1. Introduction
A wound is defined as a fault or a break of the skin caused by physical, chemical, and thermal injury [
1]. Subsequent wound infections and inflammation caused by various bacterial strains often lead to challenging complications for both personal and public health during wound care management [
2]. The dressing is an important component contributing to wound healing in the shortest possible time and to protection against bacterial infection, with minimal pain and discomfort to the patients, leading to tissue repair and regeneration [
3,
4].
Hydrogel based dressings prepared from natural and synthetic biocompatible polymers can prevent microbial infection, absorb large wound exudates, and can be used as vehicles for the simultaneous release of various anti-inflammatory or analgesic drugs to the wound site. Besides, hydrogel wound dressings will not stick to the wound, offering further benefits [
5]. Hydrogels are 3D cross-linked networks having high water retention capacity obtained through various techniques, including radiation cross-linking [
6,
7], freeze-thawing cycles [
8,
9], and chemical synthesis [
10].
The radiation cross-linking is a well-established technique for developing sterile hydrogel wound dressings. The e-beam synthesis of the hydrogel gives homogeneity in the hydrogel network structure, while the degree of cross-linking, which strongly determines the extent of swelling, can be easily controlled by varying the absorbed dose [
11,
12].
Similar to other radiation technologies, the e-beam radiation initiates chemical reactions in dilute or semi-dilute aqueous solutions which give rise to a series of reactive species as the main results of the water radiolysis. Of these, radical species and molecular products as
, HO•, H•, H
2, H
2O
2, and H
3O
+, with different reactivities are produced. In deoxygenated or Ar-saturated polymers solution, the hydroxyl radical (HO•) and
presents the highest chemical yields [
13]. The HO• and H• radicals react rapidly with polymer chains through hydrogen abstraction which produces several polymer macroradicals depending on the initial concentration of the polymers. The resulted macroradicals participate in intra-and intermolecular free-radical recombination reactions, finally forming a crosslinked polymeric networks with stable and permanent structure [
14].
Several natural and synthetic polymers can be used for biomedical hydrogels production designed for wound treatments. Recently, biopolymers based wound dressings have been widely used, among them being chitin/chitosan [
15,
16,
17], collagen [
18], cellulose [
19], and gelatin [
20] due to their biocompatibility, biodegradability, nontoxic, analgesic, and moisture retention capacity. Chitosan (CS) is a derivative of chitin (poly-
N acetyl glucosamine), which is the second-most abundant biopolymer after cellulose [
21]. CS and its derivatives have been widely used as a natural source for hydrogels in many fields, including wound dressing [
22,
23], tissue engineering [
24], drug delivery applications [
25], and gene delivery [
26]. Poly(
N-vinyl pyrrolidone) (PVP) is a linear synthetic polymer, is non-toxic and biocompatible, and it has been used as the main component of temporary skin covers or wound dressing [
27].
The miscibility of chitosan and PVP has been reported as a result of the interaction between the C = O groups of the pyrrolidone rings of PVP and the amino and OH groups of CS, by forming H-bonds and thus producing material with novel characteristic [
28]. Gamma-irradiation of CS-PVP pH-sensitive hydrogels was reported by Dergunov et al. [
29]. In that study, the adsorption of bovine serum albumin in CS-PVP hydrogels and their release behavior have been investigated and it has been found that the adsorption capacity of hydrogels increases with increasing CS content in the gel system. Risbud et al. developed a pH-sensitive CS-PVP hydrogel for the controlled release of antibiotics in the gastric environment [
30].
The interaction of CS, PVP, and polyethylene glycol (PEG) has been reported by several groups in the last decades. Mahmud et al. reported the characterization of hydrogels produced from CS and PVP via γ-radiation and evaluated the effect of PEG on hydrogels in terms of cross-linking density, gel fraction, swelling degree, syneresis effect at a different temperature, and morphological structure [
31]. Also, Das et al. [
32] showed that the presence of PEG in CS-PVP hydrogels increases the release rate of active ingredients such as drugs embedded in the hydrogel networks. Li et al. investigated the physical and functional properties of CS films prepared with different content of PVP and poly(ethylene oxide) (PEO) and showed that the blending of CS-PVP does not affect the physical and chemical properties of CS significantly [
33]. Rasool et al. prepared CS-PVP-Poly(acrylic acid) (PAA) for the controlled release of Ag-sulfadiazine and demonstrated that such polymeric systems can successfully be used as wound healing and wound dressing for drug delivery [
34].
Mozalewska et al. [
35] developed a hydrogel wound dressing based on chitosan-PVP-agar by radiation-initiated cross-linking. The studies showed antimicrobial character of the CS-based hydrogel towards Gram-positive bacterial strain. Zhao et al. prepared hydrogels using carboxymethylated chitosan (CM-chitosan) by e-beam irradiation at room temperature and studied the mechanical properties, gel fraction, and swelling behavior [
36].
The present work reports on the preparation and evaluation of CS-PVP-PEG-PAA or CS-PVP-PEO-Poly(lactic acid) (PAL) hydrogels via e-beam cross-linking. To the best of our knowledge, no study has been carried out on such hydrogel quaternary system to be used as a wet dressing for rapid healing and pain release of infected skin wounds and at the same time to be used as release vehicles for anti-inflammatory drugs as is ibuprofen.
The hydrogel properties were evaluated following sol-gel analysis, swelling studies, moisture retention capability, water vapor transmission rate, and network parameters, as well as FTIR and XPS analyses showed the efficient interaction of the hydrogel components upon irradiation. Further, the synthesized hydrogel was explored for its encapsulation efficiency, drug loading capacity and in vitro release of ibuprofen (IBU). These novel hydrogels showed potential for use in biomedical applications, particularly as wound healing dressings.
2. Results and Discussion
2.1. The Reaction Mechanism of CS-PVP-PEG-PAA Hydrogels
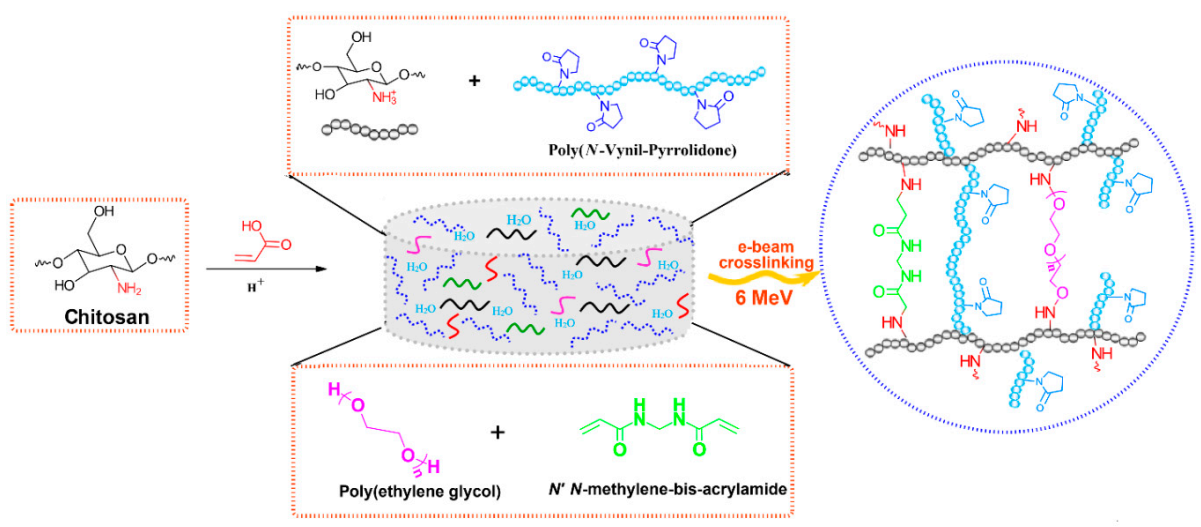
In
Scheme 1 it is presented the reaction mechanism of e-beam radiation cross-linking of CS-PVP-PEG-PAA hydrogels. High absorbed dose and dose rate generated throughout e-beam irradiation process of complex aqueous polymers solutions lead to the formation of considerable amounts of macroradicals which subsequently participate in radical recombination reactions. As a result of the recombination reactions, new crosslinked bonds within polymers macroradicals are generated, forming a new and more stabilized and compact hydrogel structure. The reaction mechanism describes the structures of substrates and the resulted structure of the e-beam crosslinked hydrogel.
2.2. Sol-Gel Analysis
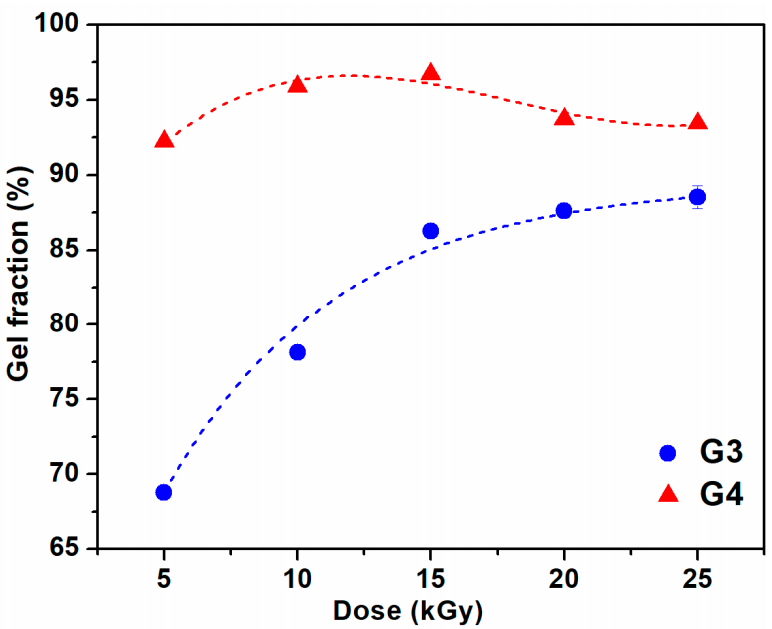
The variation of gel fraction of G3 and G4 hydrogels synthesized by e-beam cross-linking at various doses is illustrated in
Figure 1. The result indicates that the gel fraction of G3 hydrogels constantly increased with absorbed dose and reaches a maximum value of 87% at the dose of 25 kGy. For this composition, lower doses below 15 kGy lead to a hydrogel with reduced gel content, below 85%. In contrast, for the G4 composition even at doses below 15 kGy, the gel content reached above 90%, having a maximum of 96% at 15 kGy. Above 15 kGy, the G4 hydrogels present a slight decrease of the gel fraction which could be correlated with the degradation of the hydrogel structure which becomes more dominant than cross-linking at high doses. Previous studies on CS-PVP hydrogels reported a gel fraction of about 80% for hydrogels prepared from chitosan and PEG diacrylate (PEGDA) [
37] and PVP-CM-chitosan hydrogels [
38].
In our case, the
has a value of 0.28 for G3 hydrogel, which suggests that cross-linking processes are predominant. The higher
value of 0.51 obtained for G4 hydrogel is indicating, besides the above-mentioned cross-linking processes, a moderate contribution of chain scission processes. The
ratios,
and
are presented in
Table 1. Similar values were obtained for hydrogel dressings based on PVP/chitosan/agar [
35] and PVP/PAA [
39].
Other significant parameters characterizing the changes induced by ionizing radiation in the polymer are the radiation yield of the cross-linking, G(X), which provides information on the amount of new bonds formed per unit of absorbed energy, and respectively the radiation yield of chain scission, G(S), which counts the number of broken bonds in the backbone of the polymer per unit of absorbed dose. Both processes occur simultaneously and their yields determine the final results of irradiation [
40]. The G(X) and G(S) (mol/J) values can be calculated with the following equations:
where
is the number average molecular weight between two successive crosslinks (kg/mol), c is the polymer concentration in irradiated solution (g/L), D is the absorbed dose (Gy) and
is the solution density (kg/m
3). The G(X) and G(S) values calculated for G3 and G4 hydrogels are presented in
Table 2.
The calculated G(X) values for G3 hydrogels ranged between 0.17–0.23 µmol/J and increased with the absorbed dose, at the same time were considerably higher than G(S) for all radiation conditions. For G4 hydrogels, systematically higher values are obtained for all samples concerning G3, and the proportion between cross-linking and degradation yield was found to be almost equal, G(X) ≈ G(S). This has been attributed to the fact that cross-linking and chain scission processes compete with each other and the final reaction will continue from cross-linking to degradation.
The maximum value of G(X) was found to be 1.22 µmol/J and increased with absorbed dose. In the same trend, also G(S) has increased with absorbed dose, however, their values were lower than correspondent G(X). The G(X) and G(S) values presented in
Table 2 are close with other results related to the degradation of chitosan via γ-irradiation, where G(S) = 1.67 µmol/J [
41] and irradiation of chitosan in solid state (0.6 µmol/J) or aqueous solution (0.19 µmol/J) [
42].
2.3. Swelling Degree and Degradation Testing
The outstanding property of hydrogels for wound dressings is their water holding capacity assuring at the same time a moist wound environment beneficial to the healing. Since the blood contains about 90% water, and the excellent swelling properties of the hydrogels make the gel to absorb a large amount of water from the blood and stop bleeding [
17]. The absorption property of the hydrogels is also influenced by the nature of the polymers from which the hydrogel is prepared, such as chitosan, which swells readily in biological fluids.
The maximum swelling capacity of G3 and G4 hydrogels in phosphate buffer saline (PBS, pH = 7.4) and different temperature conditions, similar to normal body temperature (37 °C) and in conditions of body hyperthermia (39–41) °C is shown in
Figure 2a,b.
A fast swelling of at least 500% in the first 3 h is observed for all the prepared hydrogels, pointing out towards their porous structure, while at later stages slower modification of swelling is noticed, until the maximum capacity is attained (see also
Supplementary Materials Figure S1). The swelling degree (SD%) reached a value above 1800% (PBS, 37 °C) for the G3 hydrogel obtained with 15 kGy. A lower value of 1300% was obtained for G4 hydrogel. For the formulation of G3 hydrogel we used PEG with lower molecular weight, in the case of G4 hydrogels, we used PEO with higher molecular weight, which most probably produces an increase in the viscosity of the polymeric system, but also an increase in the cross-linking density [
43]. This behavior is also supported by the results presented in the network studies, where it was shown that the cross-linking density was considerably higher. Similar trends were found for hydrogels based on collagen/PVP/PAA/PEO [
44].
Regarding the evolution of SD% with absorbed dose and temperature, this parameter decreased as absorbed dose increased. The reduction in swelling ability is caused by the formation of a cross-linked structure more tightly folded upon irradiation with a higher dose. When the degree of cross-linking increases significantly, the macromolecular network of the hydrogel becomes denser, which prevents the diffusion of the solvent molecules from the swelling medium. When the temperature of the swelling has increased in simulated hyperthermia conditions, a reduction of the SD% was observed by a percentage of 10% at 41 °C.
Healthy and healing skin has a slightly acidic pH (5.5–6.5), while in contrast, the infected chronic wounds have a pH ranging between 7.2 and 9 due to the alkaline secondary products that appeared from the proliferation of bacterial colonies [
45]. Therefore, it is important to test the swelling behavior of G3 and G4 hydrogels as a function of pH (5.4–9.4). The results obtained at 37 °C are presented in
Figure 2c,d. In this study, it was observed that the G3 and G4 hydrogels exhibited almost the same SD% value at the pH = 5.4, 6.4, and 9.4, having a maximum swelling of 1200%, respectively 850% for the hydrogels prepared at 15 kGy (see also
Supplementary Materials, Figure S2 for the time evolution of swelling at various pH). For both types of hydrogels, it was observed a higher SD% of 1800 % (G3), respectively 1300% (G4) when the swelling media had pH = 7.4. Since the hydrogels contain both NH
2 and COOH moieties derived from CS, NMBA, and acrylic and lactic acids used for CS solubilization, these functional groups can be found in a protonated/deprotonated state. It is well established that hydrogels with a high concentration of charged ionic groups will swell considerably due to osmosis and charge repulsions [
46]. When pH > 9, the screening effect of Na+ contained in the swelling environment hinders the SD% of hydrogels [
47].
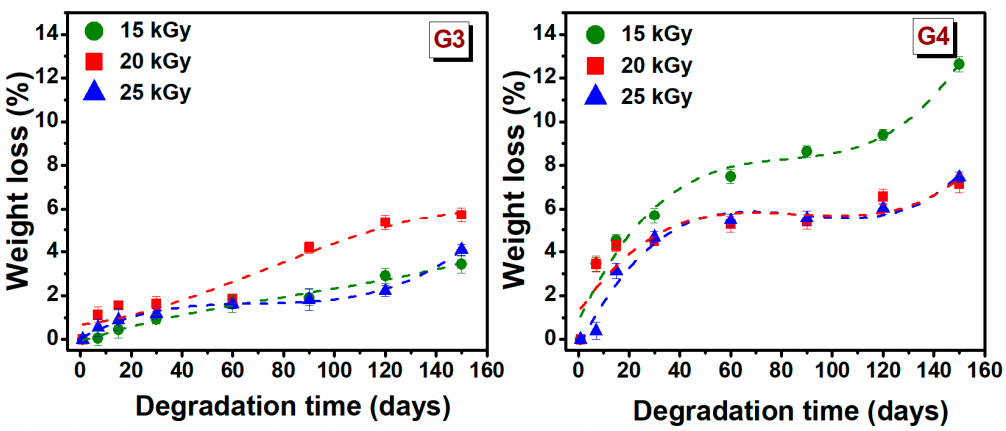
The degradation testing of hydrogels, during incubation for 150 days, based on the weight loss of the material, was monitored in PBS (pH = 7.4) at 37 °C. Degradation testing can help determine the efficiency of cross-linking and the stability of hydrogel. The weight loss (%) of hydrogels depends on the initial concentration of polymer solution, molecular weight, crosslinking agents and swelling properties [
48]. The weight loss of G3 and G4 hydrogels at a specific time point is shown in
Figure 3.
The weight loss for G3 hydrogels is significantly lower than that of the G4 hydrogels. In the first 30 days, the degradation percentage was 1.2 ± 0.4% for G3 hydrogels, respectively 5 ± 0.6% for the G4 hydrogel. The low rate of degradation of G3 hydrogel can be associated with the permanent crosslinked network formed upon e-beam irradiation as rheological experiments have shown, while a higher degree of degradation of G4 can be correlated with a lower crosslinked hydrogel network. For G3 hydrogel, the free −OH and −NH2 groups of the chitosan backbone, are the principal reactive site where the cross-linking take place, so at the end of reaction such groups are no longer available for other interactions. Taking this into account in the degradation process the interaction ability with water will be reduced.
After 150 days of study, it is observed that weight loss gradually increased over time, hydrogels had only lost 5.7% of their initial weight for G3 hydrogel and 12.6% for G4 hydrogel. These results indicated that the hydrogels exhibited a stable structure. However, the trend indicates that at a given moment, the hydrogels will disintegrate almost completely.
2.4. Hydrogel Crosslinked Network
Some important network parameters used to assess the cross-linked network structure of hydrogel have been determined. They include polymer volume fraction in the swollen state (
), the average molecular weight between two successive cross-links (
), cross-link density (
) and mesh size (ξ). Cross-link density (
) is one of the most significant structural parameters of a hydrogel and establishes the weights of fluids that can be retained in its structure. Using the G’ values determined from rheological experiments and based on the rubber elasticity theory,
can be determined using the following equation [
14].
where
R is universal gas constant (8.314 m
3 Pa/mol K),
T is the absolute experimental temperature (298.15 °K);
V2r is the polymer volume fraction after e-beam cross-linking,
V2s is the polymer volume fraction of the crosslinked hydrogel in swollen state,
is the density of the polymer and the factor
A equals 1 for an affine network. The effective crosslink density,
Ve, of a crosslinked structure can be obtained from the results of rheological measurements using Equation (4):
The polymer volume fractions (
V2r and
V2s) were calculated as:
where
hydrogel and
solvent are the densities of hydrogel and solvent (kg/m
3), w
2r(s) is the weight of the hydrogel after e-beam crosslinking, respectively after swelling (g). The weight ratio of hydrogels after crosslinking (w
2r) was calculated as: w
2r = hydrogel mass after irradiation/hydrogel dry mass. The weight ratio of hydrogels after swelling (w
2s) was calculated as: w
2s = hydrogel mass after swelling/hydrogel dry mass.
The mesh size (ξ) shows the linear distance between consecutive cross-linking points and the space available between the macromolecular chains and was estimated using the following equation [
49]:
where
is the Flory characteristic ratio determined as average weight of the contained polymers (PVP = 12.3 [
50]; CS = 32.8 [
51], AA = 6.7 [
52], PEG = 4.0 [
53]).
is the molecular weight of the monomer unit, taken as a weighted average of the molecular weights of PVP = 111.14 g/mol [
50], CS = 161.2 g/mol [
54], PEG = 44.05 [
55], and AA = 72.06 g/mol [
52], l is the carbon–carbon bond length (0.154 nm). The experimental values of
G’,
,
and
ξ are summarized in
Table 3.
As shown in
Table 3, the
and ξ decreases with increasing of absorbed dose showing a more compact and cross-linked network. The G4 hydrogel crosslinked with 25 kGy, showed a
parameters larger, in contrast with the hydrogel G3 obtained at the same dose, which means the reduction of cross-linking points. The
for the investigated hydrogels comprises 2.4–64.7 × 10
3 kg/mol and 14.9–86.6 × 10
3 kg/mol, respectively. The results show that the G4 hydrogels prepared with LA and PEO show larger
and ξ values at 15 kGy and 25 kGy, which suggests that at this dose points, either the crosslinking reaction is not completed, or at doses higher than 20 kGy, the hydrogel network is affected by degradation processes.
Therefore, large values of and ξ parameters, reflect a lower cross-linking density. The values of the mesh size are in the range 11.5–67.7 nm, respectively 23.8–59.6 nm, obviously depending on the hydrogels composition and absorbed dose. For G3 and G4 hydrogels, the cross-link density was found to be 1.6–4.2 × 10−2 mol/m3, respectively 1.2–6.7 × 10−2 mol/m3 and increased with absorbed dose, only for G4 hydrogel irradiated at a dose of 25 kGy, the decreased suggesting network degradation. The above results are very well supported by the experimental data obtained in the degradation study and XPS analysis.
2.5. Retention Capability
Being an important factor of the wound dressing, moisture retention capability of hydrogels was investigated and is shown in
Table 4. Results indicated high moisture retention capability for both hydrogels compositions. At 2 h, there were no significant differences in moisture retention capacity between the hydrogels, with a range of 96.7%–97.6%. It can be seen in
Table 5 that G3 and G4 hydrogels prepared e-beam radiation cross-linking with 15–25 kGy, retain after 24 h between 80.59% and 82% of humidity. Maintaining increased humidity in wounds coated with such a hydrogel ensures a suitable environment for faster healing.
2.6. Water Vapor Transmission Rate (WVTR)
In clinical situations, hydrogels are usually used as a primary dressing, attached to the wound area, and then covered by a secondary dressing. The most difficult problem in treating a burned person was the fact that the victim may have lost most of its body liquid due to evaporation and exudation. The WVTR of normal skin is 204 g m
−2 day
−1, whereas for injured skin it varies a lot from 279 g m
−2 day
−1 to 5138 g m
−2 day
−1 depending upon the type of wound [
56]. Based on
Table 5, it can be seen that the WVTR values of hydrogels are around 167–273 g m
−2day
−1. When the irradiation dose increased, the WVTR increased. The hydrogels which were irradiated at 25 kGy have the highest WVTR (272.67 and 245.94 g m
−2 day
−1), presenting values of WVTR higher than that of normal skin. Such values cause a faster drying of the wound. On the other hand, the G3 hydrogel, irradiated at 15 kGy has the lowest WVTR (167 g m
−2 day
−1). These values of WVTR are beneficial to provide intermittent conditions for a faster wound healing process.
2.7. ATR-FTIR
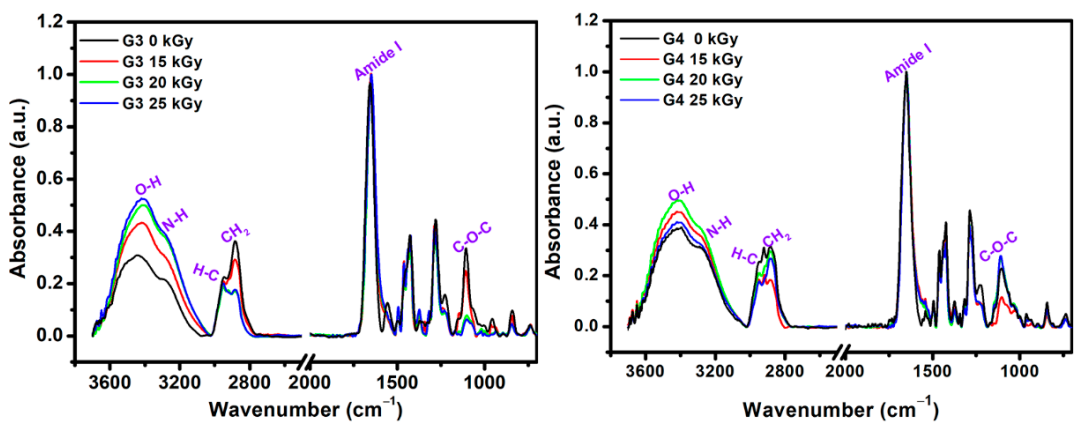
The structural changes that appeared after e-beam irradiation in the hydrogels were investigated by ATR–FTIR. Characteristic FTIR spectra of unirradiated and irradiated G3 and G4 hydrogels are shown in
Figure 4, normalized to the highest intensity peak at 1654 cm
−1.
As expected, the FTIR spectra of the obtained materials contain features originating from each of their component. The main FTIR characteristic absorption bands corresponding to chitosan are: 3356 cm
−1 and 3293 cm
−1 (O-H stretch overlapped with N–H stretching), 2800–2900 cm
−1 (C-H stretch), 1647 cm
−1 (amide I band, C-O stretch of an acetyl group), 1573 cm
−1 (amide II band and N-H stretch), 1375 cm
−1 (amide III, asymmetric C-H bending of CH
2 group),1149 cm
−1 (antisymmetric elongation of the C-O-C bridge), and 1062 cm
−1 indicate skeletal vibration involving the bridge C-O stretch of glucosamine residue [
57].
The specific absorption bands corresponding to PVP are: 3470 cm
−1 (O-H stretching); 2870–2950 cm
−1(C-H and CH
2 stretching); 1651 cm
−1 (C = O and C-N stretching vibration); 1370/1420/1459/1490 cm
−1 (C-H and CH
2 deformation vibrations of pyrrolic ring); and 1265/1280 cm
−1 (stretching vibrations of amide III band) [
58]. The main FTIR peaks of PEO are: 2875 cm
−1 stretching vibration of CH
2 groups; 1094 cm
−1 band is derived from the association of CH
2 groups with the etheric ones of the C-O-C group; 1464 cm
−1/1342 cm
−1 deformation vibrations outside the plane (wagging) of CH
2 groups; 954 cm
−1 and 840 cm
−1 rocking plane vibrations of the CH
2 groups [
59].
In the FTIR spectra of the G3 (0 kGy) pre-hydrogel, the main peaks were found at 3440 cm−1, assigned to the N-H groups from PVP molecule; 3290 cm−1 assigned to the overlapped N-H and O-H groups of chitosan. The peaks in the 2880–2945 cm−1 range show stretching vibration of C-H and CH2 bonds common of PVP, chitosan and PEO. The band observed at 1654 cm−1 caused by the intermolecular inter-polymers H bonds of O-H, N-H of chitosan and C = O of PVP. The band from 1556 cm−1 is specific to the amide II of the chitosan molecule. At 1427 cm−1 are present the deformation vibrations of CH2 bond common of PVP ring. The peak at 1280 cm−1 corresponds to the amide III of the PVP molecule and the peak situated at 1105 cm−1 corresponding both to the C-O-C and C-O group of chitosan and PEO. In the FTIR spectra of the G3 cross-linked hydrogel, within 3700–3000 cm−1 range the increase of bands intensity and the shift to lower wavenumbers up to 3407 cm−1 as function of absorbed dose was observed. Moreover, the intensity of C-H and CH2 groups has decreased after e-beam cross-linking. The stretching vibration peak at of 1654 cm−1 is getting broader, while the contribution of amide II peak in chitosan appears just as a shoulder, suggesting a strong participation of O-H and N-H groups in the chemical reaction under e-beam irradiation. On the other hand, the decrease in the intensity of the C-O-C peak from 1105 cm−1 could be correlated with the shortening of polymeric chain length as a consequence of a higher absorbed dose. The above observation is also supported by XPS investigations.
The unirradiated (0 kGy) G4 pre-hydrogel showed a wide absorption band from 3600 to 2800 cm−1. In this region, the main peaks were identified at 3394 and 3284 cm−1 (O-H and N-H from chitosan and PVP), 2922 cm−1, and 2882 cm−1 (C-H and CH2 bonds common of both chitosan, PEO and PVP).
In the range, 1700–650 cm−1 the peaks corresponding to both chitosan and PVP were found: 1650 cm−1 (amide I band), 1430 cm−1 (C = O, and C-N), 1280 cm−1 (amide III), 1105 cm−1 (C-O-C).
For G4 hydrogel cross-linked with 20 kGy and 25 kGy, it was observed the loss of peak from 2920 cm−1 associated with C-H bond, most probably due to high cross-linking evidenced for this composition. Other significant changes in the FTIR spectra of G4 hydrogels were observed for the peaks situated around 1105 cm−1, whose intensities increased with the irradiation dose, suggesting a more significant contribution of some oxidative processes due to the higher absorbed dose.
2.8. X-Ray Photoelectron Spectroscopy (XPS)
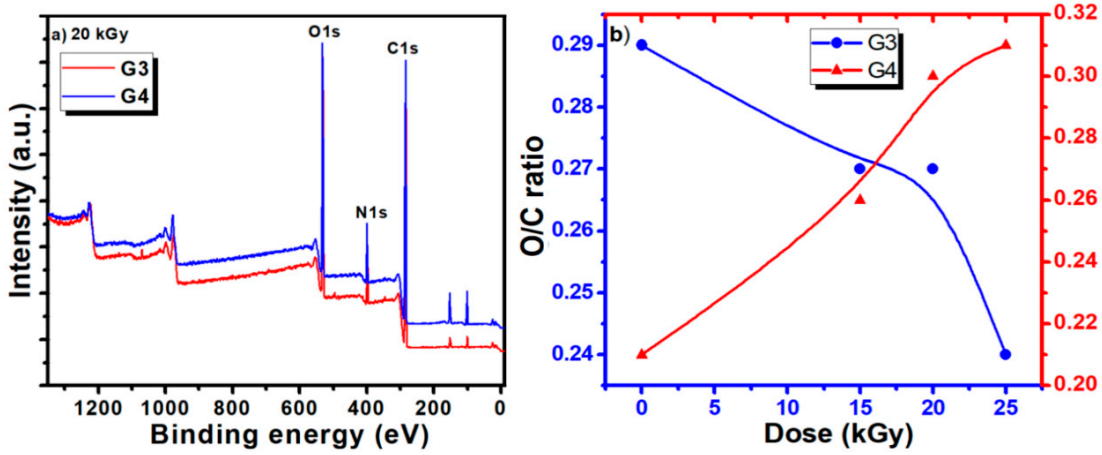
Typical survey spectra for the G3 and G4 hydrogels are presented in
Figure 5a. They present a similar aspect, revealing the presence of carbon, nitrogen, and oxygen. The elemental composition of the unirradiated CS-PVP-PEG/PEO polymeric mixtures and G3, G4 hydrogels synthesized by e-beam irradiation at various doses, as determined from the XPS survey spectra, are reported in
Table 6, evidencing only small variation of the composition.
The calculated ratios between oxygen and carbon concentration (O/C ratio) and nitrogen and carbon concentration (N/C ratio) are also included.
The evolution of the O/C ratios for the G3 and G4 hydrogels is illustrated in
Figure 5b. For the G3 hydrogel based on CS-PVP-PEG, the O/C ratio shows just a slight decrease for irradiation doses below 20 kGy, suggesting the partial preservation of the pre-hydrogel initial chemical structure, while upon higher doses a more pronounced decrease is noticed, related to cross-linking of the material.
For the G4 hydrogel based on CS-PVP-PEO, the O/C ratio presents an ascendant trend with the irradiation dose, suggesting an easy opening of epoxide ring in acid in the case of PEO conducting to more pronounced oxidation state is in this case. N/C ratio present an increasing trend for both hydrogels, for irradiation dose up to 20 kGy, while at 25 kGy a sharp decrease is noticed for the G4 gel, pointing again towards a different behavior for this experimental condition, as previously evidenced by the gel fraction and radio-chemical yield of degradation investigations.
The C1s high resolution spectra recorded for the hydrogels obtained at various absorbed doses, for both G3 and G4, are displayed in
Figure 6. They evidence on one hand the specificity given by the different composition of the initial polymeric solution which undergoes the electron beam irradiation procedure, and on the other hand the modifications induced in the chemical bonding as function of the irradiation dose.
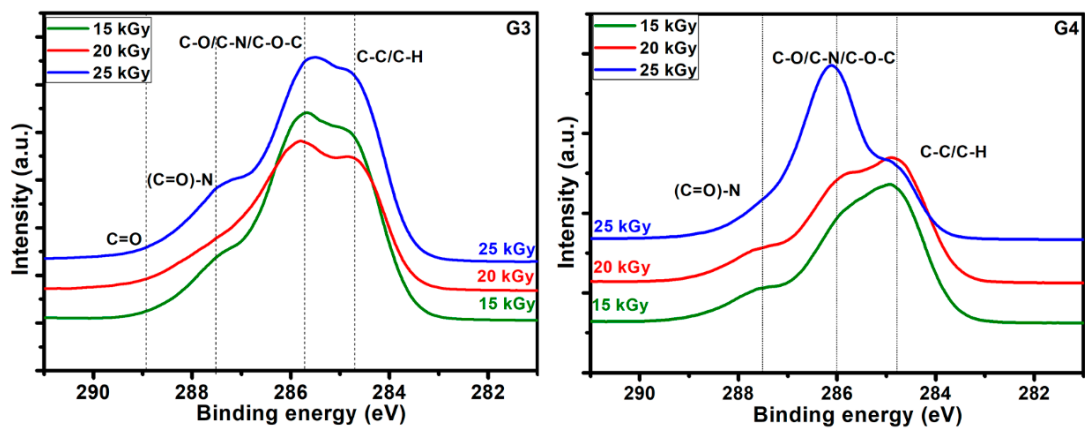
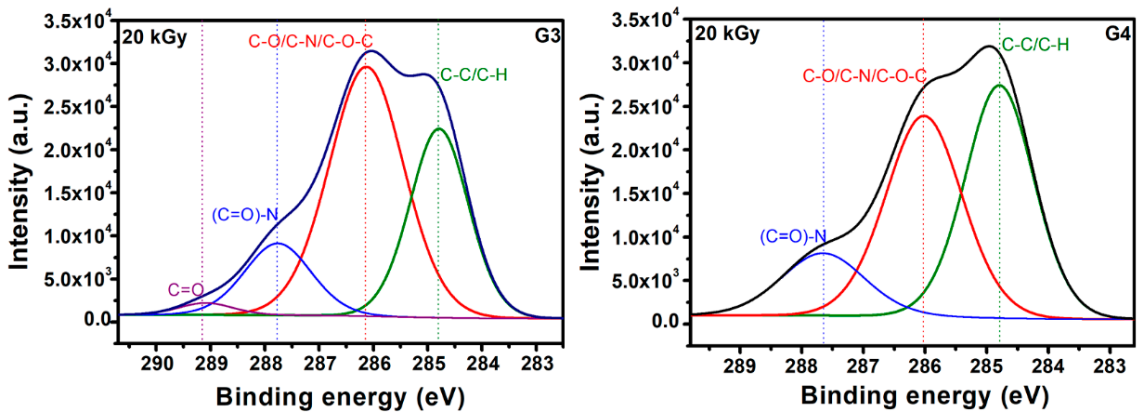
Typical deconvolutions performed to reveal various bonds present in the materials, for both G3 and G4 hydrogels are presented in
Figure 7. The fitting was considered completed when the chi values minimized, below 1. In the case of G3 hydrogels, the deconvolution of C1s spectrum was considering four components, as follows: C-C/C-H at 285 eV, C-O/C-N/C-O-C at 286 eV, (C = O)-N bonds at 288 eV and C = O bonds at 289 eV. For the G4 hydrogel, the deconvolution was performed using three components, as follows: C-C/C-H bonds at 285 eV, C-O/C-N/C-O-C bonds at 286 eV and (C = O)-N bonds at 288 eV [
60,
61].
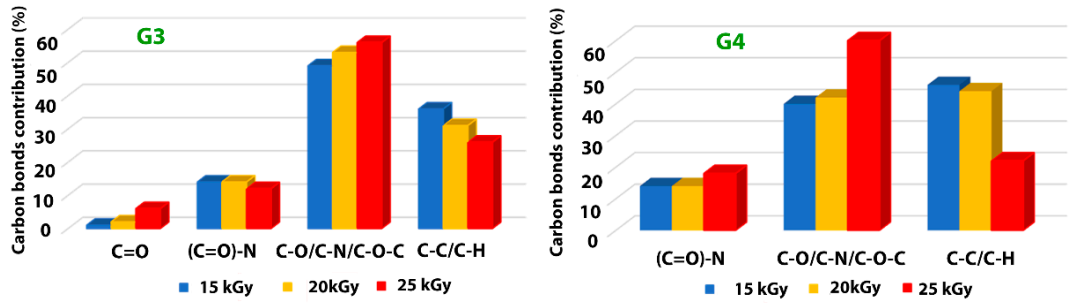
In
Table 7 are presented the contributions of various carbon bonds as function of irradiation dose for G3 and G4 hydrogels, while in
Figure 8 are represented their dependencies upon the irradiation dose.
For both investigated hydrogels, the amount of C-C/C-H bonds decreases with irradiation dose suggesting a breaking of the polymeric chains due to e-beam irradiation. This is accompanied by the increase of the contribution of C-O/C-N/C-O-C-bonds with the absorbed dose, which points out towards a cross-linking of the broken chains to the surrounding oxygen or nitrogen atoms. For the G3 hydrogels, the amount of carbon bonded in C = O bond is also increasing with irradiation dose, while the contribution of (C = O)-N reveals slight decreasing which is in accordance with the N/C ratio trend. In the case of G4 hydrogel, one can notice the drastic decrease of C-C/C-H bonds at large irradiation dose of 25 kGy with an increase of the contribution counted for C-O/C-N/C-O-C-bonds, as well as that of (C = O)-N bonds, suggesting more accentuated oxidative degradation in this case.
The results indicate, once again, following specific hydrogel investigations revealed in paragraph 2.1 and FTIR analysis, that the chemical structure of the G4 hydrogel based on CS-PVP-PEO is significantly degraded under irradiation dose of 25 kGy.
2.9. Evaluation of Encapsulation Efficiency, Drug Loading Capacity, and In Vitro Drug Release Study
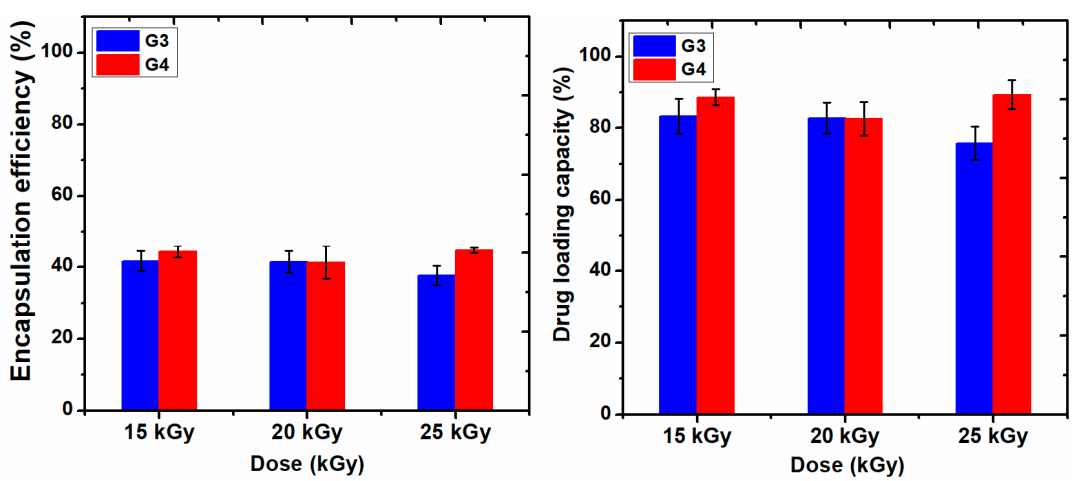
Figure 9 shows the data of encapsulation efficiency (EE) and drug loading capacity (LC) of ibuprofen in the G3 and G4 hydrogels in correlation with absorbed dose.
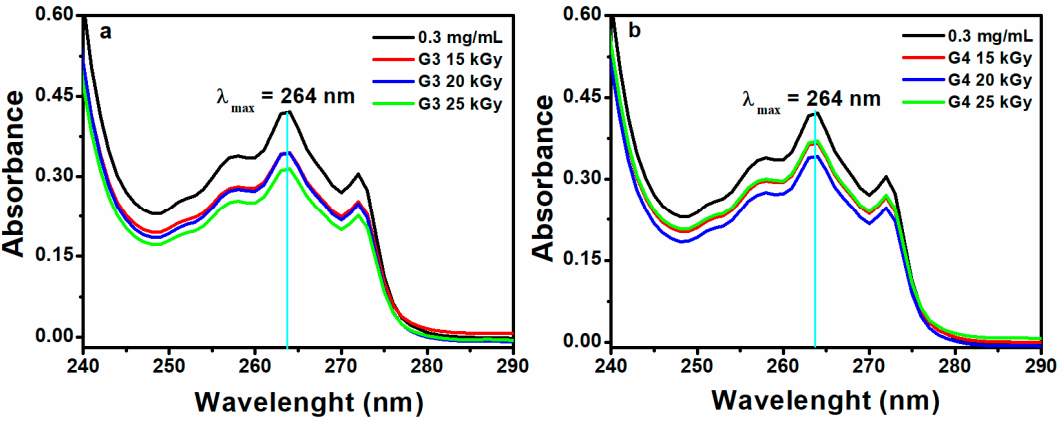
Figure 10a,b show typical UV-Vis absorption spectra of IBU in G3 and G4 hydrogels. The corresponding calibration curve of IBU solubilized in EtOH within 0.05–0.6 mg/mL and UV-Vis absorption spectra are presented in the
Supplementary Materials, Figure S3a,b.
Ibuprofen is stabilized in the hydrogel matrix through H bonding or polar interactions between drug molecules with the ionized groups of the polymer chain and other hydrophobic interaction between the hydrophobic moieties of the drug and hydrogel. Since the release of a drug from hydrogel is essentially a mesh-controlled diffusion, this process will be influenced by mesh size and cross-link density of the hydrogels as well as by the size of the released molecules. As can be seen in
Figure 9, the EE (%) of IBU decreased with the absorbed dose for G3 hydrogels. A decrease of 5% was observed for the hydrogel cross-linked with a dose of 25 kGy, most likely due to the increase of cross-linking degree, thus the active substance is effectively prevented from entering throughout the macromolecular network of the hydrogel. The typical UV-Vis absorption spectra of IBU in G3 and G4 hydrogels are presented in
Figure 10a,b.
The drug loading capacity of IBU was reduced below 80% in the case of G3 hydrogels when the absorbed dose is 25 kGy, on the contrary, the drug loading capacity in the G4 hydrogels increased at the dose of 25 kGy, probably due to the breaking of polymer chains due to e-beam irradiation.
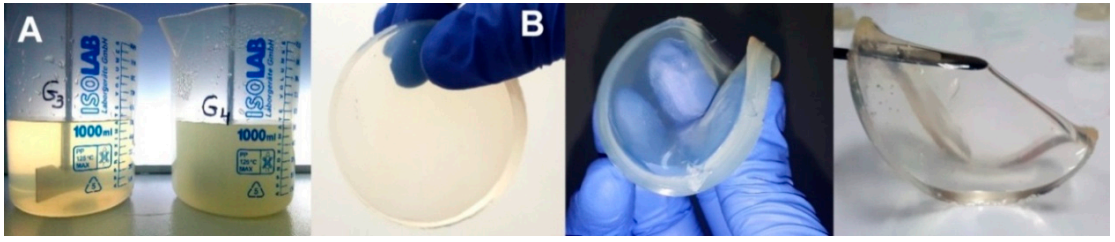
In
Figure 11 it is presented the encapsulation process of IBU in G3 hydrogels and the appearance of G3 hydrogel loaded with IBU and a piece of hydrogel after IBU complete releasing. The release profile of the IBU was afterwards investigated at pH = 7.4 and 9.4, corresponding to non-infected and infected wounds, respectively.
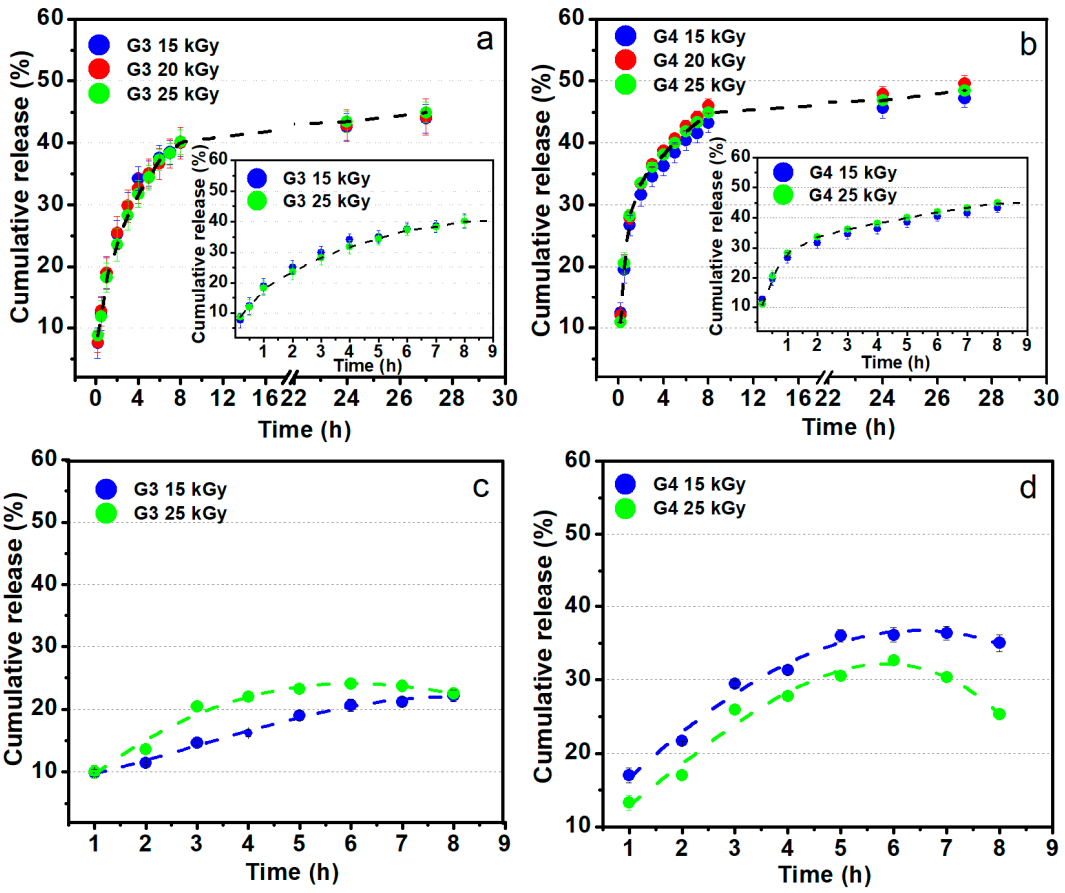
Figure 12a,b shows the release profiles of the ibuprofen loaded in G3 and G4 hydrogels up to 27 h in PBS (pH = 7.4, 37 °C), pointing out similar release behavior regardless of absorbed dose. An inset showing the release behavior in the first 8 h is also included. One can notice that about 30–45% of ibuprofen is released in the first 5 h for both types of compositions, equivalent to around 30 mg. This behavior points out that the drug release rate from the IBU loaded hydrogel is abrupt at the starting time, afterwards, the absorbed IBU is still released from hydrogel up to 30 h. Prolonged-release of IBU from a hydrogel-type polymeric matrix may be beneficial if it is considered that the normal half-life is 1–3 h, thus can help to maintain an optimum concentration in the body [
62]. A faster release rate of IBU during the first 3 h with a linear dependence was observed for both hydrogels composition. At the starting time, the release process is controlled by the swelling capacity of the hydrogel while as the polymer networks become more hydrated, the release of IBU takes place through a diffusion process. This behavior was pointed out by other studies based on the evaluation of IBU release from different hydrogel matrix having in composition chitosan or other formulation typically designed for topical administration of ibuprofen [
63]. Djekic et al. showed that the maximum amount of IBU that can be released from a reference gel, namely Nurofen
® gel was approximately about 50% after 6 h [
64]. Comparing with the above-mentioned study, our hydrogels can release within 40–50% of IBU after 8 h. More than that, our data are very close to that of other hydrogels formulation with similar composition [
65].
Figure 12c,d shows the ibuprofen loaded in G3 and G4 hydrogels behavior regarding the release profiles for up to 8 h in sodium carbonate–sodium bicarbonate buffer (pH = 9.4, 37 °C). These data simulate the drug release on the site of an infected wound. In contrast with the experiments at pH 7.4, in the weak alkaline media, the IBU release is lower, reaching 25–35% in the first 5 h. Such behavior could be the result of lower swelling degree at pH 9.4 as previously presented in the swelling experiments (
Figure 2).
4. Conclusions
In this study, chitosan-poly(vinyl-pyrrolidone)-poly (ethylene glycol)/poly (ethylene oxide)-poly(acrylic)/poly(lactic) acid hydrogels were successfully synthesized by one step e-beam cross-linking to be used for rapid healing and pain release of infected skin wounds. Investigations of chemical composition by FTIR and XPS confirmed that hydrogels were successfully prepared, with deterioration with respect to the optimum hydrogel properties only for irradiation doses above 20 kGy.
The hydrogels showed high gel fractions (96%) even at lower absorbed dose. The swelling capacity decreased with the absorbed dose, the hydrogels reaching equilibrium after 8 h of immersion in PBS at various pH and temperatures. The hydrogels showed good stability, lower rate of degradation and superabsorbent properties in simulated hyperthermia conditions (37–41 °C) and different pH intervals. The shape of the hydrogels was not affected even after 48 h in the above conditions, as well as no evidence of hydrogel dissolution in studied media.
The network parameters (, and ξ) indicated the formation of a cross-linked structure with a nanostructured mesh with typical dimension in the range 11–67 nm. The WVTR values ranged from 167.21 and 272.67 g m−2 day−1 confirming that all hydrogels can be used to control body fluid loss and maintain a moist environment for infected skin wounds. This behavior is accompanied by the favorable maintenance of humidity for more than 6 h and considering that this time represents the normal frequency of changing a dressing, ensure a favorable environment for wound healing.
Moreover, the synthesized hydrogels have a significant loading capacity for IBU, in the range of 75–89% depending on the absorbed dose, ensuring the possibility of incorporating a therapeutic dose needed for severe pains. The IBU was released from the macromolecular network of the hydrogels in a proportion below 30% in the first 2 h, reaching a maximum after 8 h, demonstrating the controlled release capacity, both for pHs of 7.4 and 9.4, corresponding to the case of non-infected and infected wounds, respectively. The results point out that the optimum irradiation doses for obtaining hydrogels with the best biomedical properties are between 15–20 kGy.
Further research in order to evidence the antimicrobial response of the hydrogels for various strains, including Gram-negative and Gram-positive bacteria and yeasts are considered. The investigations will be continued as well with the evaluation of viability, cell proliferation, and migration for various cell lines of fibroblasts and keratinocytes. This information will allow the determination of the optimum formulation of the hydrogels dressings based on chitosan-poly(vinyl-pyrrolidone)-poly (ethylene glycol)/poly (ethylene oxide)-poly(acrylic)/poly (lactic) acid for wound healing applications.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}