Role of Nucleotide Excision Repair in Cisplatin Resistance


Abstract
:
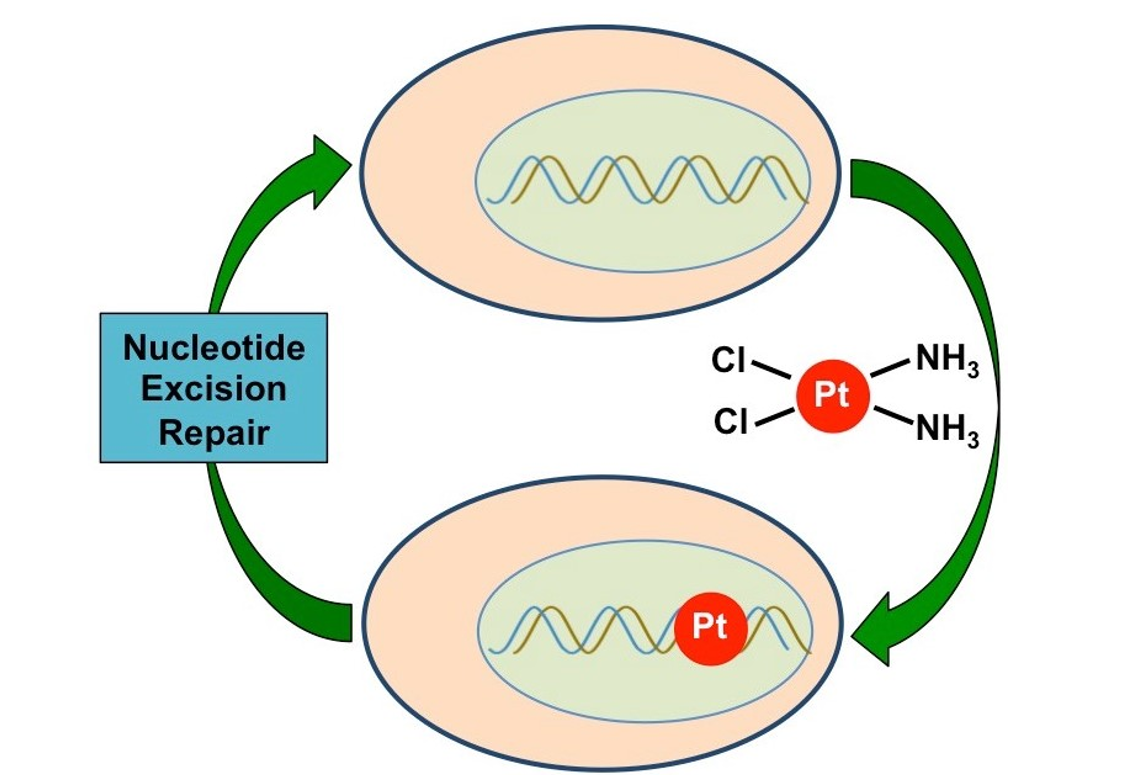
1. Introduction
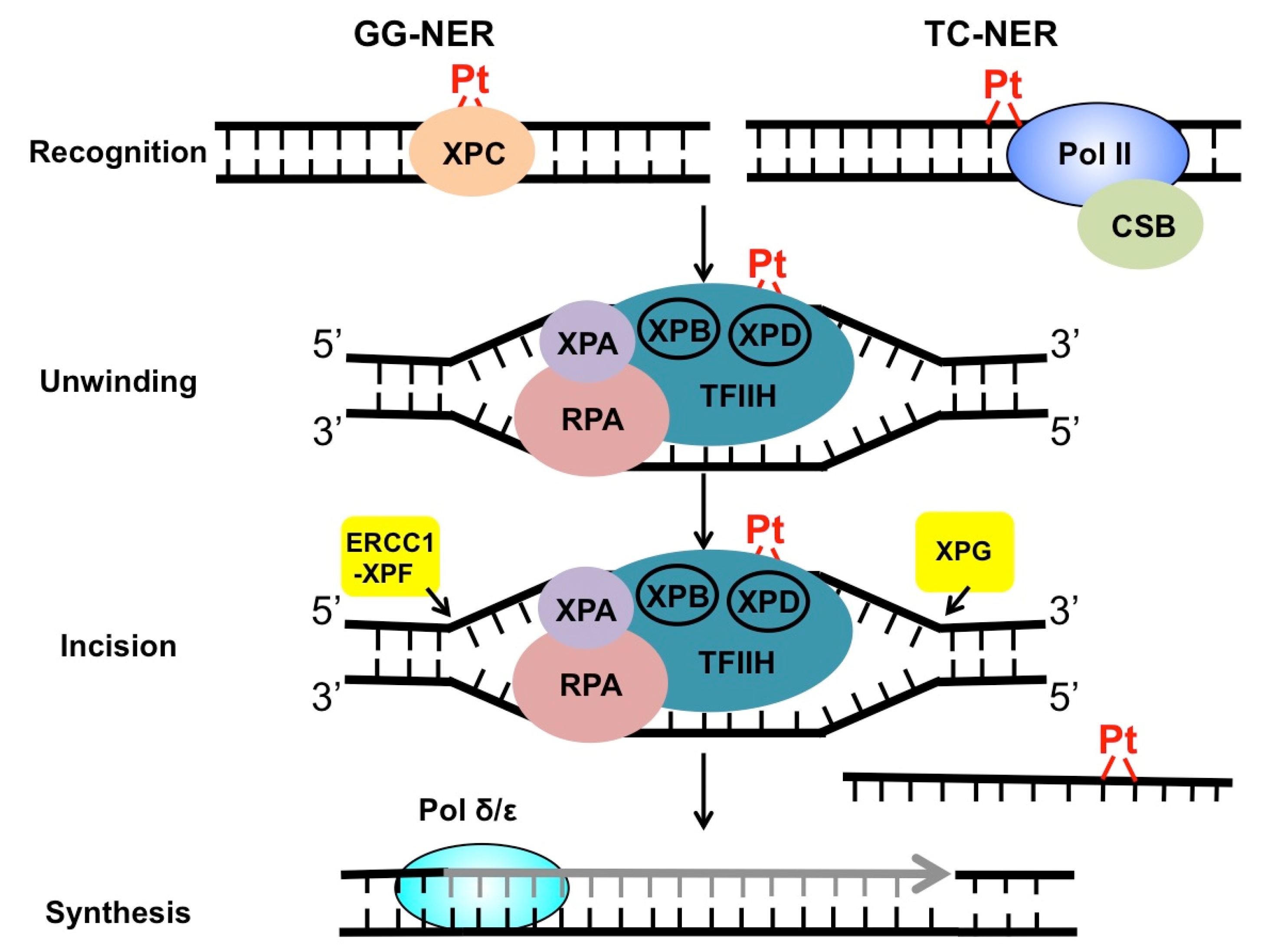
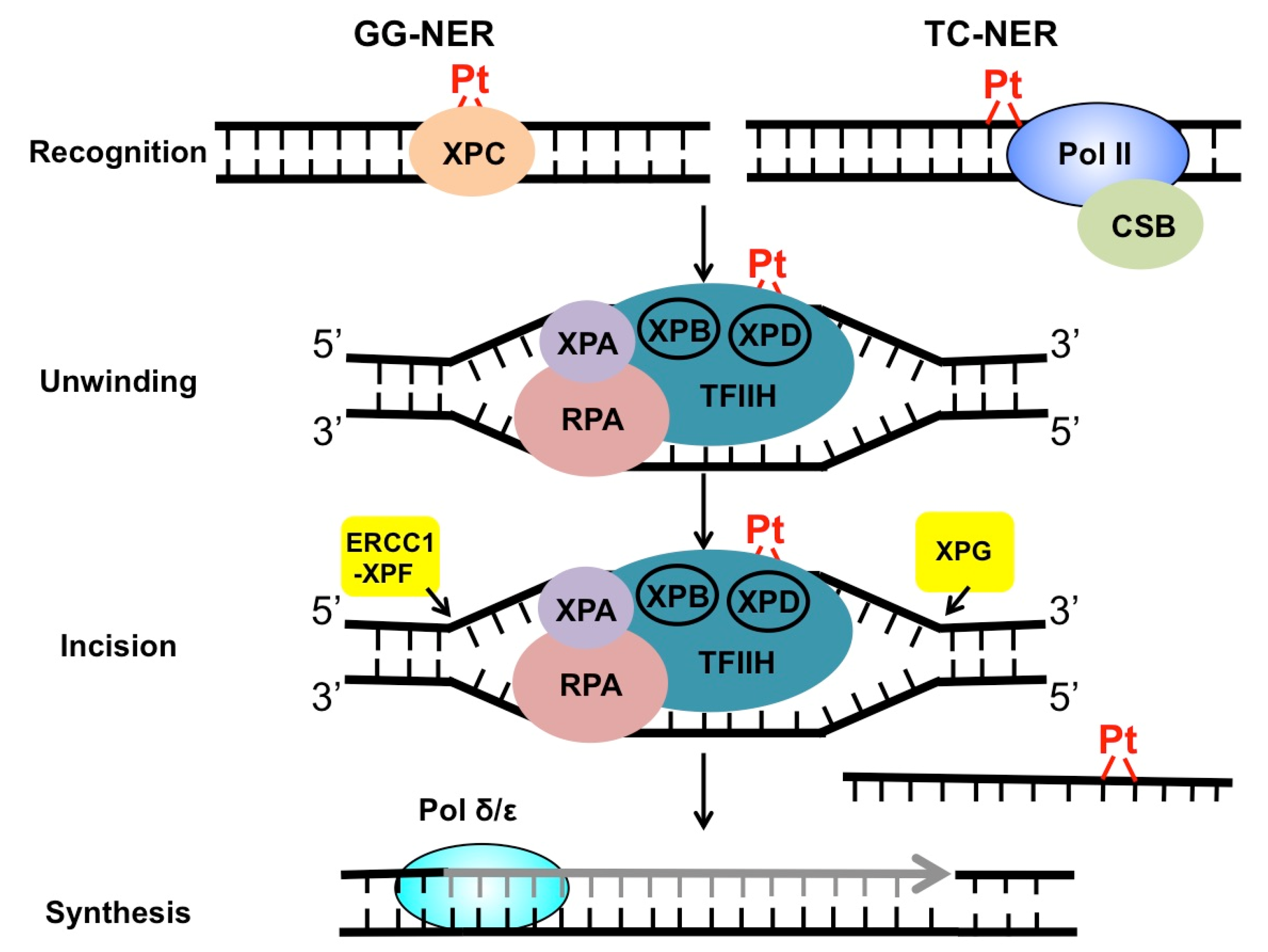
2. Mechanism of NER in the Repair of Cisplatin Adducts
3. Roles of GG-NER Factors in Cisplatin Resistance
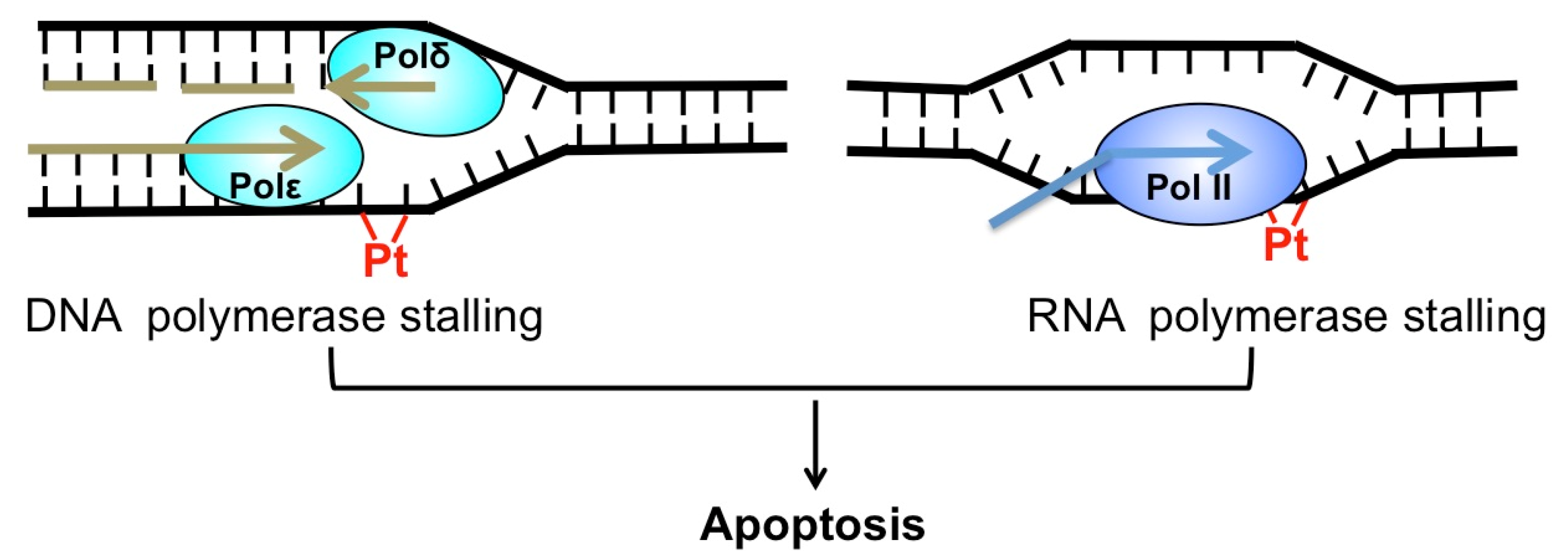
4. Roles of TC-NER Factors in Cisplatin Resistance
5. NER Inhibitors and Their Roles in Suppressing DNA Repair
6. Discussion and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichtinger-Schepman, A.M.J.; Van der Veer, J.L.; Den Hartog, J.H.J.; Lohman, P.H.M.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: Formation, identification, and quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Horwich, A.; Shipley, J.; Huddart, R. Testicular germ-cell cancer. Lancet 2006, 367, 754–765. [Google Scholar] [CrossRef]
- Köberle, B.; Masters, J.R.; Hartley, J.A.; Wood, R.D. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol. 1999, 9, 273–276. [Google Scholar] [CrossRef] [Green Version]
- Einhorn, L.H. Curing metastatic testicular cancer. PNAS 2002, 99, 4592–4595. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [Green Version]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.H.W.; Kuo, M.T. Role of Glutathione in the Regulation of Cisplatin Resistance in Cancer Chemotherapy. Met. Based Drugs 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Hamilton, T.C.; Chaney, S.G. The Role of hMLH1, hMSH3, and hMSH6 Defects in Cisplatin and Oxaliplatin Resistance: Correlation with Replicative Bypass of Platinum-DNA Adducts. Cancer Res. 1998, 58, 3579–3585. [Google Scholar] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics (Sao Paulo) 2018, 73. [Google Scholar] [CrossRef] [PubMed]
- Zamble, D.B.; Mu, D.; Reardon, J.T.; Sancar, A.; Lippard, S.J. Repair of Cisplatin−DNA Adducts by the Mammalian Excision Nuclease. Biochemistry 1996, 35, 10004–10013. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.T.; Vaisman, A.; Chaney, S.G.; Sancar, A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999, 59, 3968–3971. [Google Scholar] [PubMed]
- Hu, J.; Lieb, J.D.; Sancar, A.; Adar, S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 11507–11512. [Google Scholar] [CrossRef] [Green Version]
- Welsh, C.; Day, R.; McGurk, C.; Masters, J.R.W.; Wood, R.D.; Köberle, B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer 2004, 110, 352–361. [Google Scholar] [CrossRef]
- Masters, J.R.W.; Köberle, B. Curing metastatic cancer: Lessons from testicular germ-cell tumours. Nat. Rev. Cancer 2003, 3, 517–525. [Google Scholar] [CrossRef]
- Ferry, K.V.; Hamilton, T.C.; Johnson, S.W. Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: Role of ercc1–xpf. Biochem. Pharmacol. 2000, 60, 1305–1313. [Google Scholar] [CrossRef]
- Rosell, R.; Taron, M.; Barnadas, A.; Scagliotti, G.; Sarries, C.; Roig, B. Nucleotide Excision Repair Pathways Involved in Cisplatin Resistance in Non-Small-Cell Lung Cancer. Cancer Control. 2003, 10, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Damish, A.W.; Frazier, Z.; Liu, D.; Reznichenko, E.; Kamburov, A.; Bell, A.; Zhao, H.; Jordan, E.J.; Gao, S.P.; et al. ERCC2 Helicase Domain Mutations Confer Nucleotide Excision Repair Deficiency and Drive Cisplatin Sensitivity in Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2019, 25, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cao, J.; Meng, Y.; Qu, C.; Shen, F.; Xu, L. Overexpression of xeroderma pigmentosum group C decreases the chemotherapeutic sensitivity of colorectal carcinoma cells to cisplatin. Oncol. Lett. 2018, 15, 6336–6344. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo-Lozano, N.; Bargiela-Iparraguirre, J.; Dominguez, G.; Quiroga, A.G.; Perona, R.; Sanchez-Perez, I. XPA, XPC, and XPD Modulate Sensitivity in Gastric Cisplatin Resistance Cancer Cells. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [Green Version]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef]
- Gregersen, L.H.; Svejstrup, J.Q. The Cellular Response to Transcription-Blocking DNA Damage. Trends Biochem. Sci. 2018, 43, 327–341. [Google Scholar] [CrossRef]
- Huang, J.C.; Zamble, D.B.; Reardon, J.T.; Lippard, S.J.; Sancar, A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc. Natl. Acad. Sci. USA 1994, 91, 10394–10398. [Google Scholar] [CrossRef] [Green Version]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Ohndorf, U.-M.; Rould, M.A.; He, Q.; Pabo, C.O.; Lippard, S.J. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature 1999, 399, 708–712. [Google Scholar] [CrossRef]
- Awuah, S.G.; Riddell, I.A.; Lippard, S.J. Repair shielding of platinum-DNA lesions in testicular germ cell tumors by high-mobility group box protein 4 imparts cisplatin hypersensitivity. PNAS 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamble, D.B.; Mikata, Y.; Eng, C.H.; Sandman, K.E.; Lippard, S.J. Testis-specific HMG-domain protein alters the responses of cells to cisplatin. J. Inorg. Biochem. 2002, 91, 451–462. [Google Scholar] [CrossRef]
- Hu, J.; Adar, S.; Selby, C.P.; Lieb, J.D.; Sancar, A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 2015, 29, 948–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, J.M.Y.; Vermeulen, W.; van der Horst, G.T.J.; Bergink, S.; Sugasawa, K.; Vrieling, H.; Hoeijmakers, J.H.J. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 2003, 17, 1630–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hey, T.; Lipps, G.; Sugasawa, K.; Iwai, S.; Hanaoka, F.; Krauss, G. The XPC−HR23B Complex Displays High Affinity and Specificity for Damaged DNA in a True-Equilibrium Fluorescence Assay. Biochemistry 2002, 41, 6583–6587. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-H.; Pavletich, N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449, 570–575. [Google Scholar] [CrossRef]
- Okuda, M.; Nakazawa, Y.; Guo, C.; Ogi, T.; Nishimura, Y. Common TFIIH recruitment mechanism in global genome and transcription-coupled repair subpathways. Nucleic Acids Res. 2017, 45, 13043–13055. [Google Scholar] [CrossRef]
- Yokoi, M.; Masutani, C.; Maekawa, T.; Sugasawa, K.; Ohkuma, Y.; Hanaoka, F. The Xeroderma Pigmentosum Group C Protein Complex XPC-HR23B Plays an Important Role in the Recruitment of Transcription Factor IIH to Damaged DNA. J. Biol. Chem. 2000, 275, 9870–9875. [Google Scholar] [CrossRef] [Green Version]
- Teng, X.; Fan, X.-F.; Li, Q.; Liu, S.; Wu, D.-Y.; Wang, S.-Y.; Shi, Y.; Dong, M. XPC inhibition rescues cisplatin resistance via the Akt/mTOR signaling pathway in A549/DDP lung adenocarcinoma cells. Oncol. Rep. 2019, 41, 1875–1882. [Google Scholar] [CrossRef]
- Barakat, B.M.; Wang, Q.-E.; Han, C.; Milum, K.; Yin, D.-T.; Zhao, Q.; Wani, G.; Arafa, E.-S.A.; El-Mahdy, M.A.; Wani, A.A. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int J. Cancer 2010, 127, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Coin, F.; Oksenych, V.; Egly, J.-M. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell 2007, 26, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Kuper, J.; Braun, C.; Elias, A.; Michels, G.; Sauer, F.; Schmitt, D.R.; Poterszman, A.; Egly, J.-M.; Kisker, C. In TFIIH, XPD Helicase Is Exclusively Devoted to DNA Repair. PLoS Biol. 2014, 12, e1001954. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, A.R. The xeroderma pigmentosum group D (XPD) gene: One gene, two functions, three diseases. Genes Dev. 2001, 15, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Mouw, K.W.; Kim, P.; Iyer, G.; Wagle, N.; Al-Ahmadie, H.; Zhu, C.; Ostrovnaya, I.; Kryukov, G.V.; O’Connor, K.W.; et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014, 4, 1140–1153. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Plimack, E.R.; Hoffman-Censits, J.; Garraway, L.A.; Bellmunt, J.; Van Allen, E.; Rosenberg, J.E. Clinical Validation of Chemotherapy Response Biomarker ERCC2 in Muscle-Invasive Urothelial Bladder Carcinoma. JAMA Oncol. 2016, 2, 1094–1096. [Google Scholar] [CrossRef] [Green Version]
- Sugitani, N.; Sivley, R.M.; Perry, K.E.; Capra, J.A.; Chazin, W.J. XPA: A key scaffold for human nucleotide excision repair. DNA Repair 2016, 44, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Roginskaya, M.; Colis, L.C.; Basu, A.K.; Shell, S.M.; Liu, Y.; Musich, P.R.; Harris, C.M.; Harris, T.M.; Zou, Y. Specific and Efficient Binding of Xeroderma Pigmentosum Complementation Group A to Double-Strand/Single-Strand DNA Junctions with 3′- and/or 5′-ssDNA Branches. Biochemistry 2006, 45, 15921–15930. [Google Scholar] [CrossRef] [Green Version]
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Pulzova, L.B.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer 2020, 20, 17. [Google Scholar] [CrossRef]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Schärer, O.D.; Wagner, G.; Ellenberger, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsodikov, O.V.; Enzlin, J.H.; Schärer, O.D.; Ellenberger, T. Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF–ERCC1. PNAS 2005, 102, 11236–11241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; André, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef]
- Jun, H.J.; Ahn, M.J.; Kim, H.S.; Yi, S.Y.; Han, J.; Lee, S.K.; Ahn, Y.C.; Jeong, H.-S.; Son, Y.-I.; Baek, J.-H.; et al. ERCC1 expression as a predictive marker of squamous cell carcinoma of the head and neck treated with cisplatin-based concurrent chemoradiation. Br. J. Cancer 2008, 99, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Wang, Y.; Chen, L.; Gan, Y.; Wu, Q. High ERCC1 expression is associated with platinum-resistance, but not survival in patients with epithelial ovarian cancer. Oncol. Lett. 2016, 12, 857–862. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Shao, C.-J.; Chen, F.-R.; Kwan, A.-L.; Chen, Z.-P. Role of ERCC1 promoter hypermethylation in drug resistance to cisplatin in human gliomas. Int. J. Cancer 2010, 126, 1944–1954. [Google Scholar] [CrossRef]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF–ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair (Amst) 2010, 9, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Booton, R.; Ward, T.; Ashcroft, L.; Morris, J.; Heighway, J.; Thatcher, N. ERCC1 mRNA Expression Is Not Associated with Response and Survival after Platinum-Based Chemotherapy Regimens in Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2007, 2, 902–906. [Google Scholar] [CrossRef]
- Friboulet, L.; Olaussen, K.A.; Pignon, J.-P.; Shepherd, F.A.; Tsao, M.-S.; Graziano, S.; Kratzke, R.; Douillard, J.-Y.; Seymour, L.; Pirker, R.; et al. ERCC1 Isoform Expression and DNA Repair in Non–Small-Cell Lung Cancer. N. Eng. J. Med. 2013, 368, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Friboulet, L.; Postel-Vinay, S.; Sourisseau, T.; Adam, J.; Stoclin, A.; Ponsonnailles, F.; Dorvault, N.; Commo, F.; Saulnier, P.; Salome-Desmoulez, S.; et al. ERCC1 function in nuclear excision and interstrand crosslink repair pathways is mediated exclusively by the ERCC1-202 isoform. Cell Cycle 2013, 12, 3298–3306. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.S.; Ogawa, S.; Karahashi, H.; Scoles, D.R.; Pavelka, J.C.; Tran, H.; Miller, C.W.; Kawamata, N.; Ginther, C.; Dering, J.; et al. ERCC5 is a novel biomarker of ovarian cancer prognosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2952–2958. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, M.A.; Marabese, M.; Ganzinelli, M.; Caiola, E.; Geroni, C.; Broggini, M. Down-regulation of the Nucleotide Excision Repair gene XPG as a new mechanism of drug resistance in human and murine cancer cells. Mol. Cancer 2010, 9, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D. A panorama of transcription-coupled repair in yeast chromatin. Proc. Natl. Acad. Sci. USA 2020, 117, 20991–20993. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Selvam, K.; Wyrick, J.J.; Mao, P. Genome-wide role of Rad26 in promoting transcription-coupled nucleotide excision repair in yeast chromatin. Proc. Natl. Acad. Sci. USA 2020, 202003868. [Google Scholar] [CrossRef]
- Stubbert, L.J.; Smith, J.M.; McKay, B.C. Decreased transcription-coupled nucleotide excision repair capacity is associated with increased p53- and MLH1-independent apoptosis in response to cisplatin. BMC Cancer 2010, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Damsma, G.E.; Alt, A.; Brueckner, F.; Carell, T.; Cramer, P. Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat. Struct. Mol. Biol. 2007, 14, 1127–1133. [Google Scholar] [CrossRef]
- Ljungman, M.; Zhang, F. Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene 1996, 13, 823–831. [Google Scholar]
- Caputo, M.; Frontini, M.; Velez-Cruz, R.; Nicolai, S.; Prantera, G.; Proietti-De-Santis, L. The CSB repair factor is overexpressed in cancer cells, increases apoptotic resistance, and promotes tumor growth. DNA Repair (Amst) 2013, 12, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Selby, C.P.; Sancar, A. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. USA 1997, 94, 11205–11209. [Google Scholar] [CrossRef] [Green Version]
- Rainey, R.N.; Ng, S.; Llamas, J.; van der Horst, G.T.J.; Segil, N. Mutations in Cockayne Syndrome-Associated Genes (Csa and Csb) Predispose to Cisplatin-Induced Hearing Loss in Mice. J. Neurosci. 2016, 36, 4758–4770. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, W.; Fousteri, M. Mammalian Transcription-Coupled Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012625. [Google Scholar] [CrossRef] [PubMed]
- Saijo, M. The role of Cockayne syndrome group A (CSA) protein in transcription-coupled nucleotide excision repair. Mech. Ageing Dev. 2013, 134, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, M.; Vermeulen, W.; Van Zeeland, A.A.; Mullenders, L.H.F. Cockayne Syndrome A and B Proteins Differentially Regulate Recruitment of Chromatin Remodeling and Repair Factors to Stalled RNA Polymerase II In Vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Sasaki, K.; Mitsutake, N.; Matsuse, M.; Shimada, M.; Nardo, T.; Takahashi, Y.; Ohyama, K.; Ito, K.; Mishima, H.; et al. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 2012, 44, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.W.; Raams, A.; van der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.J.; Demmers, J.A.A.; Fousteri, M.; Vermeulen, W.; et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef]
- Barakat, K.; Tuszynski, J. Nucleotide Excision Repair Inhibitors: Still a Long Way to Go. New Res. Dir. DNA Repair 2013. [Google Scholar] [CrossRef]
- Alekseev, S.; Ayadi, M.; Brino, L.; Egly, J.-M.; Larsen, A.K.; Coin, F. A Small Molecule Screen Identifies an Inhibitor of DNA Repair Inducing the Degradation of TFIIH and the Chemosensitization of Tumor Cells to Platinum. Chem. Biol. 2014, 21, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Matsuura, K.; Kawai, H.; Wakasugi, M.; Matsunaga, T. Spironolactone-induced XPB degradation depends on CDK7 kinase and SCFFBXL18 E3 ligase. Genes Cells 2019, 24, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M.G.; Krishnamurthy, S.; Kent, M.N.; Schumacher, D.L.; Sharma, P.; Excoffon, K.J.D.A.; Travers, J.B. Spironolactone Depletes the XPB Protein and Inhibits DNA Damage Responses in UVB-Irradiated Human Skin. J. Invest. Dermatol. 2019, 139, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Neher, T.M.; Shuck, S.C.; Liu, J.; Zhang, J.-T.; Turchi, J.J. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem. Biol. 2010, 5, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Yang, L.-Y. Cell Cycle Checkpoint Abrogator UCN-01 Inhibits DNA Repair: Association with Attenuation of the Interaction of XPA and ERCC1 Nucleotide Excision Repair Proteins. Cancer Res. 1999, 59, 4529–4534. [Google Scholar] [PubMed]
- Barakat, K.H.; Jordheim, L.P.; Perez-Pineiro, R.; Wishart, D.; Dumontet, C.; Tuszynski, J.A. Virtual Screening and Biological Evaluation of Inhibitors Targeting the XPA-ERCC1 Interaction. PLoS ONE 2012, 7, e51329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Heyza, J.; Zhang, H.; Kalman-Maltese, V.; Tillison, K.; Floyd, A.M.; Chalfin, E.M.; Bepler, G.; Patrick, S.M. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget 2016, 7, 75104–75117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordheim, L.P.; Barakat, K.H.; Heinrich-Balard, L.; Matera, E.-L.; Cros-Perrial, E.; Bouledrak, K.; El Sabeh, R.; Perez-Pineiro, R.; Wishart, D.S.; Cohen, R.; et al. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol. Pharmacol. 2013, 84, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Elmenoufy, A.H.; Gentile, F.; Jay, D.; Karimi-Busheri, F.; Yang, X.; Soueidan, O.M.; Weilbeer, C.; Mani, R.S.; Barakat, K.H.; Tuszynski, J.A.; et al. Targeting DNA Repair in Tumor Cells via Inhibition of ERCC1-XPF. J. Med. Chem. 2019, 62, 7684–7696. [Google Scholar] [CrossRef]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Tiao, G.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.D.; et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Name | Function | Role in Cisplatin Resistance | References |
|---|---|---|---|
| XPC | Damage recognition | Overexpression of XPC in cancer cells increases cisplatin resistance. | [23,24,40] |
| UV-DDB | Damage recognition | Overexpression of DDB2 sensitizes ovarian cancer cells to cisplatin. | [41] |
| XPB | 3′-5′ helicase | Degradation of XPB by spironolactone increases cisplatin sensitivity. | [77] |
| XPD | 5′-3′ helicase | Somatic mutations of XPD increase cisplatin sensitivity in cells and in bladder cancer patients. | [22,46,47] |
| XPA | Scaffolding protein | Low XPA expression correlates with increased cisplatin sensitivity in testicular tumors. | [7,50] |
| ERCC1 | Partner of XPF | ERCC1 expression in tumor cells may predict cisplatin efficacy. | [18,53,54] |
| XPF | Endonuclease (5′ to lesion) | Low XPF expression correlates with high cisplatin sensitivity. | [18] |
| XPG | Endonuclease (3′ to lesion) | Low XPG expression is associated with good cisplatin response in ovarian patients. | [61] |
| CSB | TC-NER initiation | Knockdown of CSB sensitizes cancer cells to cisplatin. | [25,65] |
| CSA | TC-NER; E3 ligase protein | Knockout of CSA gene sensitizes human cells to oxaliplatin. | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, M.; Ulibarri, J.; Liu, K.J.; Mao, P. Role of Nucleotide Excision Repair in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 9248. https://doi.org/10.3390/ijms21239248
Duan M, Ulibarri J, Liu KJ, Mao P. Role of Nucleotide Excision Repair in Cisplatin Resistance. International Journal of Molecular Sciences. 2020; 21(23):9248. https://doi.org/10.3390/ijms21239248
Chicago/Turabian StyleDuan, Mingrui, Jenna Ulibarri, Ke Jian Liu, and Peng Mao. 2020. "Role of Nucleotide Excision Repair in Cisplatin Resistance" International Journal of Molecular Sciences 21, no. 23: 9248. https://doi.org/10.3390/ijms21239248
APA StyleDuan, M., Ulibarri, J., Liu, K. J., & Mao, P. (2020). Role of Nucleotide Excision Repair in Cisplatin Resistance. International Journal of Molecular Sciences, 21(23), 9248. https://doi.org/10.3390/ijms21239248