transit. Met. Chem. 2000, 25, 491–504. [Google Scholar] [CrossRef]
- Mamardashvili, G.M.; Mamardashvili, N.Z.; Koifman, O.I. Self-assembling systems based on porphyrins. Russ. Chem. Rev. 2008, 77, 59–75. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Novakova, V.; Zimcik, P.; Stuzhin, P.A. tetrapyrazinoporphyrazines and their metal derivatives. Part I: Synthesis and basic structural information. Coord. Chem. Rev. 2016, 309, 107–179. [Google Scholar] [CrossRef]
- Lomova, T.N. Axial Coordinated Metal Porphyrins in Science and Practice; URSS: Moscow, Russia, 2018. [Google Scholar]
- Khelevina, O.G.; Malyasova, A.S. 40 years with porphyrazines. J. Porphyr. Phthalocyanines 2019, 23, 1251–1264. [Google Scholar] [CrossRef]
- Calborean, A.; Morari, C.; Maldivi, P. Combined molecular and periodic DFT analysis of the adsorption of co macrocycles on graphene. J. Comput. Chem. 2018, 39, 130–138. [Google Scholar] [CrossRef]
- Patel, A.; Abozeid, S.M.; Cullen, P.A.; Morrow, J.R. Co(II) Macrocyclic Complexes Appended with Fluorophores as paraCEST and cellCEST Agents. Inorg. Chem. 2020, 59, 16531–16544. [Google Scholar] [CrossRef]
- Reynolds, P.A.; Figgis, B.N.; Kucharski, E.S.; Mason, S.A. Neutron diffraction at 115 K to 1.09 Å-1 from cobalt phthalocyanine. Acta Cryst. B 1991, 47, 899–904. [Google Scholar] [CrossRef]
- Ballirano, P.; Caminiti, R.; Ercolani, C.; Maras, A.; Orru, M.A. X-ray Powder Diffraction Structure Reinvestigation of the α and β Forms of Cobalt Phthalocyanine and Kinetics of the α→β Phase Transition. J. Am. Chem. Soc. 1998, 120, 12798–12807. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Density Functional Theory Modeling of Molecular Structures of Heteroligand 3d M(IV) Complexes with Porphyrazine and Oxo Anion. Russ. J. Inorg. Chem. 2020, 65, 1019–1024. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Quantum-chemical calculation of molecular structures of (5656)macrotetracyclic 3d-metal complexes “self-assembled” in quaternary systems M(II) ion-ethanedithioamide- formaldehyde- ammonia by the density functional theory method. Russ. J. Inorg. Chem. 2014, 59, 218–223. [Google Scholar] [CrossRef]
- Mikhailov, O.V. Synthesis of 3d-element metalmacrocyclic chelates into polypeptide biopolymer medium and their molecular structures. Inorg. Chim. Acta 2013, 394, 664–684. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Structure of (5656)macrotetracyclic chelates in the ternary systems M(II)-ethanedithioamide-acetone (M = Mn, Fe, Co, Ni, Cu, Zn) according to DFT calculations. Russ. J. Inorg. Chem. 2013, 58, 1073–1078. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Novel oxidation state—Zinc(III) in chelate with 3,7,11,15-tetraazaporphine and one fluorine ligand: Quantum-chemical modeling. J. Porphyr. Phthalocyanines 2019, 23, 685–689. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. M(VI) Oxidation State Stabilization in Iron, Cobalt and Nickel Heteroligand Metal Chelates Containing 3,7,11,15-Tetraazaporphine and Two Axial Oxo Ligands: Quantum-Chemical Simulation. Int. J. Mol. Sci. 2020, 21, 1494. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schaefer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Paulsen, H.; Duelund, L.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Free Energy of Spin-Crossover Complexes Calculated with Density Functional Methods. Inorg. Chem. 2001, 40, 2201–2203. [Google Scholar] [CrossRef] [PubMed]
- Swart, M.; Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Validation of Exchange−Correlation Functionals for Spin States of Iron Complexes. J. Phys. Chem. A 2004, 108, 5479–5483. [Google Scholar] [CrossRef]
- Swart, M.; Ehlers, A.W.; Lammertsma, K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. [Google Scholar] [CrossRef]
- Swart, M. Metal–ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V. Gaussian 09; Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | [CoL1(O)2] | [CoL2(O)2] | [CoL3(O)2] | |||
|---|---|---|---|---|---|---|
| Structural parameter | Calculated by DFT | Calculated by DFT | Calculated by DFT | |||
| OPBE/TZVP | B3PW91/TZVP | OPBE/TZVP | B3PW91/TZVP | OPBE/TZVP | B3PW91/TZVP | |
| Co–N bond lengths in chelate node MN4, pm | ||||||
| Co1N1 | 192.2 | 192.2 | 195.2 | 193.2 | 193.6 | 194.0 |
| Co1N2 | 193.2 | 191.5 | 194.8 | 192.4 | 193.6 | 195.3 |
| Co1N3 | 194.2 | 192.2 | 195.2 | 193.2 | 193.6 | 194.0 |
| Co1N4 | 193.2 | 191.5 | 192.8 | 192.4 | 193.6 | 195.3 |
| Co–O bond lengths, pm | ||||||
| Co1O1 | 168.9 | 173.1 | 168.7 | 174.5 | 175.4 | 171.5 |
| Co1O2 | 168.9 | 176.7 | 168.7 | 174.5 | 175.4 | 179.0 |
| C–N bond lengths in 6-numbered chelate rings, pm | ||||||
| N1C3 | 136.4 | 135.4 | 136.7 | 135.7 | 136.2 | 136.8 |
| N1C4 | 136.4 | 135.4 | 136.6 | 135.7 | 136.2 | 136.8 |
| N2C1 | 136.3 | 135.7 | 136.7 | 135.1 | 136.2 | 136.3 |
| N2C2 | 136.2 | 135.7 | 136.7 | 135.1 | 136.2 | 136.3 |
| N3C7 | 136.7 | 135.4 | 136.6 | 135.7 | 136.2 | 136.8 |
| N3C8 | 136.7 | 135.4 | 136.7 | 135.7 | 136.2 | 136.8 |
| N4C5 | 136.2 | 135.7 | 136.4 | 135.1 | 136.2 | 136.3 |
| N4C6 | 136.3 | 135.7 | 136.4 | 135.1 | 136.2 | 136.3 |
| N5C2 | 132.4 | 131.3 | 132.3 | 131.8 | 132.0 | 131.3 |
| N5C3 | 132.4 | 132.7 | 131.9 | 131.8 | 132.0 | 131.3 |
| N6C6 | 132.4 | 131.3 | 132.4 | 131.8 | 132.0 | 131.3 |
| N6C7 | 132.3 | 132.7 | 131.9 | 131.8 | 132.0 | 131.3 |
| N7C4 | 132.4 | 132.7 | 131.9 | 131.8 | 132.0 | 131.3 |
| N7C5 | 132.4 | 131.3 | 132.4 | 131.8 | 132.0 | 131.3 |
| N8C1 | 132.4 | 131.3 | 132.3 | 131.8 | 132.0 | 131.3 |
| N8C8 | 132.3 | 132.7 | 131.9 | 131.8 | 132.0 | 131.3 |
| C–C bond lengths in 5-numbered chelate ring (N1C4C9C10C3), pm | ||||||
| C4C9 | 145.2 | 146.2 | 145.8 | 146.4 | 146.4 | 145.0 |
| C9C10 | 135.7 | 134.4 | 140.2 | 139.4 | 139.9 | 139.7 |
| C10C3 | 145.2 | 146.2 | 145.8 | 146.4 | 146.4 | 145.0 |
| Bond angles in chelate node CoN4, deg | ||||||
| (N1Co1N2) | 90.1 | 90.0 | 89.9 | 90.0 | 90.0 | 90.0 |
| (N2Co1N3) | 89.9 | 89.9 | 89.9 | 90.0 | 90.0 | 90.0 |
| (N3Co1N4) | 89.9 | 90.0 | 90.1 | 90.0 | 90.0 | 90.0 |
| (N4Co1N1) | 90.1 | 89.9 | 90.1 | 90.0 | 90.0 | 90.0 |
| Bond angles sum (BAS), deg | 360.0 | 359.8 | 360.0 | 360.0 | 360.0 | 360.0 |
| Non-bond angles between N atoms in N4 grouping, deg | ||||||
| (N1N2N3) | 90.0 | 90.2 | 90.2 | 90.2 | 90.0 | 89.6 |
| (N2N3N4) | 89.8 | 89.7 | 89.6 | 89.8 | 90.0 | 90.4 |
| (N3N4N1) | 90.0 | 90.2 | 90.6 | 90.2 | 90.0 | 89.6 |
| (N4N1N2) | 90.2 | 89.7 | 89.6 | 89.8 | 90.0 | 90.4 |
| Non-bond angles sum (NBAS), deg | 360.0 | 359.8 | 360.0 | 360.0 | 360.0 | 360.0 |
| Bond angles in 6-numbered chelate ring (Co1N1C4N7C5N4), deg | ||||||
| (Co1N1C4) | 126.1 | 126.3 | 125.3 | 125.6 | 125.8 | 125.5 |
| (N1C4N7) | 128.3 | 127.9 | 128.4 | 128.4 | 128.5 | 128.3 |
| (C4N7C5) | 121.4 | 121.1 | 121.7 | 121.7 | 121.4 | 122.6 |
| (N7C5N4) | 128.1 | 128.4 | 128.3 | 128.0 | 128.5 | 128.1 |
| (C5N4Co1) | 126.1 | 126.3 | 126.2 | 126.3 | 125.8 | 125.5 |
| (N4Co1N1) | 90.0 | 89.9 | 90.1 | 90.0 | 90.0 | 90.0 |
| Bond angles sum (BAS61), deg | 720.0 | 719.9 | 720.0 | 720.0 | 720.0 | 720.0 |
| Bond angles in 6-numbered chelate ring (Co1N4C6N6C7N3), deg | ||||||
| (Co1N4C6) | 126.1 | 126.3 | 126.2 | 126.3 | 125.8 | 125.6 |
| (N4C6N6) | 128.5 | 128.4 | 128.3 | 128.0 | 128.5 | 128.0 |
| (C6N6C7) | 121.4 | 121.1 | 121.7 | 121.7 | 121.4 | 122.6 |
| (N6C7N3) | 128.2 | 127.9 | 128.4 | 128.4 | 128.5 | 128.2 |
| (C7N3Co1) | 125.9 | 126.3 | 125.3 | 125.6 | 125.8 | 125.6 |
| (N3Co1N4) | 89.9 | 89.9 | 90.1 | 90.0 | 90.0 | 90.0 |
| Bond angles sum (BAS62), deg | 720.0 | 719.9 | 720.0 | 720.0 | 720.0 | 720.0 |
| Bond angles in 5-numbered ring (C3N1C4C9C10), deg | ||||||
| (C3N1C4) | 107.8 | 107.3 | 109.3 | 108.8 | 108.4 | 108.9 |
| (N1C4C9) | 109.1 | 109.7 | 108.9 | 109.5 | 109.7 | 109.0 |
| (C4C9C10) | 107.0 | 106.6 | 106.4 | 106.1 | 106.1 | 106.6 |
| (C9C10C3) | 107.0 | 106.6 | 106.5 | 106.1 | 106.1 | 106.6 |
| (C10C3N1) | 109.1 | 109.7 | 108.9 | 109.5 | 109.7 | 108.9 |
| Bond angles sum (BAS51), deg | 540.0 | 539.9 | 540.0 | 540.0 | 540.0 | 540.0 |
| Bond angles in 5-numbered ring (C1N2C2C12C11), deg | ||||||
| (C1N2C2) | 107.8 | 107.5 | 108.0 | 107.4 | 108.4 | 108.9 |
| (N2C2C12) | 109.2 | 109.4 | 108.9 | 109.8 | 109.7 | 109.1 |
| (C2C12C11) | 106.9 | 106.8 | 107.1 | 106.5 | 106.1 | 106.4 |
| (C12C11C1) | 107.0 | 106.8 | 107.1 | 106.5 | 106.1 | 106.4 |
| (C11C1N2) | 109.1 | 109.4 | 108.9 | 109.8 | 109.7 | 109.1 |
| Bond angles sum (BAS52), deg | 540.0 | 539.9 | 540.0 | 540.0 | 540.0 | 540.0 |
| Bond angles between O, Co and N atoms, deg | ||||||
| O1Co1N1 | 94.9 | 91.1 | 90.0 | 90.0 | 90.0 | 91.5 |
| O1Co1N2 | 90.0 | 92.7 | 85.0 | 90.0 | 90.0 | 91.0 |
| O1Co1N3 | 85.1 | 91.1 | 90.0 | 90.0 | 90.0 | 91.5 |
| O1Co1N4 | 90.0 | 92.7 | 95.0 | 90.0 | 90.0 | 91.0 |
| O2Co1N1 | 94.9 | 88.9 | 90.0 | 90.0 | 90.0 | 88.5 |
| O2Co1N2 | 90.0 | 87.3 | 85.0 | 90.0 | 90.0 | 89.0 |
| O2Co1N3 | 85.1 | 88.9 | 90.0 | 90.0 | 90.0 | 88.5 |
| O2Co1N4 | 90.0 | 87.3 | 85.0 | 90.0 | 90.0 | 89.0 |
| Bond angles between Co and two O atoms, deg | ||||||
| O1Co1O2 | 170.1 | 180.0 | 169.9 | 180.0 | 180.0 | 180.0 |
| Complex | Effective Charge of Atom, in Units of Electron Charge (ē) | <S**2> | |||
|---|---|---|---|---|---|
| Co1 | N1 (N3) | N2 (N4) | O1 (O2) | ||
| [CoL1(O)2] | +0.0647 | −0.2989 (−0.2938) | −0.2905 (−0.2905) | −0.0895 (−0.0895) | 0.7609 |
| [CoL2(O)2] | +0.0859 | −0.2699 (−0.2699) | −0.2982 (−0.3065) | −0.1673 (−0.1673) | 0.7627 |
| [CoL3(O)2] | +0.0881 | −0.2669 (−02669) | −0.2669 (−02669) | −0.4071 (−0.4071) | 3.7715 |
| Complex | ΔH0f, 298, kJ/mole | S0f, 298, J/mole ∙K | ΔG0f, 298, kJ/mole |
|---|---|---|---|
| [CoL1(O)2] | 616.3 | 759.8 | 842.1 |
| [CoL2(O)2] | 567.7 | 949.8 | 814.5 |
| [CoL3(O)2] | 530.1 | 1168.9 | 789.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, O.V.; Chachkov, D.V. DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands. Int. J. Mol. Sci. 2020, 21, 9085. https://doi.org/10.3390/ijms21239085
Mikhailov OV, Chachkov DV. DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands. International Journal of Molecular Sciences. 2020; 21(23):9085. https://doi.org/10.3390/ijms21239085
Chicago/Turabian StyleMikhailov, Oleg V., and Denis V. Chachkov. 2020. "DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands" International Journal of Molecular Sciences 21, no. 23: 9085. https://doi.org/10.3390/ijms21239085
APA StyleMikhailov, O. V., & Chachkov, D. V. (2020). DFT Quantum-Chemical Modeling Molecular Structures of Cobalt Macrocyclic Complexes with Porphyrazine or Its Benzo-Derivatives and Two Oxygen Acido Ligands. International Journal of Molecular Sciences, 21(23), 9085. https://doi.org/10.3390/ijms21239085