Structural, Pro-Inflammatory and Calcium Handling Remodeling Underlies Spontaneous Onset of Paroxysmal Atrial Fibrillation in JDP2-Overexpressing Mice

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
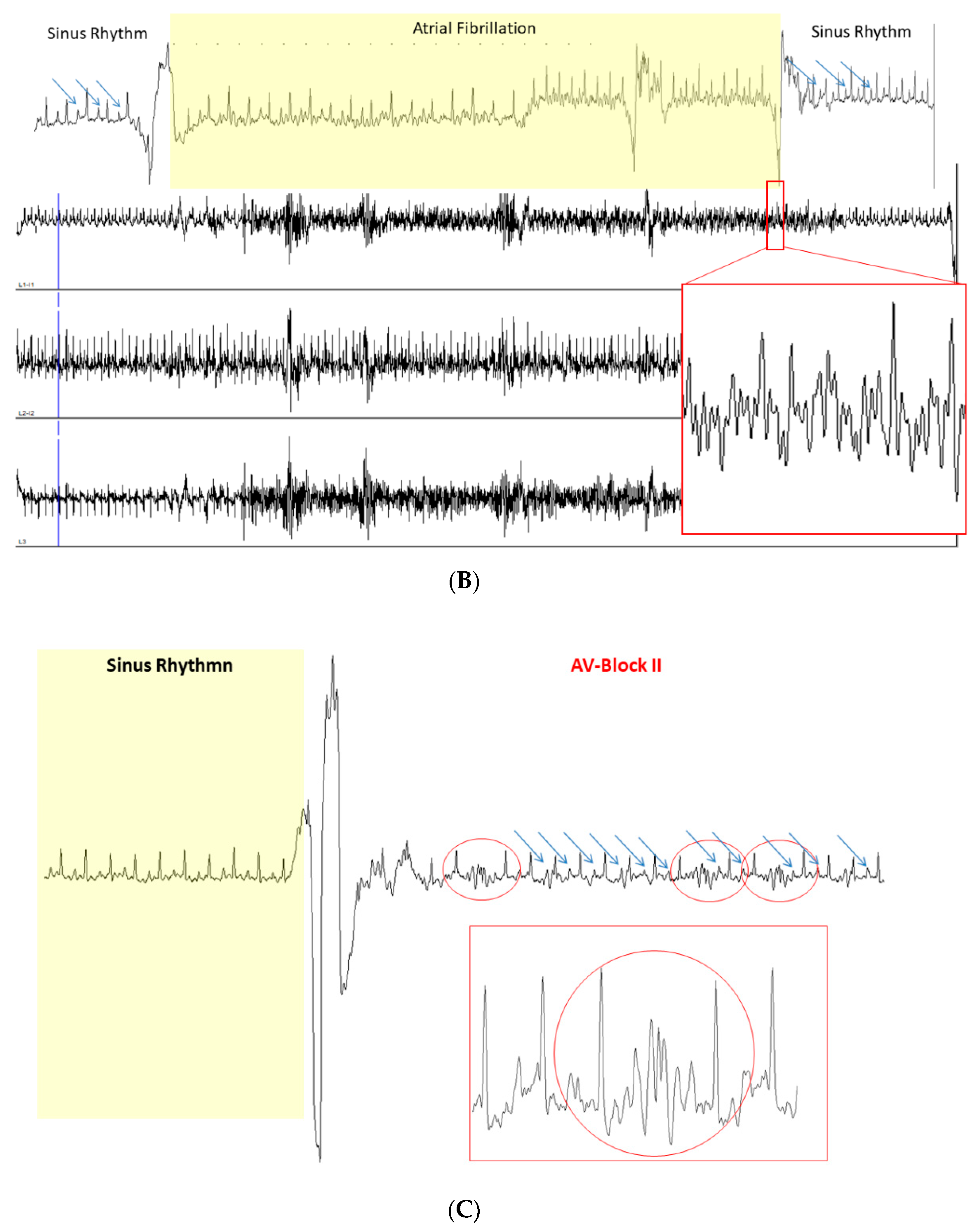
2.1. Conduction Defects and Arrhythmias in JDP2 Mice
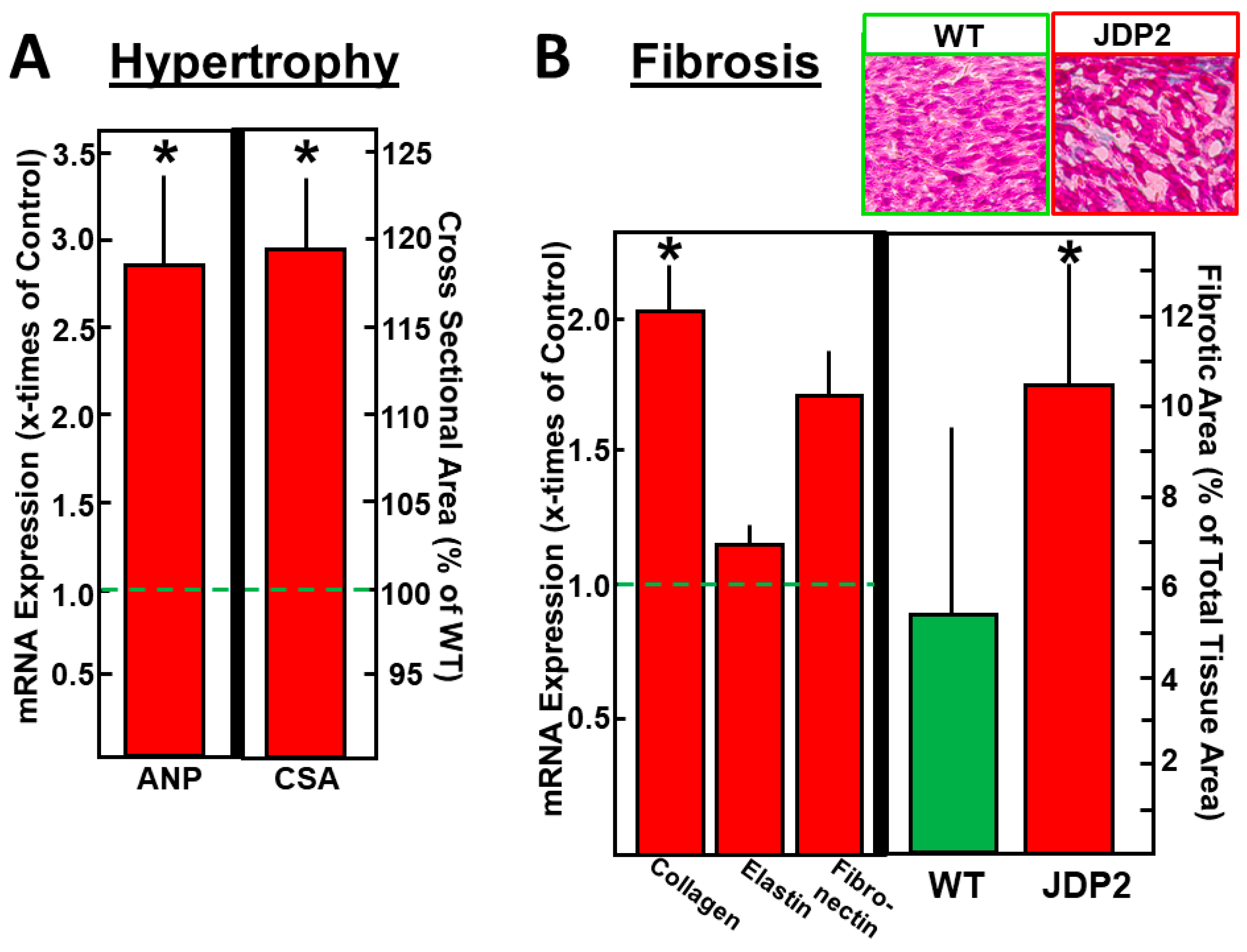
2.2. Atrial Hypertrophy
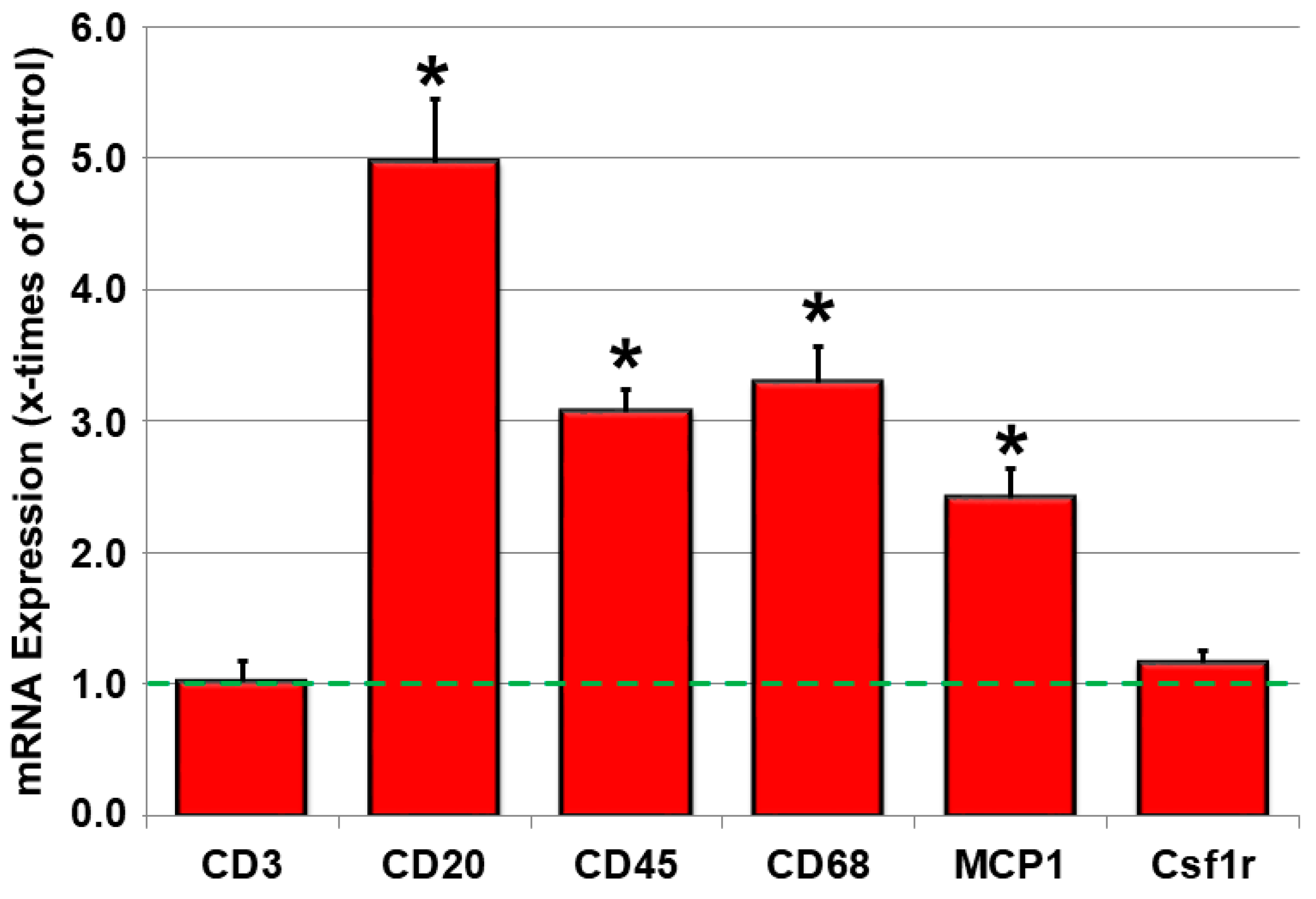
2.3. Inflammatory Parameters Are Increased in JDP2 Mice
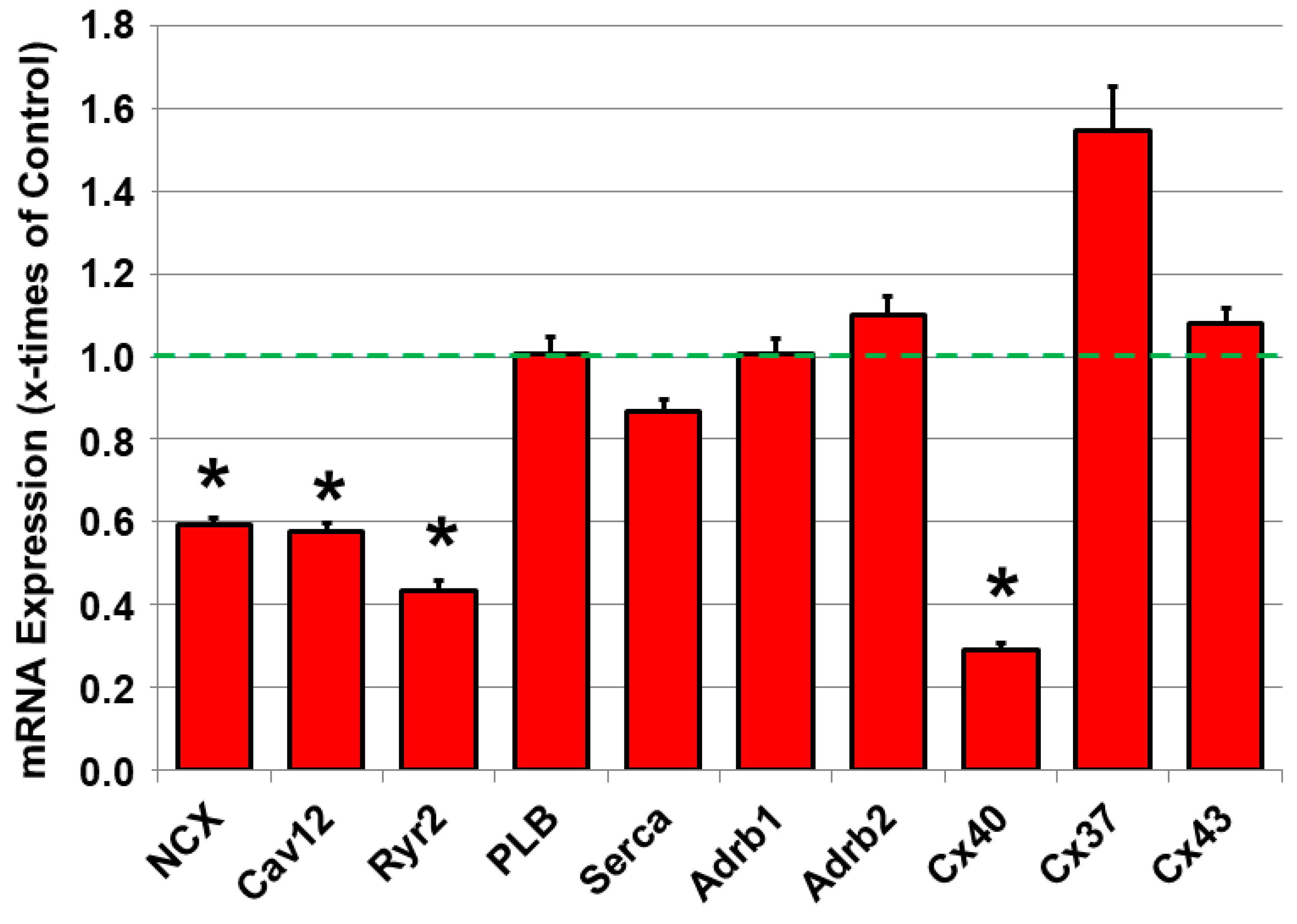
2.4. Dysregulated mRNA Expression of Calcium-Handling Proteins and Connexin40 in JDP2 Mice
2.5. Remodeling of Sarcoplasmic Reticulum Calcium Handling in JDP2 Mice
3. Discussion
4. Limitations
5. Methods
5.1. JDP2-Overexpressing Mice
5.2. Electrocardiography Recordings
5.3. Atrial Tissue Preparation
5.4. Isolation of Atrial Cardiomyocytes and Determination of Cell Size
5.5. Real-Time RT-PCR
5.6. Western Blots
5.7. Analysis of Fibrosis
5.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur. Heart. J. 2020, 1–125. [Google Scholar] [CrossRef]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Greiser, M.; Lederer, W.J.; Schotten, U. Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc. Res. 2011, 89, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Xintarakou, A.; Tzeis, S.; Psarras, S.; Asvestas, D.; Vardas, P. Atrial fibrosis as a dominant factor for the development of atrial fibrillation: Facts and gaps. EP Eur. 2020, 22, 342–351. [Google Scholar] [CrossRef]
- Hu, Y.-F.; Chen, Y.-J.; Lin, Y.-J.; Chen, S.-A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef]
- Riley, G.; Syeda, F.; Kirchhof, P.; Fabritz, L. An introduction to murine models of atrial fibrillation. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Kalfon, R.; Friedman, T.; Aronheim, A. JDP2 and ATF3—bZIP repressors in cardiac remodeling. Int. J. Cardiol. 2018, 257, 228. [Google Scholar] [CrossRef]
- Kehat, I.; Heinrich, R.; Ben-Izhak, O.; Miyazaki, H.; Gutkind, J.S.; Aronheim, A. Inhibition of basic leucine zipper transcription is a major mediator of atrial dilatation. Cardiovasc. Res. 2006, 70, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Heger, J.; Bornbaum, J.; Würfel, A.; Hill, C.; Brockmann, N.; Gáspár, R.; Pálóczi, J.; Varga, Z.V.; Sárközy, M.; Bencsik, P.; et al. JDP2 overexpression provokes cardiac dysfunction in mice. Sci. Rep. 2018, 8, 7647–7657. [Google Scholar] [CrossRef]
- Fan, D.; Yang, Z.; Liu, F.Y.; Tang, N.; Deng, W.; Tang, Q.Z. A potential therapeutic approach to cardiac remodeling: JDP2. Int. J. Cardiol. 2018, 254, 283. [Google Scholar] [CrossRef]
- Ozcan, C.; Battaglia, E.; Young, R.; Suzuki, G. LKB1 knockout mouse develops spontaneous atrial fibrillation and provides mechanistic insights into human disease process. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Magnani, J.W.; Johnson, V.M.; Sullivan, L.M.; Gorodeski, E.Z.; Schnabel, R.B.; Lubitz, S.A.; Levy, D.; Ellinor, P.T.; Benjamin, E.J. P wave duration and risk of longitudinal atrial fibrillation in persons ≥ 60 years old (from the Framingham Heart Study). Am. J. Cardiol. 2001, 107, 917–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, F.U.; Lewin, G.; Baba, H.A.; Bokník, P.; Fabritz, L.; Kirchhefer, U.; Kirchhof, P.; Loser, K.; Matus, M.; Neumann, J.; et al. Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J. Biol. Chem. 2005, 280, 6906–6914. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.U.; Neumann, J.; Schmitz, W. Transcriptional regulation by cAMP in the heart. Mol. Cell. Biochem. 2000, 212, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Darlyuk-Saadon, I.; Weidenfeld-Baranboim, K.; Yokoyama, K.K.; Hai, T.; Aronheim, A. The bZIP repressor proteins, c-Jun dimerization protein 2 and activating transcription factor 3, recruit multiple HDAC members to the ATF3 promoter. Biochim. Biophys. Acta 2012, 1819, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Stümpel, F.T.; Stein, J.; Himmler, K.; Scholz, B.; Seidl, M.D.; Skryabin, B.V.; Müller, F.U. Homozygous CREM-IbΔC-X Overexpressing Mice Are a Reliable and Effective Disease Model for Atrial Fibrillation. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Nattel, S. How does fibrosis promote atrial fibrillation persistence: In silico findings, clinical observations, and experimental data. Cardiovasc. Res. 2016, 110, 295–297. [Google Scholar] [CrossRef] [Green Version]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef] [Green Version]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.Y.; Li, N.; Karck, M.; Wehrens, X.H.T.; Nattel, M.; Dobrev, D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Pluteanu, F.; Nikonova, Y.; Holzapfel, A.; Herzog, B.; Scherer, A.; Preisenberger, J.; Plačkić, J.; Scheer, K.; Ivanova, T.; Bukowska, A.; et al. Progressive Impairment of Atrial Myocyte Function During Left Ventricular Hypertrophy and Heart Failure. J. Mol. Cell. Cardiol. 2018, 114, 253–263. [Google Scholar] [CrossRef]
- Shahid, F.; Lip, G.Y.H.; Shantsila, E. Role of Monocytes in Heart Failure and Atrial Fibrillation. J. Am. Heart. Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Sekiguchi, A.; Iwasaki, Y.K.; Date, T.; Sagara, K.; Tanabe, H.; Suma, H.; Sawada, H.; Aizawa, T. Recruitment of immune cells across atrial endocardium in human atrial fibrillation. Circ. J. 2010, 74, 262–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silbernagel, N.; Walecki, M.; Schäfer, M.K.; Kessler, M.; Zobeiri, M.; Rinné, S.; Kiper, A.K.; Komadowski, M.A.; Vowinkel, K.S.; Wemhöner, K.; et al. The VAMP-associated protein VAPB is required for cardiac and neuronal pacemaker channel function. FASEB J. 2018, 32, 6159–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignier, N.; Mougenot, N.; Bonne, G.; Muchir, A. Effect of genetic background on the cardiac phenotype in a mouse model of Emery-Dreifuss muscular dystrophy. Biochem. Biophys. Rep. 2019, 19. [Google Scholar] [CrossRef]
- Parahuleva, M.S.; Lipps, C.; Parviz, B.; Hölschermann, H.; Schieffer, B.; Schulz, R.; Euler, G. MicroRNA expression profile of human advanced coronary atherosclerotic plaques. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECG Recordings | 1 Week | 5 Weeks | ||
|---|---|---|---|---|
| WT | JDP2 | WT | JDP2 | |
| P-wave duration | 10.0 ± 0.9 ms | 10.6 ± 0.8 ms | 10.0 ± 1.5 ms | 11.6 ± 1.1 ms * |
| PQ-interval | 32.0 ± 5.1 ms | 32.3 ± 2.6 ms | 30.3 ± 7.0 ms | 35.1 ± 3.6 ms |
| QRS-complex | 14.3 ± 1.2 ms | 13.0 ± 1.2 ms | 13.9 ± 1.7 ms | 15.8 ± 2.3 ms * |
| RR-interval | 86.8 ± 5.0 ms | 84.9 ± 4.0 ms | 83.3 ± 4.1 ms | 84.5 ± 3.1 ms |
| HR-variability | 0.031 ± 0.013 | 0.042 ± 0.034 | 0.021 ± 0.01 | 0.027 ± 0.01 |
| QT-interval | 29.1 ± 0.7 ms | 29.1 ± 1.4 ms | 29.5 ± 1.8 ms | 31.3 ± 1.9 ms |
| n | 6 | 7 | 10 | 11 |
| Primer | Sequences of Primer |
|---|---|
| Adrb1 | Qiagen, QT00258692 |
| Adrb2 | 5′-TGGTACCGTGCCACCCACAA-3′ |
| 5′-AAGACCATCACCACCAGGGGCA-3′ | |
| ANP | 5′-CTGCTAATCAGCCATGCAAA-3′ |
| 5′-GATGGAGACCATCCTGGCTA-3′ | |
| Cav1.2 | 5′-CAGCCACTCTCCAGTCACTC-3′ |
| 5′-CTGGAGTAGGGATGTGCTCG-3′ | |
| CD3 | 5′-ATGCGGTGGAACACTTTCTGG-3′ |
| 5′-GCACGTCAACTCTACACTGGT-3′ | |
| CD11b | 5′-CTGAGACTGGAGGCAACCAT-3′ |
| 5′-GATATCTCCTTCGCGCAGAC-3′ | |
| CD20 | 5′-CCTTTCCCAGCAGAGCCTAC-3′ |
| 5′-TCATGATTTGGACAGCCCCC-3′ | |
| CD45 | 5′-ATGGTCCTCTGAATAAAGCCCA-3′ |
| 5′-TCAGCACTATTGGTAGGCTCC-3′ | |
| CD68 | 5′-ACTTCGGGCCATGTTTCTCT-3′ |
| 5′-GCTGGTAGGTTGATTGTCGT-3′ | |
| Collagen1 | 5′-TTCTCCTGGRAAAGATGGTGC-3′ |
| 5′-GGACCAGCATCACCTTTAACA-3′ | |
| Csf1r | 5′-TCCACCGGGACGTAGCA-3′ |
| 5′-CCAGTCCAAAGTCCCCAATCT-3′ | |
| Cx37 | 5′-ATAAAGGCACGAAGGGACCA-3′ |
| 5′-GTCAAGTTGGCCCAGTTCTG-3′ | |
| Cx40 | 5′-AGGGCTGAGCTTGCTTCTTA-3′ |
| 5′-TTAGTGCCAGTGTCGGGAAT-3′ | |
| Cx43 | 5′-GAAACAATTCCTCCTGCCGC-3′ |
| 5′-AGTTGGAGATGGTGCTTCCG-3′ | |
| Elastin | 5′-CTGCTGCTAAGGCTGCTAAG-3′ |
| 5′-CCACCAACACCAGGAATGC-3′ | |
| Fibronectin | 5′-ACAGAGCTCAACCTCCCTGA-3′ |
| 5′-TGTGCTCTCCTGGTTCTCCT-3′ | |
| JDP2 | 5′-ATGATGCCTGGGCAGATCCCA-3′ |
| 5′-TCACTTCTTGTCCAGCTGCTCC-3′ | |
| MCP1 | 5′-CCACAACCACCTCAAGCA-3′ |
| 5′-TGAAAGGGAATACCATAACATC-3′ | |
| NCX | 5′-CTACCAGGTCCTAAGTCAACAG-3′ |
| 5′-TGCGTGCCTCTTCAAGATG-3′ | |
| PLB | 5′-GCAATACCTCACTCGCTCGGCTATC-3′ |
| 5′-TGGAGATTCTGACGTGCTTGCTGAG-3′ | |
| RyR | 5′-AGCTGGAAGACCCTGCAATC-3′ |
| 5′-ACCAGGCTGAAATATCCCCG-3′ | |
| SERCA | 5′-TGACTGGTGATGGTGTGAATG-3′ |
| 5′-GATGAGGTAGCGGATGAACTG-3′ | |
| 18SrRNA | 5′-TTGACGGAAGGGCACCACCA-3′ |
| 5′-AGAACGGCCATGCACCACCA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parahuleva, M.S.; Kockskämper, J.; Heger, J.; Grimm, W.; Scherer, A.; Bühler, S.; Kreutz, J.; Schulz, R.; Euler, G. Structural, Pro-Inflammatory and Calcium Handling Remodeling Underlies Spontaneous Onset of Paroxysmal Atrial Fibrillation in JDP2-Overexpressing Mice. Int. J. Mol. Sci. 2020, 21, 9095. https://doi.org/10.3390/ijms21239095
Parahuleva MS, Kockskämper J, Heger J, Grimm W, Scherer A, Bühler S, Kreutz J, Schulz R, Euler G. Structural, Pro-Inflammatory and Calcium Handling Remodeling Underlies Spontaneous Onset of Paroxysmal Atrial Fibrillation in JDP2-Overexpressing Mice. International Journal of Molecular Sciences. 2020; 21(23):9095. https://doi.org/10.3390/ijms21239095
Chicago/Turabian StyleParahuleva, Mariana S., Jens Kockskämper, Jacqueline Heger, Wolfram Grimm, Anna Scherer, Sarah Bühler, Julian Kreutz, Rainer Schulz, and Gerhild Euler. 2020. "Structural, Pro-Inflammatory and Calcium Handling Remodeling Underlies Spontaneous Onset of Paroxysmal Atrial Fibrillation in JDP2-Overexpressing Mice" International Journal of Molecular Sciences 21, no. 23: 9095. https://doi.org/10.3390/ijms21239095
APA StyleParahuleva, M. S., Kockskämper, J., Heger, J., Grimm, W., Scherer, A., Bühler, S., Kreutz, J., Schulz, R., & Euler, G. (2020). Structural, Pro-Inflammatory and Calcium Handling Remodeling Underlies Spontaneous Onset of Paroxysmal Atrial Fibrillation in JDP2-Overexpressing Mice. International Journal of Molecular Sciences, 21(23), 9095. https://doi.org/10.3390/ijms21239095