PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer


Abstract
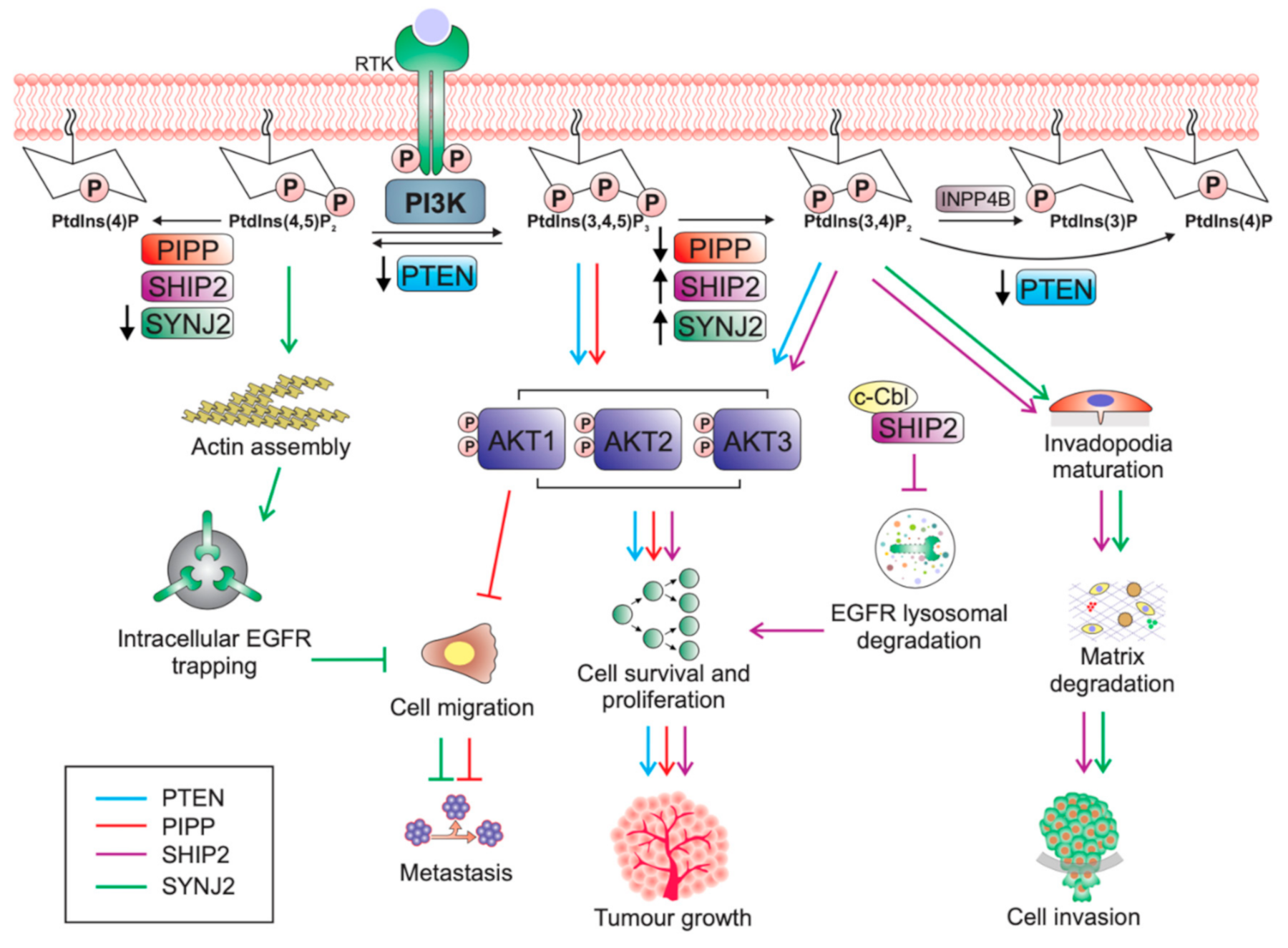
1. Introduction
2. The Phosphoinositide-3-Kinase (PI3K) Signalling Pathway
2.1. AKT Isoforms
2.2. PI3K Pathway Regulation
3. PTEN
PTEN Protein Phosphatase Activity in Tumourigenesis
4. Inositol Polyphosphate 5-Phosphatases
5. The Role of PIPP in Breast Cancer Suppression
PIPP Functions as a Tumour Suppressor in Other Cancers
6. The Pro-Tumourigenic Role of SHIP2 in Breast Cancer
The Negative Role SHIP2 May Play in Cancer Progression
7. SYNJ2 Promotes Breast Tumourigenesis
8. The PtdIns(3,4,5)P3 Phosphatases in Breast Cancer: Oncogenes Versus Tumour Suppressors?
8.1. PtdIns(3,4)P2 Is An Activator of AKT Signalling
8.2. PtdIns(4,5)P2 and Cancer
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Rabbani, S.A.; Mazar, A.P. Evaluating distant metastases in breast cancer: From biology to outcomes. Cancer Metastasis Rev. 2007, 26, 663–674. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869. [Google Scholar] [CrossRef]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Eroles, P.; Bosch, A.; Pérez-Fidalgo, J.A.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef]
- Voduc, K.D.; Cheang, M.C.U.; Tyldesley, S.; Gelmon, K.; Nielsen, T.O.; Kennecke, H. Breast cancer subtypes and the risk of local and regional relapse. J. Clin. Oncol. 2010, 28, 1684–1691. [Google Scholar] [CrossRef]
- López-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.A.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Gen. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Rodgers, S.J.; Ferguson, D.T.; Mitchell, C.A.; Ooms, L.M. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci. Rep. 2017, 37, BSR20160432. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, N.; Chung, G.H.C.; Lete, M.G.; Alonso, A.; Byrne, R.D.; Calleja, V.; Larijani, B. Endomembrane PtdIns(3,4,5)P3 activates the PI3K–Akt pathway. J. Cell Sci. 2015, 128, 3456. [Google Scholar] [CrossRef]
- Chopra, S.S.; Cantley, L.C. PI3K-Akt-mTOR Signaling in Cancer and Cancer Therapeutics. In PI3K-mTOR in Cancer and Cancer Therapy; Dey, N., De, P., Leyland-Jones, B., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–25. [Google Scholar]
- Clark, A.R.; Toker, A. Signalling specificity in the Akt pathway in breast cancer. Biochem. Soc. Trans. 2014, 42, 1349–1355. [Google Scholar] [CrossRef]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef]
- Chen, W.S.; Xu, P.Z.; Gottlob, K.; Chen, M.L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef]
- Konishi, H.; Kuroda, S.; Tanaka, M.; Matsuzaki, H.; Ono, Y.; Kameyama, K.; Haga, T.; Kikkawa, U. Molecular cloning and characterization of a new member of the RAC protein kinase family: Association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem. Biophys. Res. Commun. 1995, 216, 526–534. [Google Scholar] [CrossRef]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw Iii, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Tschopp, O.; Yang, Z.Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase Bγ (PKBγ/Akt3) in postnatal brain developmental but not in glucose homeostasis. Development 2005, 132, 2943–2954. [Google Scholar] [CrossRef]
- Dillon, R.L.; Marcotte, R.; Hennessy, B.T.; Woodgett, J.R.; Mills, G.B.; Muller, W.J. Akt1 and Akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009, 69, 5057–5064. [Google Scholar] [CrossRef]
- Hutchinson, J.N.; Jin, J.; Cardiff, R.D.; Woodgett, J.R.; Muller, W.J. Activation of Akt-1 (PKB-α) Can Accelerate ErbB-2-Mediated Mammary Tumorigenesis but Suppresses Tumor Invasion. Cancer Res. 2004, 64, 3171–3178. [Google Scholar] [CrossRef]
- Young, C.D.; Nolte, E.C.; Lewis, A.; Serkova, N.J.; Anderson, S.M. Activated Akt1 accelerates MMTV-c-ErbB2 mammary tumourigenesis in mice without activation of ErbB3. Breast Cancer Res. 2008, 10, R70. [Google Scholar] [CrossRef]
- Ju, X.; Katiyar, S.; Wang, C.; Liu, M.; Jiao, X.; Li, S.; Zhou, J.; Turner, J.; Lisanti, M.P.; Russell, R.G.; et al. Akt1 governs breast cancer progression in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 7438–7443. [Google Scholar] [CrossRef]
- Maroulakou, I.G.; Oemler, W.; Naber, S.P.; Tsichlis, P.N. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007, 67, 167–177. [Google Scholar] [CrossRef]
- Chen, X.; Ariss, M.M.; Ramakrishnan, G.; Nogueira, V.; Blaha, C.; Putzbach, W.; Islam, A.; Frolov, M.V.; Hay, N. Cell-Autonomous versus Systemic Akt Isoform Deletions Uncovered New Roles for Akt1 and Akt2 in Breast Cancer. Mol. Cell 2020, 80, 87–101.e105. [Google Scholar] [CrossRef]
- Eramo, M.J.; Mitchell, C.A. Regulation of PtdIns(3,4,5)P3/Akt signalling by inositol polyphosphate 5-phosphatases. Biochem. Soc. Trans. 2016, 44, 240–252. [Google Scholar] [CrossRef]
- Zardavas, D.; Phillips, W.A.; Loi, S. PIK3CA mutations in breast cancer: Reconciling findings from preclinical and clinical data. Breast Cancer Res. 2014, 16, 201. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Kirsch, K.H.; Kaloudis, P.; Yang, H.; Pavletich, N.P.; Hanafusa, H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000, 60, 7033–7038. [Google Scholar]
- Ijuin, T. Phosphoinositide phosphatases in cancer cell dynamics—Beyond PI3K and PTEN. Semin. Cancer Biol. 2019, 59, 50–65. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, D. PI(3)king apart PTEN’s role in cancer. Clin. Cancer Res. 2010, 16, 4325–4330. [Google Scholar] [CrossRef]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.M.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacoke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Marsh, D.J.; Dahia, P.L.; Caron, S.; Kum, J.B.; Frayling, I.M.; Tomlinson, I.P.; Hughes, K.S.; Eeles, R.A.; Hodgson, S.V.; Murday, V.A.; et al. Germline PTEN mutations in Cowden syndrome-like families. J. Med. Genet. 1998, 35, 881–885. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Liang, F.; Jia, Z.-L.; Song, S.-T.; Jiang, Z.-F. PTEN mutation, methylation and expression in breast cancer patients. Oncol. Lett. 2013, 6, 161–168. [Google Scholar] [CrossRef]
- Li, S.; Shen, Y.; Wang, M.; Yang, J.; Lv, M.; Li, P.; Chen, Z.; Yang, J. Loss of PTEN expression in breast cancer: Association with clinicopathological characteristics and prognosis. Oncotarget 2017, 8, 32043–32054. [Google Scholar] [CrossRef]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef]
- Suzuki, A.; de la Pompa, J.L.; Stambolic, V.; Elia, A.J.; Sasaki, T.; Barrantes, I.d.B.; Ho, A.; Wakeham, A.; ltie, A.; Khoo, W.; et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr. Biol. 1998, 8, 1169–1178. [Google Scholar] [CrossRef]
- Cristofano, A.D.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Stambolic, V.; Tsao, M.-S.; Macpherson, D.; Suzuki, A.; Chapman, W.B.; Mak, T.W. High Incidence of Breast and Endometrial Neoplasia Resembling Human Cowden Syndrome in pten+/− Mice. Cancer Res. 2000, 60, 3605. [Google Scholar]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Post, K.L.; Belmadani, M.; Ganguly, P.; Meili, F.; Dingwall, R.; McDiarmid, T.A.; Meyers, W.M.; Herrington, C.; Young, B.P.; Callaghan, D.B.; et al. Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat. Commun. 2020, 11, 2073. [Google Scholar] [CrossRef]
- Heinrich, F.; Chakravarthy, S.; Nanda, H.; Papa, A.; Pandolfi, P.P.; Ross, A.H.; Harishchandra, R.K.; Gericke, A.; Lösche, M. The PTEN Tumor Suppressor Forms Homodimers in Solution. Structure 2015, 23, 1952–1957. [Google Scholar] [CrossRef]
- Li, G.; Robinson, G.W.; Lesche, R.; Martinez-Diaz, H.; Jiang, Z.; Rozengurt, N.; Wagner, K.-U.; Wu, D.-C.; Lane, T.F.; Liu, X.; et al. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development 2002, 129, 4159. [Google Scholar]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.-L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef]
- Malek, M.; Kielkowska, A.; Chessa, T.; Anderson, K.E.; Barneda, D.; Pir, P.; Nakanishi, H.; Eguchi, S.; Koizumi, A.; Sasaki, J.; et al. PTEN Regulates PI(3,4)P(2) Signaling Downstream of Class I PI3K. Mol. Cell 2017, 68, 566–580.e510. [Google Scholar] [CrossRef]
- deGraffenried, L.A.; Fulcher, L.; Friedrichs, W.E.; Grünwald, V.; Ray, R.B.; Hidalgo, M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann. Oncol. 2004, 15, 1510–1516. [Google Scholar] [CrossRef]
- Yuan, Y.; Wen, W.; Yost, S.E.; Xing, Q.; Yan, J.; Han, E.S.; Mortimer, J.; Yim, J.H. Combination therapy with BYL719 and LEE011 is synergistic and causes a greater suppression of p-S6 in triple negative breast cancer. Sci. Rep. 2019, 9, 7509. [Google Scholar] [CrossRef]
- Wee, S.; Wiederschain, D.; Maira, S.-M.; Loo, A.; Miller, C.; deBeaumont, R.; Stegmeier, F.; Yao, Y.-M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057. [Google Scholar] [CrossRef]
- Hosford, S.R.; Dillon, L.M.; Bouley, S.J.; Rosati, R.; Yang, W.; Chen, V.S.; Demidenko, E.; Morra, R.P., Jr.; Miller, T.W. Combined Inhibition of Both p110α and p110β Isoforms of Phosphatidylinositol 3-Kinase Is Required for Sustained Therapeutic Effect in PTEN-Deficient, ER(+) Breast Cancer. Clin. Cancer Res. 2017, 23, 2795–2805. [Google Scholar] [CrossRef]
- Chen, M.-L.; Xu, P.-Z.; Peng, X.-d.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006, 20, 1569–1574. [Google Scholar] [CrossRef]
- Xu, P.Z.; Chen, M.L.; Jeon, S.M.; Peng, X.d.; Hay, N. The effect Akt2 deletion on tumor development in Pten(+/−) mice. Oncogene 2012, 31, 518–526. [Google Scholar] [CrossRef]
- Chin, Y.R.; Yuan, X.; Balk, S.P.; Toker, A. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer Discov. 2014, 4, 945–955. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943. [Google Scholar] [CrossRef]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.H.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 1997, 94, 9052. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Whisstock, J.C.; Romero, S.; Gurung, R.; Nandurkar, H.; Ooms, L.M.; Bottomley, S.P.; Mitchell, C.A. The inositol polyphosphate 5-phosphatases and the apurinic/apyrimidinic base excision repair endonucleases share a common mechanism for catalysis. J. Biol. Chem. 2000, 275, 37055–37061. [Google Scholar] [CrossRef]
- Tsujishita, Y.; Guo, S.; Stolz, L.E.; York, J.D.; Hurley, J.H. Specificity Determinants in Phosphoinositide Dephosphorylation: Crystal Structure of an Archetypal Inositol Polyphosphate 5-Phosphatase. Cell 2001, 105, 379–389. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.-i.; Parsons, R.; Yamada, K.M. Inhibition of Cell Migration, Spreading, and Focal Adhesions by Tumor Suppressor PTEN. Science 1998, 280, 1614. [Google Scholar] [CrossRef]
- Wozniak, D.J.; Kajdacsy-Balla, A.; Macias, V.; Ball-Kell, S.; Zenner, M.L.; Bie, W.; Tyner, A.L. PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat. Commun. 2017, 8, 1508. [Google Scholar] [CrossRef]
- Tibarewal, P.; Zilidis, G.; Spinelli, L.; Schurch, N.; Maccario, H.; Gray, A.; Perera, N.M.; Davidson, L.; Barton, G.J.; Leslie, N.R. PTEN Protein Phosphatase Activity Correlates with Control of Gene Expression and Invasion, a Tumor-Suppressing Phenotype, But Not with AKT Activity. Sci. Signal. 2012, 5, ra18. [Google Scholar] [CrossRef]
- Yip, H.Y.K.; Chee, A.; Ang, C.-S.; Shin, S.-Y.; Ooms, L.M.; Mohammadi, Z.; Phillips, W.A.; Daly, R.J.; Cole, T.J.; Bronson, R.T.; et al. Control of Glucocorticoid Receptor Levels by PTEN Establishes a Failsafe Mechanism for Tumor Suppression. Mol. Cell 2020, 80, 279–295.e278. [Google Scholar] [CrossRef]
- Billcliff, P.G.; Lowe, M. Inositol lipid phosphatases in membrane trafficking and human disease. Biochem. J. 2014, 461, 159–175. [Google Scholar] [CrossRef]
- Osborn, D.P.S.; Pond, H.L.; Mazaheri, N.; Dejardin, J.; Munn, C.J.; Mushref, K.; Cauley, E.S.; Moroni, I.; Pasanisi, M.B.; Sellars, E.A.; et al. Mutations in INPP5K Cause a Form of Congenital Muscular Dystrophy Overlapping Marinesco-Sjögren Syndrome and Dystroglycanopathy. Am. J. Hum. Genet. 2017, 100, 537–545. [Google Scholar] [CrossRef]
- Wiessner, M.; Roos, A.; Munn, C.J.; Viswanathan, R.; Whyte, T.; Cox, D.; Schoser, B.; Sewry, C.; Roper, H.; Phadke, R.; et al. Mutations in INPP5K, Encoding a Phosphoinositide 5-Phosphatase, Cause Congenital Muscular Dystrophy with Cataracts and Mild Cognitive Impairment. Am. J. Hum. Genet. 2017, 100, 523–536. [Google Scholar] [CrossRef]
- Matzaris, M.; Jackson, S.P.; Laxminarayan, K.M.; Speed, C.J.; Mitchell, C.A. Identification and characterization of the phosphatidylinositol-(4, 5)-bisphosphate 5-phosphatase in human platelets. J. Biol. Chem. 1994, 269, 3397–3402. [Google Scholar]
- Jackson, S.P.; Schoenwaelder, S.M.; Matzaris, M.; Brown, S.; Mitchell, C.A. Phosphatidylinositol 3,4,5-trisphosphate is a substrate for the 75 kDa inositol polyphosphate 5-phosphatase and a novel 5-phosphatase which forms a complex with the p85/p110 form of phosphoinositide 3-kinase. EMBO J. 1995, 14, 4490–4500. [Google Scholar] [CrossRef]
- Schmid, A.C.; Wise, H.M.; Mitchell, C.A.; Nussbaum, R.; Woscholski, R. Type II phosphoinositide 5-phosphatases have unique sensitivities towards fatty acid composition and head group phosphorylation. FEBS Lett. 2004, 576, 9–13. [Google Scholar] [CrossRef]
- Kong, A.M.; Speed, C.J.; O’Malley, C.J.; Layton, M.J.; Meehan, T.; Loveland, K.L.; Cheema, S.; Ooms, L.M.; Mitchell, C.A. Cloning and characterization of a 72-kDa inositol-polyphosphate 5-phosphatase localized to the Golgi network. J. Biol. Chem. 2000, 275, 24052–24064. [Google Scholar] [CrossRef]
- Kisseleva, M.V.; Wilson, M.P.; Majerus, P.W. The isolation and characterization of a cDNA encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J. Biol. Chem. 2000, 275, 20110–20116. [Google Scholar] [CrossRef]
- Damen, J.E.; Liu, L.; Rosten, P.; Humphries, R.K.; Jefferson, A.B.; Majerus, P.W.; Krystal, G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA 1996, 93, 1689. [Google Scholar] [CrossRef]
- Lioubin, M.N.; Algate, P.A.; Tsai, S.; Carlberg, K.; Aebersold, A.; Rohrschneider, L.R. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev. 1996, 10, 1084–1095. [Google Scholar] [CrossRef]
- Taylor, V.; Wong, M.; Brandts, C.; Reilly, L.; Dean, N.M.; Cowsert, L.M.; Moodie, S.; Stokoe, D. 5′ phospholipid phosphatase SHIP-2 causes protein kinase B inactivation and cell cycle arrest in glioblastoma cells. Mol. Cell. Biol. 2000, 20, 6860–6871. [Google Scholar] [CrossRef]
- Ijuin, T.; Takenawa, T. SKIP negatively regulates insulin-induced GLUT4 translocation and membrane ruffle formation. Mol. Cell. Biol. 2003, 23, 1209–1220. [Google Scholar] [CrossRef]
- Ooms, L.M.; Fedele, C.G.; Astle, M.V.; Ivetac, I.; Cheung, V.; Pearson, R.B.; Layton, M.J.; Forrai, A.; Nandurkar, H.H.; Mitchell, C.A. The inositol polyphosphate 5-phosphatase, PIPP, is a novel regulator of phosphoinositide 3-kinase-dependent neurite elongation. Mol. Biol. Cell 2006, 17, 607–622. [Google Scholar] [CrossRef][Green Version]
- Mochizuki, Y.; Takenawa, T. Novel inositol polyphosphate 5-phosphatase localizes at membrane ruffles. J. Biol. Chem. 1999, 274, 36790–36795. [Google Scholar] [CrossRef]
- Laxminarayan, K.M.; Matzaris, M.; Speed, C.J.; Mitchell, C.A. Purification and characterization of a 43-kDa membrane-associated inositol polyphosphate 5-phosphatase from human placenta. J. Biol. Chem. 1993, 268, 4968–4974. [Google Scholar]
- Laxminarayan, K.M.; Chan, B.K.; Tetaz, T.; Bird, P.I.; Mitchell, C.A. Characterization of a cDNA encoding the 43-kDa membrane-associated inositol-polyphosphate 5-phosphatase. J. Biol. Chem. 1994, 269, 17305–17310. [Google Scholar]
- Ooms, L.M.; Binge, L.C.; Davies, E.M.; Rahman, P.; Conway, J.R.W.; Gurung, R.; Ferguson, D.T.; Papa, A.; Fedele, C.G.; Vieusseux, J.L.; et al. The Inositol Polyphosphate 5-Phosphatase PIPP Regulates AKT1-Dependent Breast Cancer Growth and Metastasis. Cancer Cell 2015, 28, 155–169. [Google Scholar] [CrossRef]
- Prasad, N.K.; Tandon, M.; Handa, A.; Moore, G.E.; Babbs, C.F.; Snyder, P.W.; Bose, S. High expression of obesity-linked phosphatase SHIP2 in invasive breast cancer correlates with reduced disease-free survival. Tumor Biol. 2008, 29, 330–341. [Google Scholar] [CrossRef]
- Ben-Chetrit, N.; Chetrit, D.; Russell, R.; Körner, C.; Mancini, M.; Abdul-Hai, A.; Itkin, T.; Carvalho, S.; Cohen-Dvashi, H.; Koestler, W.J.; et al. Synaptojanin 2 is a druggable mediator of metastasis and the gene is overexpressed and amplified in breast cancer. Sci. Signal. 2015, 8, ra7. [Google Scholar] [CrossRef]
- Bononi, A.; Pinton, P. Study of PTEN subcellular localization. Methods 2015, 77-78, 92–103. [Google Scholar] [CrossRef]
- Sharma, V.P.; Eddy, R.; Entenberg, D.; Kai, M.; Gertler, F.B.; Condeelis, J. Tks5 and SHIP2 regulate invadopodium maturation, but not initiation, in breast carcinoma cells. Curr. Biol. 2013, 23, 2079–2089. [Google Scholar] [CrossRef]
- Dyson, J.M.; O’Malley, C.J.; Becanovic, J.; Munday, A.D.; Berndt, M.C.; Coghill, I.D.; Nandurkar, H.H.; Ooms, L.M.; Mitchell, C.A. The SH2-containing inositol polyphosphate 5-phosphatase, SHIP-2, binds filamin and regulates submembraneous actin. J. Cell Biol. 2001, 155, 1065–1079. [Google Scholar] [CrossRef]
- Prasad, N.; Topping, R.S.; Decker, S.J. SH2-containing inositol 5′-phosphatase SHIP2 associates with the p130(Cas) adapter protein and regulates cellular adhesion and spreading. Mol. Cell. Biol. 2001, 21, 1416–1428. [Google Scholar] [CrossRef]
- Sleeman, M.W.; Wortley, K.E.; Lai, K.-M.V.; Gowen, L.C.; Kintner, J.; Kline, W.O.; Garcia, K.; Stitt, T.N.; Yancopoulos, G.D.; Wiegand, S.J.; et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 2005, 11, 199–205. [Google Scholar] [CrossRef]
- Kagawa, S.; Soeda, Y.; Ishihara, H.; Oya, T.; Sasahara, M.; Yaguchi, S.; Oshita, R.; Wada, T.; Tsuneki, H.; Sasaoka, T. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology 2008, 149, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Manji, S.S.M.; Williams, L.H.; Miller, K.A.; Ooms, L.M.; Bahlo, M.; Mitchell, C.A.; Dahl, H.-H.M. A mutation in synaptojanin 2 causes progressive hearing loss in the ENU-mutagenised mouse strain Mozart. PLoS ONE 2011, 6, e17607. [Google Scholar] [CrossRef]
- Gurung, R.; Tan, A.; Ooms, L.M.; McGrath, M.J.; Huysmans, R.D.; Munday, A.D.; Prescott, M.; Whisstock, J.C.; Mitchell, C.A. Identification of a novel domain in two mammalian inositol-polyphosphate 5-phosphatases that mediates membrane ruffle localization: The inositol 5-phosphatase SKIP localizes to the endoplasmic reticulum and translocates to membrane ruffles following epidermal growth factor stimulation. J. Biol. Chem. 2003, 278, 11376–11385. [Google Scholar] [CrossRef]
- Denley, A.; Gymnopoulos, M.; Kang, S.; Mitchell, C.; Vogt, P.K. Requirement of phosphatidylinositol(3,4,5)trisphosphate in phosphatidylinositol 3-kinase-induced oncogenic transformation. Mol. Cancer Res. 2009, 7, 1132–1138. [Google Scholar] [CrossRef]
- Iida, A.; Kurose, K.; Isobe, R.; Akiyama, F.; Sakamoto, G.; Yoshimoto, M.; Kasumi, F.; Nakamura, Y.; Emi, M. Mapping of a new target region of allelic loss to a 2-cM interval at 22q13.1 in primary breast cancer. Genes Chromosomes Cancer 1998, 21, 108–112. [Google Scholar] [CrossRef]
- Allione, F.; Eisinger, F.; Parc, P.; Noguchi, T.; Sobol, H.; Birnbaum, D. Loss of heterozygosity at loci from chromosome arm 22Q in human sporadic breast carcinomas. Int. J. Cancer 1998, 75, 181–186. [Google Scholar] [CrossRef]
- Takahashi, H.; Masuda, K.; Ando, T.; Kobayashi, T.; Honda, H. Prognostic predictor with multiple fuzzy neural models using expression profiles from DNA microarray for metastases of breast cancer. J. Biosci. Bioeng. 2004, 98, 193–199. [Google Scholar] [CrossRef]
- Gruvberger, S.; Ringnér, M.; Chen, Y.; Panavally, S.; Saal, L.H.; Borg, A.; Fernö, M.; Peterson, C.; Meltzer, P.S. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001, 61, 5979–5984. [Google Scholar]
- Liu, H.; Radisky, D.C.; Nelson, C.M.; Zhang, H.; Fata, J.E.; Roth, R.A.; Bissell, M.J. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc. Natl. Acad. Sci. USA 2006, 103, 4134–4139. [Google Scholar] [CrossRef]
- Yoeli-Lerner, M.; Yiu, G.K.; Rabinovitz, I.; Erhardt, P.; Jauliac, S.; Toker, A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol. Cell 2005, 20, 539–550. [Google Scholar] [CrossRef]
- Yoeli-Lerner, M.; Chin, Y.R.; Hansen, C.K.; Toker, A. The akt/pkb and gsk-3β signaling pathway regulates cell migration through the nfat1 transcription factor. Mol. Cancer Res. 2009, 7, 425–432. [Google Scholar] [CrossRef]
- Chabottaux, V.; Noel, A. Breast cancer progression: Insights into multifaceted matrix metalloproteinases. Clin. Exp. Metastasis 2007, 24, 647–656. [Google Scholar] [CrossRef]
- Jezierska, A.; Motyl, T. Matrix metalloproteinase-2 involvement in breast cancer progression: A mini-review. Med. Sci. Monit. 2009, 15, Ra32–Ra40. [Google Scholar]
- Park, B.-K.; Zeng, X.; Glazer, R.I. Akt1 Induces Extracellular Matrix Invasion and Matrix Metalloproteinase-2 Activity in Mouse Mammary Epithelial Cells. Cancer Res. 2001, 61, 7647. [Google Scholar]
- Santi, S.A.; Lee, H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. Cell Physiol. 2010, 298, C580–C591. [Google Scholar] [CrossRef]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef]
- Chew, C.L.; Lunardi, A.; Gulluni, F.; Ruan, D.T.; Chen, M.; Salmena, L.; Nishino, M.; Papa, A.; Ng, C.; Fung, J.; et al. In Vivo Role of INPP4B in Tumor and Metastasis Suppression through Regulation of PI3K-AKT Signaling at Endosomes. Cancer Discov. 2015, 5, 740–751. [Google Scholar] [CrossRef]
- Kofuji, S.; Kimura, H.; Nakanishi, H.; Nanjo, H.; Takasuga, S.; Liu, H.; Eguchi, S.; Nakamura, R.; Itoh, R.; Ueno, N.; et al. INPP4B Is a PtdIns(3,4,5)P3 Phosphatase That Can Act as a Tumor Suppressor. Cancer Discov. 2015, 5, 730–739. [Google Scholar] [CrossRef]
- Ye, Y.; Jin, L.; Wilmott, J.S.; Hu, W.L.; Yosufi, B.; Thorne, R.F.; Liu, T.; Rizos, H.; Yan, X.G.; Dong, L.; et al. PI(4,5)P2 5-phosphatase A regulates PI3K/Akt signalling and has a tumour suppressive role in human melanoma. Nat. Commun. 2013, 4, 1508. [Google Scholar] [CrossRef]
- Lin, C.; Liu, A.; Zhu, J.; Zhang, X.; Wu, G.; Ren, P.; Wu, J.; Li, M.; Li, J.; Song, L. miR-508 sustains phosphoinositide signalling and promotes aggressive phenotype of oesophageal squamous cell carcinoma. Nat. Commun. 2014, 5, 4620. [Google Scholar] [CrossRef]
- Rohrschneider, L.R.; Fuller, J.F.; Wolf, I.; Liu, Y.; Lucas, D.M. Structure, function, and biology of SHIP proteins. Genes Dev. 2000, 14, 505–520. [Google Scholar]
- Mercurio, F.A.; Di Natale, C.; Pirone, L.; Iannitti, R.; Marasco, D.; Pedone, E.M.; Palumbo, R.; Leone, M. The Sam-Sam interaction between Ship2 and the EphA2 receptor: Design and analysis of peptide inhibitors. Sci. Rep. 2017, 7, 17474. [Google Scholar] [CrossRef]
- Helgason, C.D.; Damen, J.E.; Rosten, P.; Grewal, R.; Sorensen, P.; Chappel, S.M.; Borowski, A.; Jirik, F.; Krystal, G.; Humphries, R.K. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998, 12, 1610–1620. [Google Scholar] [CrossRef]
- Liu, Q.; Sasaki, T.; Kozieradzki, I.; Wakeham, A.; Itie, A.; Dumont, D.J.; Penninger, J.M. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999, 13, 786–791. [Google Scholar] [CrossRef]
- Niemann, C.U.; Wiestner, A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin. Cancer Biol. 2013, 23, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.M.; Yoshida, H.; Komura, S.; Ohishi, N.; Pan, L.; Shigeno, K.; Hanamura, I.; Miura, K.; Iida, S.; Ueda, R.; et al. Possible dominant-negative mutation of the SHIP gene in acute myeloid leukemia. Leukemia 2003, 17, 1–8. [Google Scholar] [CrossRef]
- Brooks, R.; Fuhler, G.M.; Iyer, S.; Smith, M.J.; Park, M.Y.; Paraiso, K.H.; Engelman, R.W.; Kerr, W.G. SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J. Immunol. 2010, 184, 3582–3589. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C.; Toker, A. Direct Regulation of the Akt Proto-Oncogene Product by Phosphatidylinositol-3,4-bisphosphate. Science 1997, 275, 665. [Google Scholar] [CrossRef]
- Klippel, A.; Kavanaugh, W.M.; Pot, D.; Williams, L.T. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol. Cell. Biol. 1997, 17, 338–344. [Google Scholar] [CrossRef]
- Clément, S.; Krause, U.; Desmedt, F.; Tanti, J.-F.; Behrends, J.; Pesesse, X.; Sasaki, T.; Penninger, J.; Doherty, M.; Malaisse, W.; et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 2001, 409, 92–97. [Google Scholar] [CrossRef]
- Prasad, N.K.; Tandon, M.; Badve, S.; Snyder, P.W.; Nakshatri, H. Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis 2007, 29, 25–34. [Google Scholar] [CrossRef]
- Zhou, J.; Di, M.; Han, H. Upregulation of SHIP2 participates in the development of breast cancer via promoting Wnt/β-catenin signaling. Onco Targets Ther. 2019, 12, 7067–7077. [Google Scholar] [CrossRef]
- Han, H.; Wang, B.; Zhao, J.; Xu, G.; Wang, X.; Cao, F. Analysis of SHIP2 Expression and its Correlation with VEGF and HER-2 Expression in Breast Cancer. J. Clin. Exp. Pathol. 2017, 7. [Google Scholar] [CrossRef]
- Fu, C.-H.; Lin, R.-J.; Yu, J.; Chang, W.-W.; Liao, G.-S.; Chang, W.-Y.; Tseng, L.-M.; Tsai, Y.-F.; Yu, J.-C.; Yu, A.L. A Novel Oncogenic Role of Inositol Phosphatase SHIP2 in ER-Negative Breast Cancer Stem Cells: Involvement of JNK/Vimentin Activation. Stem Cells 2014, 32, 2048–2060. [Google Scholar] [CrossRef]
- Prasad, N.K.; Decker, S.J. SH2-containing 5′-inositol phosphatase, SHIP2, regulates cytoskeleton organization and ligand-dependent down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 13129–13136. [Google Scholar] [CrossRef]
- Ghosh, S.; Scozzaro, S.; Ramos, A.R.; Delcambre, S.; Chevalier, C.; Krejci, P.; Erneux, C. Inhibition of SHIP2 activity inhibits cell migration and could prevent metastasis in breast cancer cells. J. Cell Sci. 2018, 131, jcs216408. [Google Scholar] [CrossRef]
- Prasad, N.K. SHIP2 phosphoinositol phosphatase positively regulates EGFR-Akt pathway, CXCR4 expression, and cell migration in MDA-MB-231 breast cancer cells. Int. J. Oncol. 2009, 34, 97–105. [Google Scholar] [CrossRef]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef]
- Hoshino, D.; Jourquin, J.; Emmons, S.W.; Miller, T.; Goldgof, M.; Costello, K.; Tyson, D.R.; Brown, B.; Lu, Y.; Prasad, N.K.; et al. Network analysis of the focal adhesion to invadopodia transition identifies a PI3K-PKCα invasive signaling axis. Sci.Signal. 2012, 5, ra66. [Google Scholar] [CrossRef]
- Rajadurai, C.V.; Havrylov, S.; Coelho, P.P.; Ratcliffe, C.D.H.; Zaoui, K.; Huang, B.H.; Monast, A.; Chughtai, N.; Sangwan, V.; Gertler, F.B.; et al. 5′-Inositol phosphatase SHIP2 recruits Mena to stabilize invadopodia for cancer cell invasion. J. Cell Biol. 2016, 214, 719–734. [Google Scholar] [CrossRef]
- Ye, Y.; Ge, Y.M.; Xiao, M.M.; Guo, L.M.; Li, Q.; Hao, J.Q.; Da, J.; Hu, W.L.; Zhang, X.D.; Xu, J.; et al. Suppression of SHIP2 contributes to tumorigenesis and proliferation of gastric cancer cells via activation of Akt. J. Gastroenterol. 2016, 51, 230–240. [Google Scholar] [CrossRef]
- Elong Edimo, W.; Ghosh, S.; Derua, R.; Janssens, V.; Waelkens, E.; Vanderwinden, J.-M.; Robe, P.; Erneux, C. SHIP2 controls plasma membrane PI(4,5)P2 thereby participating in the control of cell migration in 1321 N1 glioblastoma cells. J. Cell Sci. 2016, 129, 1101. [Google Scholar] [CrossRef]
- Vandenbroere, I.; Paternotte, N.; Dumont, J.E.; Erneux, C.; Pirson, I. The c-Cbl-associated protein and c-Cbl are two new partners of the SH2-containing inositol polyphosphate 5-phosphatase SHIP2. Biochem. Biophys. Res. Commun. 2003, 300, 494–500. [Google Scholar] [CrossRef]
- Paternotte, N.; Zhang, J.; Vandenbroere, I.; Backers, K.; Blero, D.; Kioka, N.; Vanderwinden, J.M.; Pirson, I.; Erneux, C. SHIP2 interaction with the cytoskeletal protein Vinexin. FEBS J. 2005, 272, 6052–6066. [Google Scholar] [CrossRef]
- Koch, A.; Mancini, A.; El Bounkari, O.; Tamura, T. The SH2-domian-containing inositol 5-phosphatase (SHIP)-2 binds to c-Met directly via tyrosine residue 1356 and involves hepatocyte growth factor (HGF)-induced lamellipodium formation, cell scattering and cell spreading. Oncogene 2005, 24, 3436–3447. [Google Scholar] [CrossRef]
- Suwa, A.; Kurama, T.; Shimokawa, T. SHIP2 and its involvement in various diseases. Expert Opin Ther. Targets 2010, 14, 727–737. [Google Scholar] [CrossRef]
- Kato, K.; Yazawa, T.; Taki, K.; Mori, K.; Wang, S.; Nishioka, T.; Hamaguchi, T.; Itoh, T.; Takenawa, T.; Kataoka, C.; et al. The inositol 5-phosphatase SHIP2 is an effector of RhoA and is involved in cell polarity and migration. Mol Biol Cell 2012, 23, 2593–2604. [Google Scholar] [CrossRef]
- Xie, J.; Onnockx, S.; Vandenbroere, I.; Degraef, C.; Erneux, C.; Pirson, I. The docking properties of SHIP2 influence both JIP1 tyrosine phosphorylation and JNK activity. Cell Signal 2008, 20, 1432–1441. [Google Scholar] [CrossRef]
- Cremona, O.; Di Paolo, G.; Wenk, M.R.; Lüthi, A.; Kim, W.T.; Takei, K.; Daniell, L.; Nemoto, Y.; Shears, S.B.; Flavell, R.A.; et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 1999, 99, 179–188. [Google Scholar] [CrossRef]
- Rusk, N.; Le, P.U.; Mariggio, S.; Guay, G.; Lurisci, C.; Nabi, I.R.; Corda, D.; Symons, M. Synaptojanin 2 functions at an early step of clathrin-mediated endocytosis. Curr. Biol. 2003, 13, 659–663. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Gurung, R.; Kong, A.M.; Dyson, J.M.; Tan, A.; Ooms, L.M. Inositol polyphosphate 5-phosphatases: Lipid phosphatases with flair. Iubmb Life 2002, 53, 25–36. [Google Scholar] [CrossRef]
- Ramjaun, A.R.; McPherson, P. Tissue-specific Alternative Splicing Generates Two Synaptojanin Isoforms with Differential Membrane Binding Properties. J. Biol. Chem. 1996, 271, 24856–24861. [Google Scholar] [CrossRef]
- Nemoto, Y.; Arribas, M.; Haffner, C.; DeCamilli, P. Synaptojanin 2, a novel synaptojanin isoform with a distinct targeting domain and expression pattern. J. Biol. Chem. 1997, 272, 30817–30821. [Google Scholar] [CrossRef]
- Chuang, Y.-y.; Tran, N.L.; Rusk, N.; Nakada, M.; Berens, M.E.; Symons, M. Role of Synaptojanin 2 in Glioma Cell Migration and Invasion. Cancer Res. 2004, 64, 8271. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Ma, K.; Cheung, S.M.; Marshall, A.J.; Duronio, V. PI(3,4,5)P3 and PI(3,4)P2 levels correlate with PKB/akt phosphorylation at Thr308 and Ser473, respectively; PI(3,4)P2 levels determine PKB activity. Cell. Signal. 2008, 20, 684–694. [Google Scholar] [CrossRef]
- Kerr, W.G. Inhibitor and activator: Dual functions for SHIP in immunity and cancer. Ann. N. Y. Acad. Sci. 2011, 1217, 1–17. [Google Scholar] [CrossRef]
- Thapa, N.; Choi, S.; Tan, X.; Wise, T.; Anderson, R.A. Phosphatidylinositol phosphate 5-kinase Iγ and phosphoinositide 3-kinase/Akt signaling couple to promote oncogenic growth. J. Biol. Chem. 2015, 290, 18843–18854. [Google Scholar] [CrossRef]
- Davies, E.M.; Kong, A.M.; Tan, A.; Gurung, R.; Sriratana, A.; Bukczynska, P.E.; Ooms, L.M.; McLean, C.A.; Tiganis, T.; Mitchell, C.A. Differential SKIP expression in PTEN-deficient glioblastoma regulates cellular proliferation and migration. Oncogene 2015, 34, 3711–3727. [Google Scholar] [CrossRef]
- Ijuin, T.; Mochizuki, Y.; Fukami, K.; Funaki, M.; Asano, T.; Takenawa, T. Identification and characterization of a novel inositol polyphosphate 5-phosphatase. J. Biol. Chem. 2000, 275, 10870–10875. [Google Scholar] [CrossRef]
- Ijuin, T.; Yu, Y.E.; Mizutani, K.; Pao, A.; Tateya, S.; Tamori, Y.; Bradley, A.; Takenawa, T. Increased insulin action in SKIP heterozygous knockout mice. Mol. Cell. Biol. 2008, 28, 5184–5195. [Google Scholar] [CrossRef]


{kind=link}
| PtdIns(3,4,5)P3 Phosphatase | Substrate Specificity | Cellular Localisation | Role in Breast Cancer | Mouse Models |
|---|---|---|---|---|
| PTEN | PtdIns(3,4,5)P3 to PtdIns(4,5)P2; PtdIns(3,4)P2 to PtdIns(4)P [48] | Plasma membrane, nucleus, ER and mitochondrial-associated membranes [84] | Tumour suppressor | Pten-null Severe developmental defects; embryonically lethal [39] Pten+/− mice de novo tumours in multiple tissues including prostate, skin, colon adrenal and mammary gland [40] Mammary epithelial cell-specific deletion of Pten Precocious lobulo-alveolar development; excessive ductal branching; high frequency of mammary tumour formation [45] |
| PIPP | PtdIns(3,4,5)P3 to PtdIns(3,4)P2; PtdIns(4,5)P2 to PtdIns(4)P [77,78] | Ruffling membranes and cytosol [78] | Anti-tumourigenic; pro-migratory | Pipp-null Normal mammary gland development; no evidence of de novo mammary tumour formation [81] PyMT;Pipp−/− mice Enhanced mammary tumour formation; decreased number of lung metastases [81] |
| SHIP2 | PtdIns(3,4,5)P3 to PtdIns(3,4)P2; PtdIns(4,5)P2 to PtdIns(4)P [75] | Invadopodia, focal contacts, lamellipodia, membrane ruffles [85,86,87] | Pro-tumourigenic | Ship2-null Resistant to high-fat diet induced obesity; increased insulin sensitivity and glucose tolerance [88] Transgenic overexpression Increased body weight; reduced glucose tolerance [89] |
| SYNJ2 | PtdIns(3,4,5)P3 to PtdIns(3,4)P2; PtdIns(4,5)P2 to PtdIns(4)P (higher specificity for PtdIns(4,5)P2) [70] | Invadopodia, lamellipodia, membrane ruffles [83] | Pro-tumourigenic | ENU-induced mutation in catalytic domain Loss of cochlear hair cells; cochlea degeneration [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csolle, M.P.; Ooms, L.M.; Papa, A.; Mitchell, C.A. PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9189. https://doi.org/10.3390/ijms21239189
Csolle MP, Ooms LM, Papa A, Mitchell CA. PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer. International Journal of Molecular Sciences. 2020; 21(23):9189. https://doi.org/10.3390/ijms21239189
Chicago/Turabian StyleCsolle, Mariah P., Lisa M. Ooms, Antonella Papa, and Christina A. Mitchell. 2020. "PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer" International Journal of Molecular Sciences 21, no. 23: 9189. https://doi.org/10.3390/ijms21239189
APA StyleCsolle, M. P., Ooms, L. M., Papa, A., & Mitchell, C. A. (2020). PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer. International Journal of Molecular Sciences, 21(23), 9189. https://doi.org/10.3390/ijms21239189