2.1. Genome Completeness and Accuracy
For simulated reads which contained artificial error profiles, genome completeness and accuracy indicate the robustness of an assembler to tolerate a wide range of read parameters such as chimeras, low-quality regions, and systematic basecalling errors [
17]. The Canu, Miniasm/Racon, and Raven assemblies of mediocre-quality reads had numbers of contigs not significantly different (
p > 0.05) from the reference genomes (
Table 1), while Flye, Redbean, and Shasta produced significantly higher (
p < 0.05) numbers of contigs than the reference genomes. The Redbean assemblies generated genome sizes significantly larger than the reference genomes (
p < 0.05), whereas other assemblers produced assemblies with significantly lower (
p < 0.05) genome sizes than the reference genomes. The GC contents of all assemblies were not significantly different (
p > 0.05) from those of the reference genomes. The complete benchmarking universal single-copy orthologs (BUSCOs) of all assemblies were significantly lower (
p < 0.05) than those of the reference genomes. Significantly higher (
p < 0.05) complete BUSCOs were observed in the Miniasm/Racon and Raven assemblies compared to other assemblers (
Supplementary Table S1), while the complete BUSCOs of the Raven assemblies were significantly higher (
p < 0.05) than those of the Miniasm/Racon assemblies. Significantly lower (
p < 0.05) numbers of SNPs and indels were detected in the Canu, Miniasm/Racon, and Raven assemblies than other assemblies (
Supplementary Table S2). The numbers of SNPs and indels of the Canu and Raven assemblies were not significantly different (
p > 0.05) but were significantly lower (
p < 0.05) than the Miniasm/Racon assemblies.
We also tested simulated Oxford Nanopore long reads of low quality to examine whether each long-read assembler could tolerate a higher degree of errors. When assembling low-quality reads, Canu and Shasta failed to generate assemblies for all strains (
Table 2), while Flye and Redbean did not produce assemblies for three and one out of 10 strains, respectively. Similarly, Wick and Holt (2020) also reported the incomplete assembly processes of Canu, Flye, Redbean, and Shasta when simulated reads of prokaryote genomes were tested. In contrast, Miniasm/Racon and Raven generated assemblies for all strains. Compared to mediocre-quality reads, Flye, Miniasm/Racon, Redbean, and Raven produced assemblies with more fragmented contigs using low-quality reads. The numbers of contigs of the Raven and Redbean assemblies were not significantly different (
p > 0.05) from those of the reference genomes, whereas Flye and Miniasm/Racon generated significantly more (
p < 0.05) contigs than the reference genomes. The genome sizes and GC contents of the Raven assemblies were still not significantly different (
p > 0.05) from those of the reference genomes, although low-quality reads were used. Flye, Miniasm/Racon, and Redbean all produced significantly different (
p < 0.05) GC contents from the reference genomes. The complete BUSCOs of the Raven assemblies were still significantly higher (
p < 0.05) than those of the Flye, Miniasm/Racon, and Redbean assemblies (
Supplementary Table S3). As Flye and Miniasm/Racon produced assemblies with inaccurate genome sizes using low-quality reads, the numbers of SNPs and indels of the assemblies of low-quality reads were not compared with those of mediocre-quality reads. Significant increases (
p < 0.05) in the numbers of SNPs and indels were observed in the Raven assemblies of low-quality reads compared to mediocre-quality reads (
Supplementary Table S4).
Compared to when mediocre-quality reads were tested, the numbers of contigs of the Flye, Miniasm/Racon, Raven, and Redbean assemblies of low-quality reads became significantly higher (p < 0.05) when using low-quality reads. No significant differences (p > 0.05) were observed between the genome sizes of the Raven assemblies of mediocre- and low-quality reads, while the genome sizes of the Flye, Miniasm/Racon, and Redbean assemblies of low-quality reads were significantly lower (p < 0.05) than those of mediocre-quality reads. There were no significant differences (p > 0.05) in GC content between the Flye, Miniasm/Racon, Raven, and Redbean assemblies of mediocre- and low-quality reads. The complete BUSCOs of the Flye, Miniasm/Racon, Raven, and Redbean assemblies of low-quality reads were significantly lower than those of mediocre-quality reads (p < 0.05), while significantly higher (p < 0.05) numbers of SNPs and indels were observed when low-quality reads were tested.
Despite the advantages of simulated reads in allowing for a controlled evaluation of the effect of long-read quality on genome assembly, they cannot completely capture the challenges of using real Oxford Nanopore reads when assessing long-read assemblers. The genome completeness and accuracy of an assembler indicate its reliability to achieve a complete and accurate assembly given a set of real reads, which incorporate naturally occurring features of Oxford Nanopore long reads (e.g., error profiles, read lengths, quality scores) [
17]. All assemblers completed assembly processes for each strain to generate assemblies when real reads were used (
Table 3). Canu, Flye, and Raven were superior to other assemblers, and produced the most contiguous assemblies, while other assemblers produced inaccurate numbers of contigs for some strains. For example, Miniasm/Racon produced 227 and 43 contigs for
Clostridium botulinum CFSAN034200 and
C. jejuni NCTC 11168, respectively. Shasta generated 850 contigs for
C. sakazakii CFSAN068773. Redbean performed poorly in achieving contiguous assemblies, often producing inaccurate numbers of contigs. The genome sizes and GC contents of the Miniasm/Racon, Raven, Redbean, and Shasta assemblies were not significantly different (
p > 0.05) from those of the reference genomes, whereas Canu and Flye failed to produce assemblies which had genome sizes and GC contents not significantly different from the reference genomes (
p > 0.05). The complete BUSCOs of all assemblies were significantly lower (
p < 0.05) than those of the reference genomes (
Supplementary Table S5). The Miniasm/Racon and Raven assemblies had the highest (
p < 0.05) complete BUSCOs compared to other assemblers, while no significant differences (
p > 0.05) were observed between the Miniasm/Racon and Raven assemblies. The numbers of SNPs of the Canu assemblies were significantly lower (
p < 0.05) than those of the assemblies produced by other assemblers (
Supplementary Table S6), while no significant differences (
p > 0.05) were found among other assemblers, with only one exception that the Raven assemblies had the numbers of SNPs significantly lower (
p < 0.05) than the Miniasm/Racon assemblies. The Raven assemblies had the lowest (
p < 0.05) numbers of indels among all assemblers.
As predicted with the aid of the PlasmidFinder database, the assemblies of simulated reads showed inconsistent plasmid profiles (types and numbers of plasmids) with the reference genomes in some cases (
Supplementary Tables S7 and S8). All assemblies of
Bacillus anthracis Ames Ancestor showed consistent plasmid profile with the reference genome. Only the Flye assembly of
S. Typhimurium LT2 contained both IncFIB (S) and IncFII (S), which was consistent with the reference genome. The plasmid profiles of the Redbean and Shasta assemblies of
Cronobacter sakazakii ATCC 29544 were consistent with that of the reference genome. None of the assemblies of
Escherichia coli O157:H7 Sakai carried IncFIB (AP001918) that was present in the reference genome. Among the assemblies of low-quality reads, the Raven assembly of
Staphylococcus aureus TW20 was the only one that carried a plasmid, whereas plasmids were not present in other assemblies.
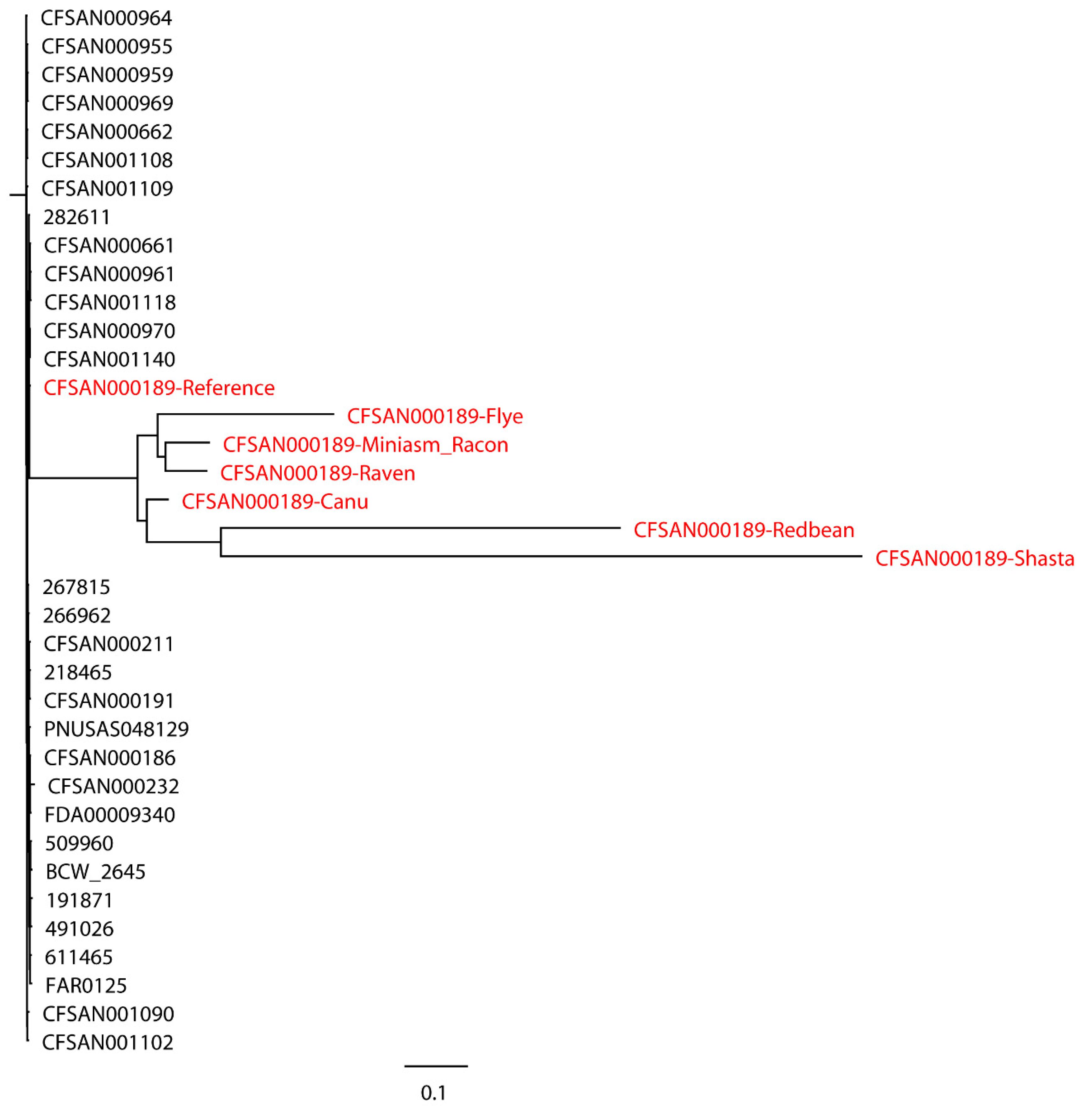
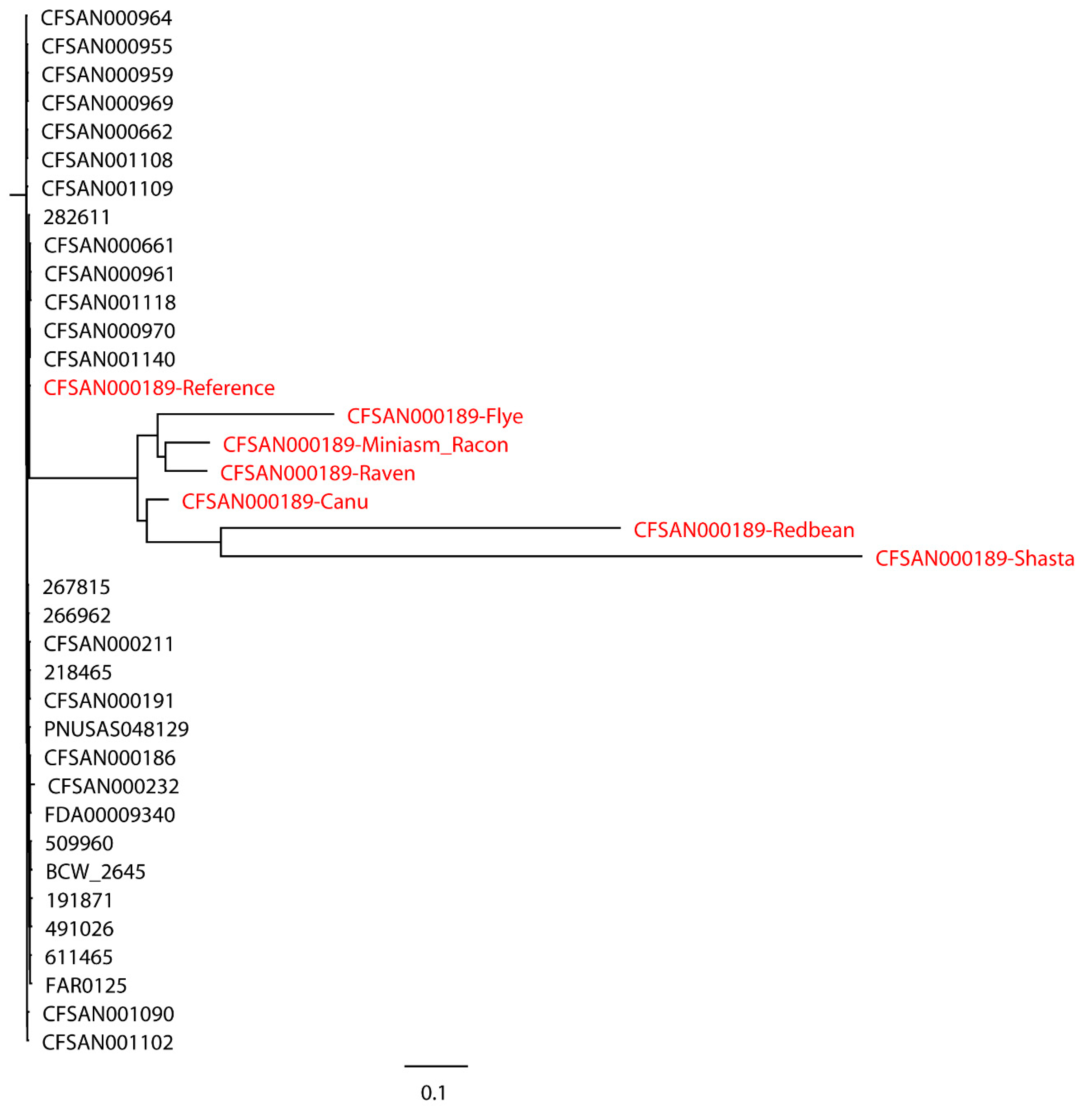
Similarly, plasmid profiles inconsistent with the reference genomes were also observed in the assemblies of real reads (
Supplementary Table S9). Compared to the reference genomes, the Shasta assembly of
S. Bareilly CFSAN000189 did not harbor IncFII (S). The Flye assembly of
S. aureus CFSAN007894 had a consistent plasmid profile with the reference genome, while inconsistent plasmid profiles were identified in other assemblies. Interestingly, similar to
Escherichia coli O157:H7 Sakai with simulated reads, none of the assemblies of
E. coli O157:H7 CFSAN076619 possessed IncFIB (AP001918) that was contained in the reference genome, which could be attributed to the loss of this plasmid during Oxford Nanopore library preparation. This finding also pointed to the suboptimal library preparation bias as a second possible explanation [
18]. Consequently, there is a current need for reliable protocols to extract high-molecular-weight genomic DNA from bacteria that are compatible with Oxford Nanopore sequencing to overcome this deficiency. The degree to which Oxford Nanopore sequencing is compatible with diverse DNA extraction chemistries remains to be validated.
According to the assemblies using both simulated and real reads, overall, Raven emerged as the best assembler that can be tolerant of both low-identity and realistic reads. Noticeably, Raven was the most robust assembler that was not significantly affected by random reads, junk reads, or chimeric reads in low-quality reads. It should be noted that one expected benefit of Oxford Nanopore sequencing is that it can identify the exact locations of mobile genetic elements (MGEs) such as plasmids. Unfortunately, since Raven does not indicate the circularity of contigs that harbor plasmids, it is not able to determine whether plasmids are integrated into the chromosome or exist extra-chromosomally in a non-integrated state. This drawback of Raven may potentially cause an imperfect representation of the genetic architectures of MGEs.
2.2. Antimicrobial Resistance Genes (ARGs)
Oxford Nanopore sequencing has great potential for accelerating AMR genotyping, as sequence data are acquired in real time within minutes of sequencing [
19,
20]. Meanwhile, Oxford Nanopore sequencing enables the identification of MGEs on which ARGs are located and also characterizes the combination of different ARGs co-located on the same MGEs. However, it is worth noting that Oxford Nanopore sequencing currently has higher error rates than Illumina sequencing, although improvements in nanopore chemistry and basecalling could potentially reduce the differential [
21]. The error rate of Oxford Nanopore long reads could also in part be compensated for by assembly algorithms through intensive computations to acquire a more accurate AMR profiling independent of other sequencing technologies such as Illumina sequencing. Therefore, we compared the AMR genotypes and phenotypes of selected bacterial pathogens, as predicted based on Oxford Nanopore long-read assemblies using different long-read assemblers.
Five genotypically antimicrobial-resistant strains with simulated reads were used (
Supplementary Table S10). All Miniasm/Racon and Raven assemblies provided AMR profiles that were consistent with the reference genomes. Canu also produced assemblies that had accurate AMR profiles, with only one exception that the Canu assembly of
S. aureus TW20 carried
erm(33) that was absent in the reference genome. Both Flye and Shasta produced assemblies with inaccurate AMR profiles for three strains, while Redbean performed the worst and produced four Redbean assemblies with inconsistent AMR profiles with the reference genomes. Noticeably, when low-quality reads were used, no ARGs were identified in the Flye, Miniasm/Racon, and Redbean assemblies (
Supplementary Table S11). Raven still produced the assembly of
Klebsiella variicola DSM 15968 that had an accurate AMR profile, while the Raven assemblies of both
P. aeruginosa PAO1 and
C. jejuni NCTC 11168 also contained three ARGs that were present in the reference genomes.
Five genotypically antimicrobial-resistant strains with real reads were used (
Supplementary Table S12). All assemblies of
P. aeruginosa CFSAN084950 had consistent AMR profiles with the reference genome. The gene associated with kanamycin resistance,
aph(3′)-III, was present in the Canu, Miniasm/Racon, Raven, and Redbean assemblies of
E. coli O157:H7 CFSAN076619, while the gene associated with ampicillin resistance,
blaOXA-61, was only identified in the Miniasm/Racon and Raven assemblies of
C. jejuni NCTC 11168. Noticeably, none of the assemblies of
S. aureus CFSAN007894 and
Campylobacter coli CFSAN032805 carried the same ARGs with the reference genomes.
Overall, compared to other assemblers, Raven was more capable of generating assemblies for accurate AMR genotypes, closely followed by Miniasm/Racon, especially when low-quality reads were used. Although it is possible to assemble Oxford Nanopore long reads into complete genomes to help implement near real-time AMR profiling, doing so would compromise the genome accuracy of bacterial pathogens and result in inaccurate AMR genotypes. Future improvements to the library preparation and basecalling of Oxford Nanopore sequencing, as well as long-read assembly algorithms, may mitigate this deficiency.
2.5. Whole-Genome Phylogeny
Taylor et al. [
27] used 23 closely related
Salmonella strains (number of SNPs < 500) to build a whole-genome phylogenetic tree with the Miniasm/Racon assembly of the Oxford Nanopore long reads of
S. Bareilly CFSAN000189, where they observed a congruent topology between trees built with the SPAdes assembly of Illumina short reads and the Miniasm/Racon assembly. In our work, both closely and distantly related strains were included when evaluating the performance of Oxford Nanopore long-read assemblies in the phylogenetic analyses. A total of 30 closely related
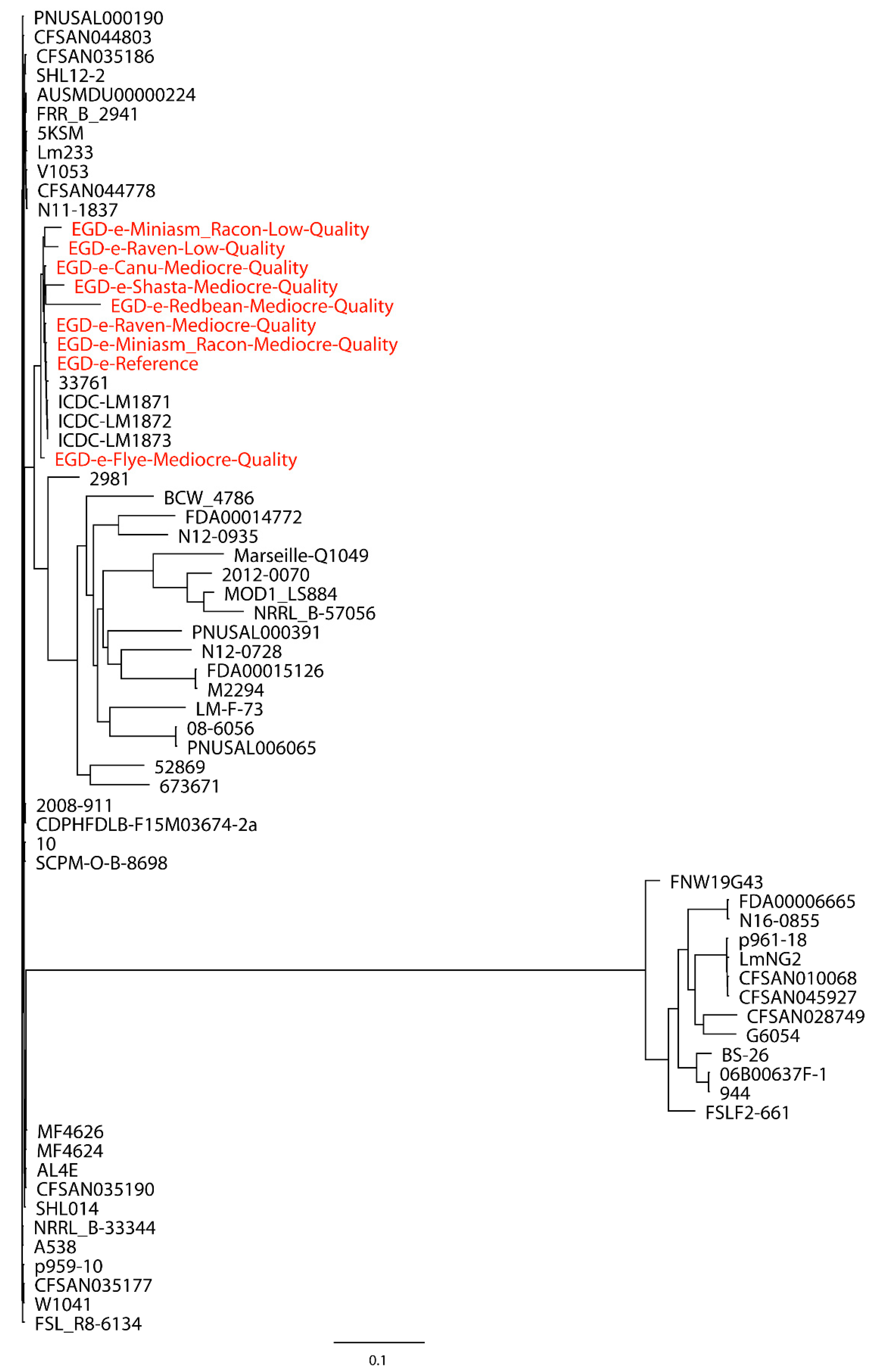
L. monocytogenes strains were first used for the phylogenetic analysis of
L. monocytogenes EGD-e with mediocre- and low-quality reads. The Redbean assembly of low-quality reads was not included due to the error produced by CSI Phylogeny when this assembly with an inaccurate genome size of 30,776 bp was processed. As shown in
Figure 1, the assemblies formed a single monophyletic clade with the reference genome and closely related strains, irrespective of the assemblies of mediocre- or low-quality reads, although the Flye assembly of mediocre-quality reads formed a dispersed paraphyletic clade relative to the major clade of the reference genome and other assemblies. The Miniasm/Racon and Raven assemblies of mediocre-quality reads had the smallest distance from the reference genome. Interestingly, the Miniasm/Racon assembly of low-quality reads with an inaccurate genome size of 542,915 bp was also on the clade where the reference genome was located. When 30 distantly related
L. monocytogenes strains were also included in the phylogenetic analysis of
L. monocytogenes EGD-e, a similar clade topology of the monophyletic clade that included the reference genome and assemblies were observed (
Figure 2), where the Miniasm/Racon and Raven assemblies still had the smallest distance from the reference genome.
S. Bareilly CFSAN000189 assemblies with real reads and its reference genome were aligned to 30 closely related
S. Bareilly strains (
Figure 3). All 30 closely related strains and the reference genome formed a single monophyletic clade, whereas the assemblies formed a dispersed paraphyletic group sister to the reference genome clade. Similar results were obtained for the tree of
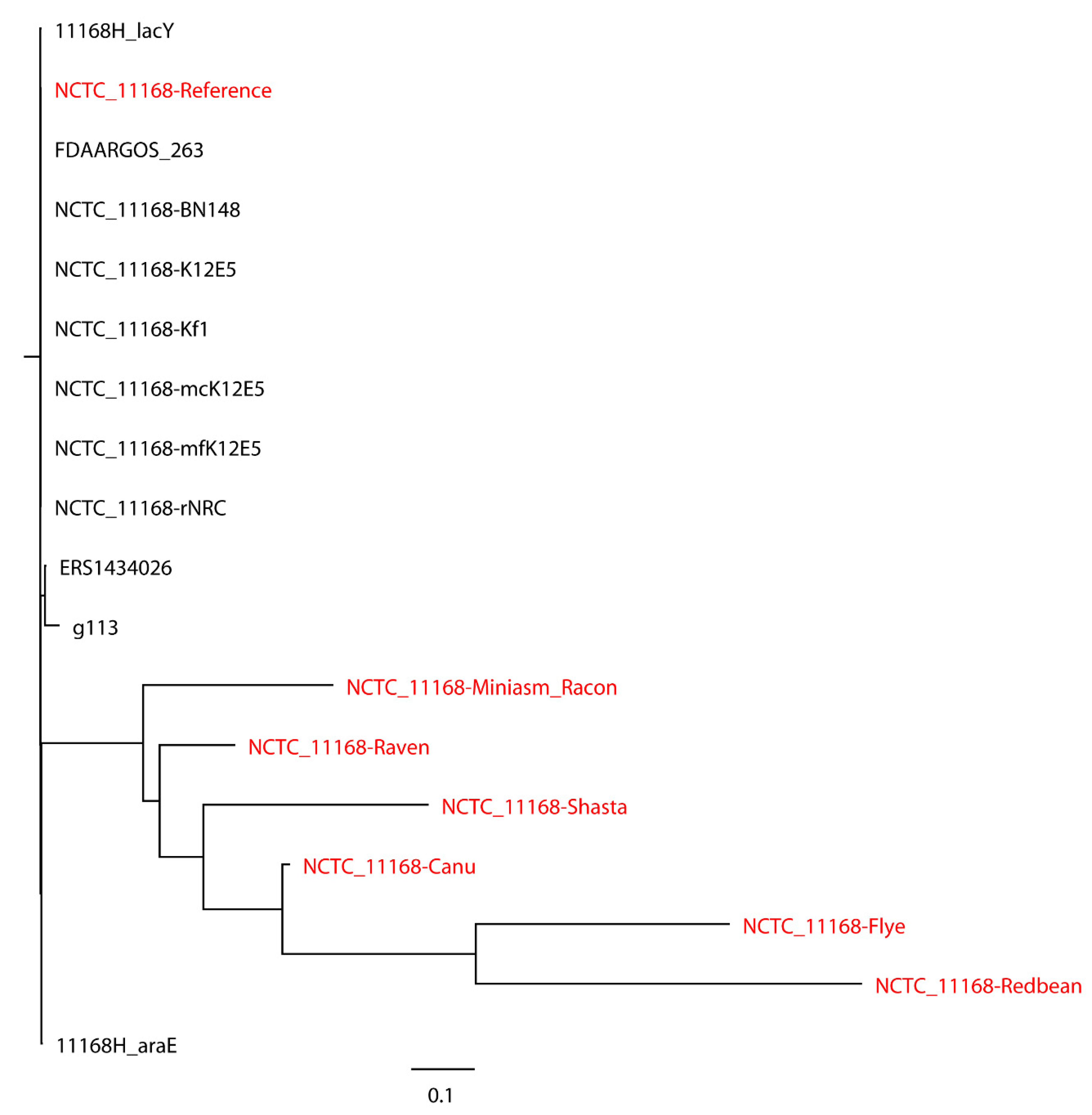
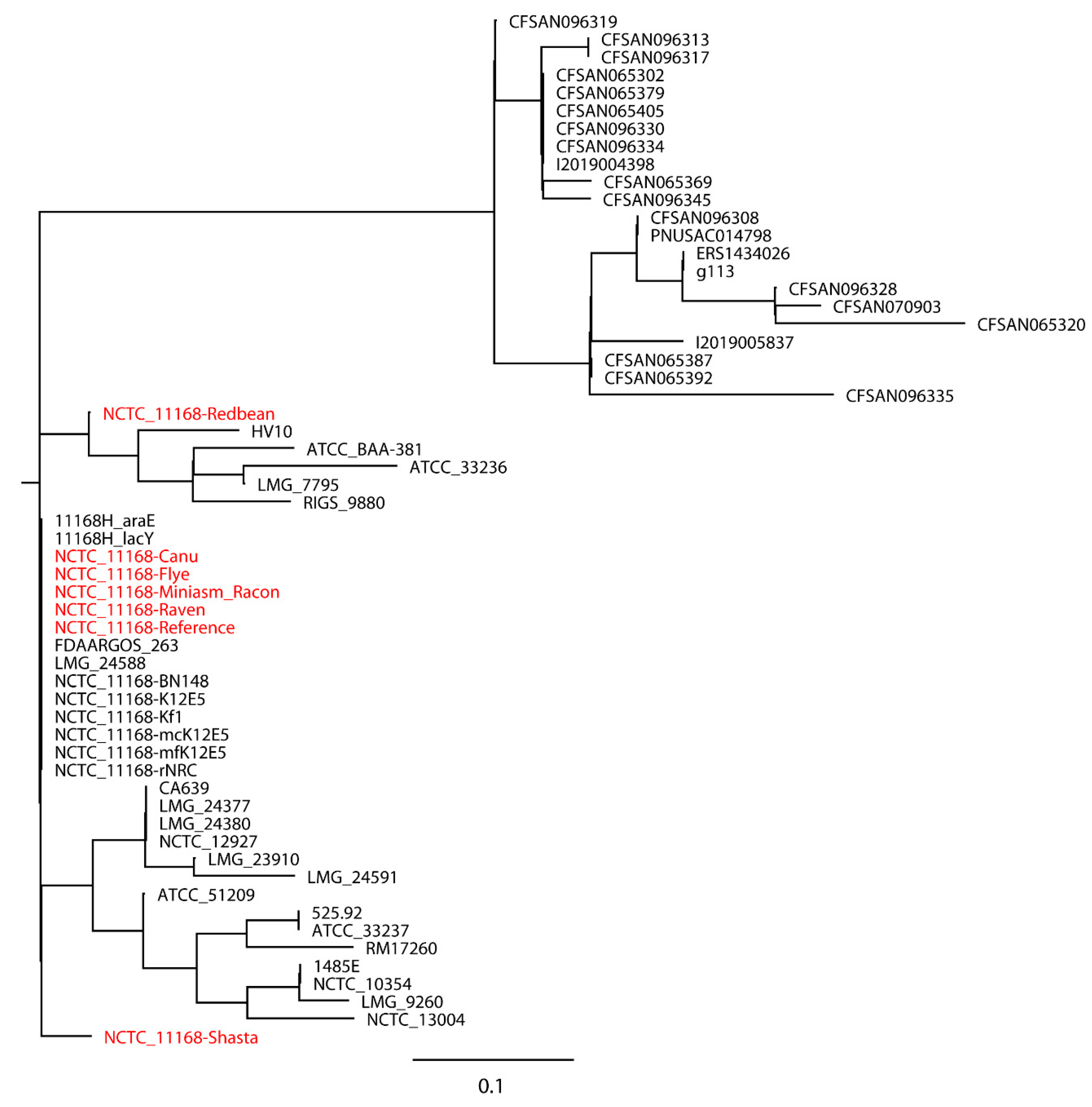
C. jejuni NCTC 11168 with real reads built with 11 closely related
C. jejuni strains selected based on the SNP strategy (
Figure 4), where the reference genome formed a monophyletic group with the closely related strains and the assemblies were a non-monophyletic group forming multiple independent clades. When 20 distantly related
C. jejuni strains were included in the phylogenetic analysis of
C. jejuni NCTC 11168, the assemblies were still placed on different clades compared to the reference genome (
Figure 5). Although biased results were observed across all assemblies of both
S. Bareilly CFSAN000189 and
C. jejuni NCTC 11168, the Miniasm/Racon and Raven assemblies always had the smallest distance from the reference genomes. We then included 20
Campylobacter strains of other species in the phylogenetic analysis of
C. jejuni NCTC 11168 (
Figure 6). Interestingly, all assemblies except the Redbean and Shasta assemblies were on the clade where the reference genome was located. The Miniasm/Racon and Raven assemblies still displayed the smallest distance from the reference genome.
The higher error rates of Oxford Nanopore long reads may confound the phylogenetic differences between the assemblies and closely related strains selected based on the SNP and core-genome MLST (cgMLST) or whole-genome MLST (wgMLST) strategies, further confirming our observations that Oxford Nanopore long-read assemblies contained SNPs (
Supplementary Tables S2, S4 and S6) and also produced inaccurate MLST results (
Table 9,
Table 10 and
Table 11) relative to the reference genomes. We identified some recurrent phylogenetic patterns of long-read assemblies that could potentially be addressed in the future, as continued improvements in nanopore chemistry and basecalling would systematically mitigate the high numbers of errors to achieve a more accurate phylogenetic inference.
2.6. Pan Genomes
A total of 20 distantly related
P. aeruginosa strains were included in the pan-genome analysis of
P. aeruginosa PAO1 with mediocre- and low-quality reads. Raven was the most effective assembler for the pan-genome analysis of
P. aeruginosa PAO1 with mediocre-quality reads, which displayed the numbers of core (core and soft-core) and accessory (shell and cloud) genes that were most similar to the reference genome (
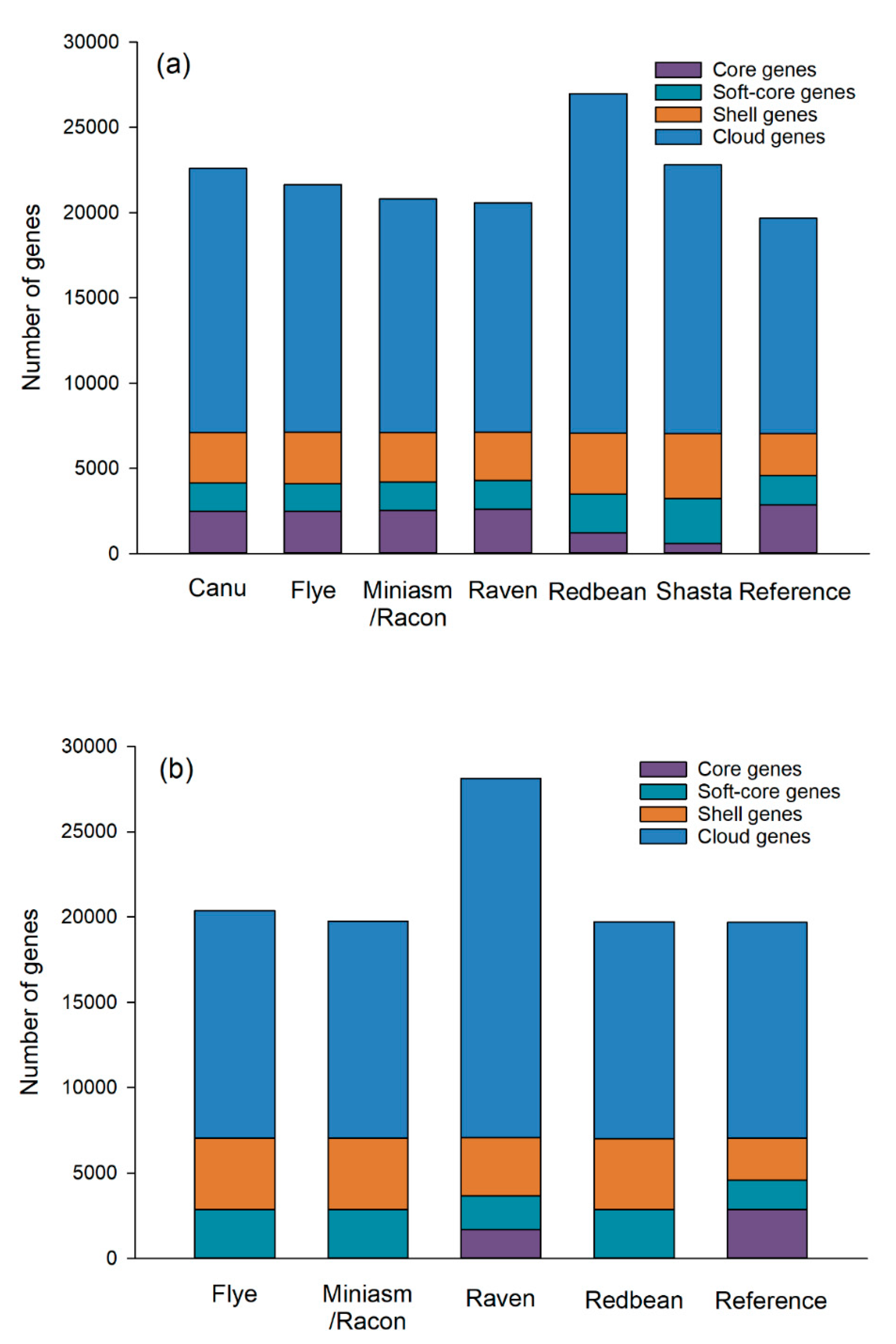
Figure 7). The pan genomes of the Raven assembly consisted of a total of 20,583 genes with 2615 core genes (12.7%) and 17,968 accessory genes (87.3%). Similarly, there were a total of 19,673 genes with 2866 core genes (14.6%) and 16,807 accessory genes (85.4%) in the pan genomes of the reference genome. Our results thus demonstrate that Raven could tolerate the inaccuracy of Oxford Nanopore long reads. Raven was closely followed by Miniasm/Racon that was also able to achieve similar pan-genome patterns with the reference genome. Canu, Flye, and Shasta were moderate performers for the pan-genome analysis, while Redbean was the least effective, producing inaccurate numbers of core genes (4.6%) and accessory genes (95.4%), respectively.
Regarding the pan-genome analysis of the assemblies of low-quality reads, the Flye, Miniasm/Racon, and Redbean assemblies did not produce accurate numbers of core genes, although their total numbers of genes were similar to the reference genome, which could be attributed to their inaccurate genome sizes. For the pan genome of the Raven assembly of low-quality reads, we observed a dramatic increase in the total number of genes (28,119) compared to that of mediocre-quality reads, with a decrease in the number of core genes (1696) and an increase in the number of accessory genes (26,423).
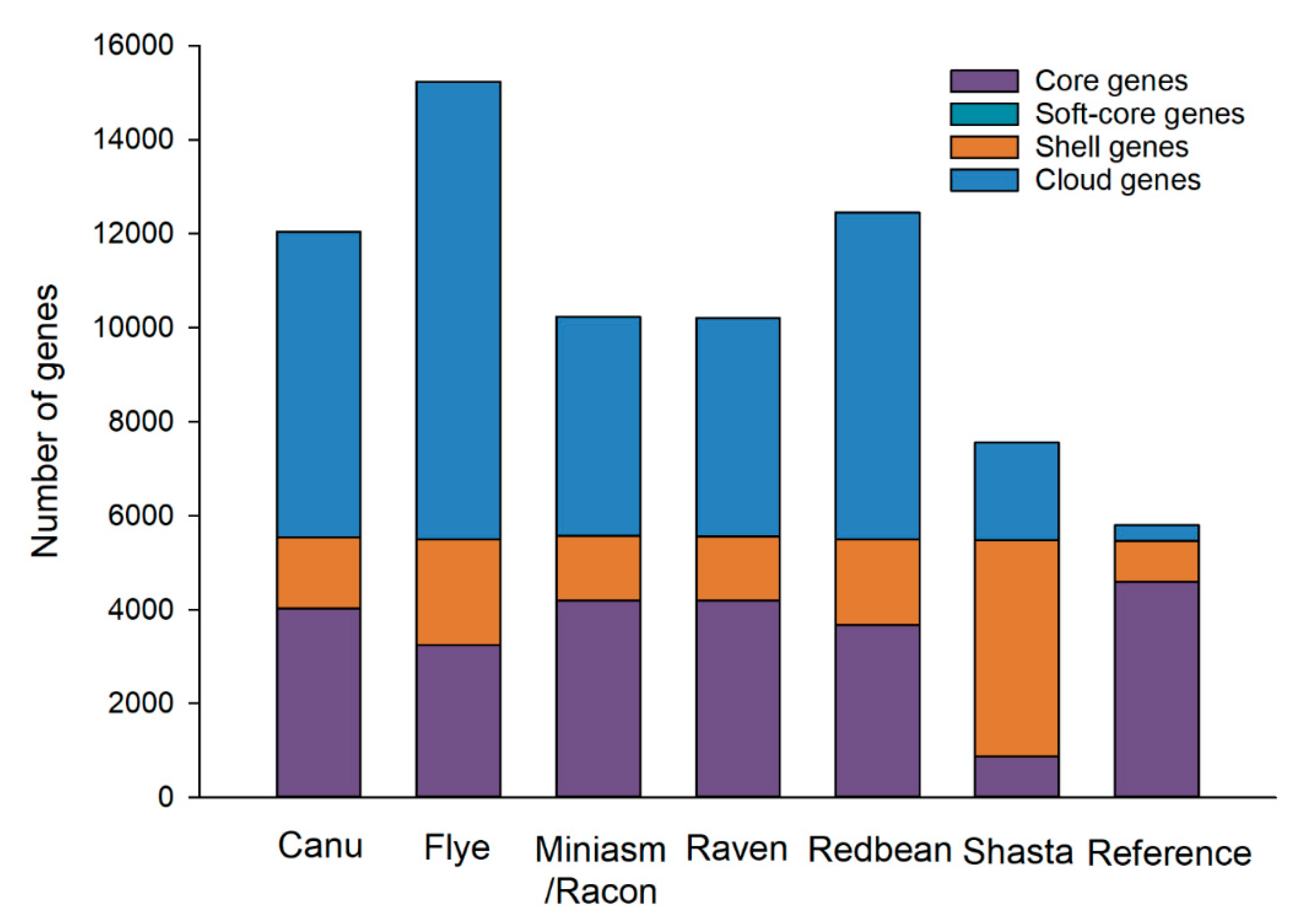
Six closely related strains of
E. coli O157:H7 CFSAN076619 with real reads were found based on the SNP and wgMLST strategies, which were thus included in its pan-genome analysis. The pan genome of the reference genome and the six strains consisted of 5790 genes with 4595 core genes (79.4%) and 1195 accessory genes (20.6%) (
Figure 8). The Miniasm/Racon and Raven assemblies had numbers of core genes most similar to the reference genome compared to other assemblies, although the pan genomes of all assemblies contained large numbers of accessory genes. One key reason for this poor performance is the inherently limited accuracy of Oxford Nanopore long reads, as also revealed by our SNP and indel analyses, demonstrating that the degree of errors in an assembly can greatly affect conclusions about gene presence and absence beyond just the inability to resolve genomic structures. SNPs and indels detected in the assemblies of Oxford Nanopore long reads could introduce truncated genes that can produce a large number of misannotated genes, which increased the numbers of accessory genes in the pan genomes [
18]. Our pan-genome analyses likely reflect the difficulty in using highly error-prone Oxford Nanopore long reads for accurately annotating genes due to gene truncation and misplaced start sites. Another aspect worth noting is that this limitation of Oxford Nanopore sequencing could become more remarkable when closely related strains were included in the pan-genome analysis. Our pan-genome analyses thus again highlight the limitation of Oxford Nanopore sequencing to produce accurate long reads. Overcoming this existing deficiency could greatly boost the annotations of many clinically and microbiologically important genomic regions in bacterial pathogens. Despite all these observations using real reads, Miniasm/Racon and Raven were comparable for the pan-genome analysis and performed the best among all assemblers tested, although both of their pan genomes still contained over 6000 accessory genes.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}