Cancer-Associated Muscle Wasting—Candidate Mechanisms and Molecular Pathways


{kind=link}
{kind=link}
Abstract
:1. Introduction
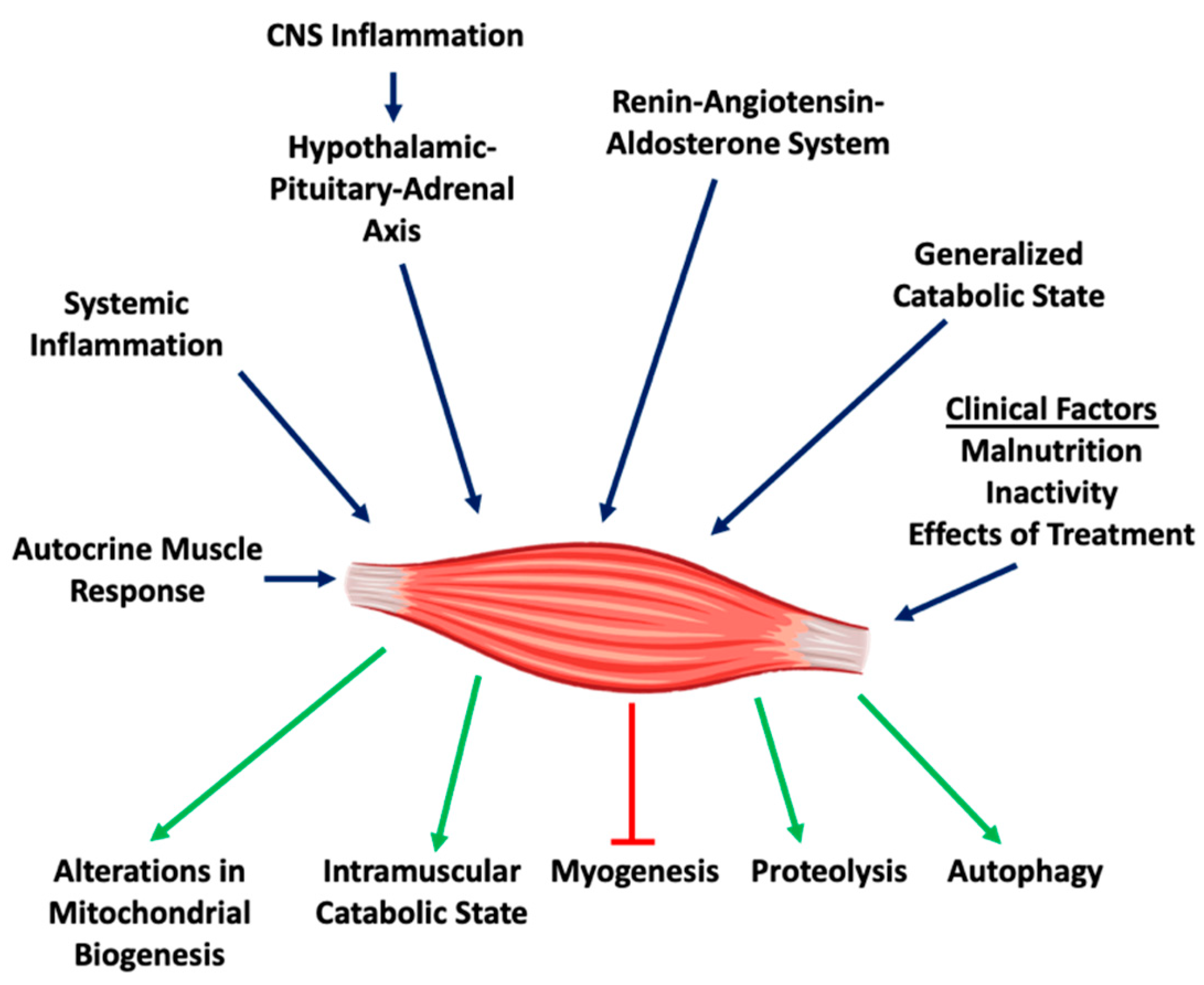
2. Pathophysiological Features of Muscle Wasting
2.1. Systemic Inflammation
2.2. Central Nervous System (CNS) Inflammation
2.3. Generalized and Intramuscular Catabolic States
2.4. Increased Muscle Protein Degradation
2.5. Inhibition of Myoblast Differentiation
2.6. Enhanced Autophagy
2.7. The Renin-Angiotensin Pathway
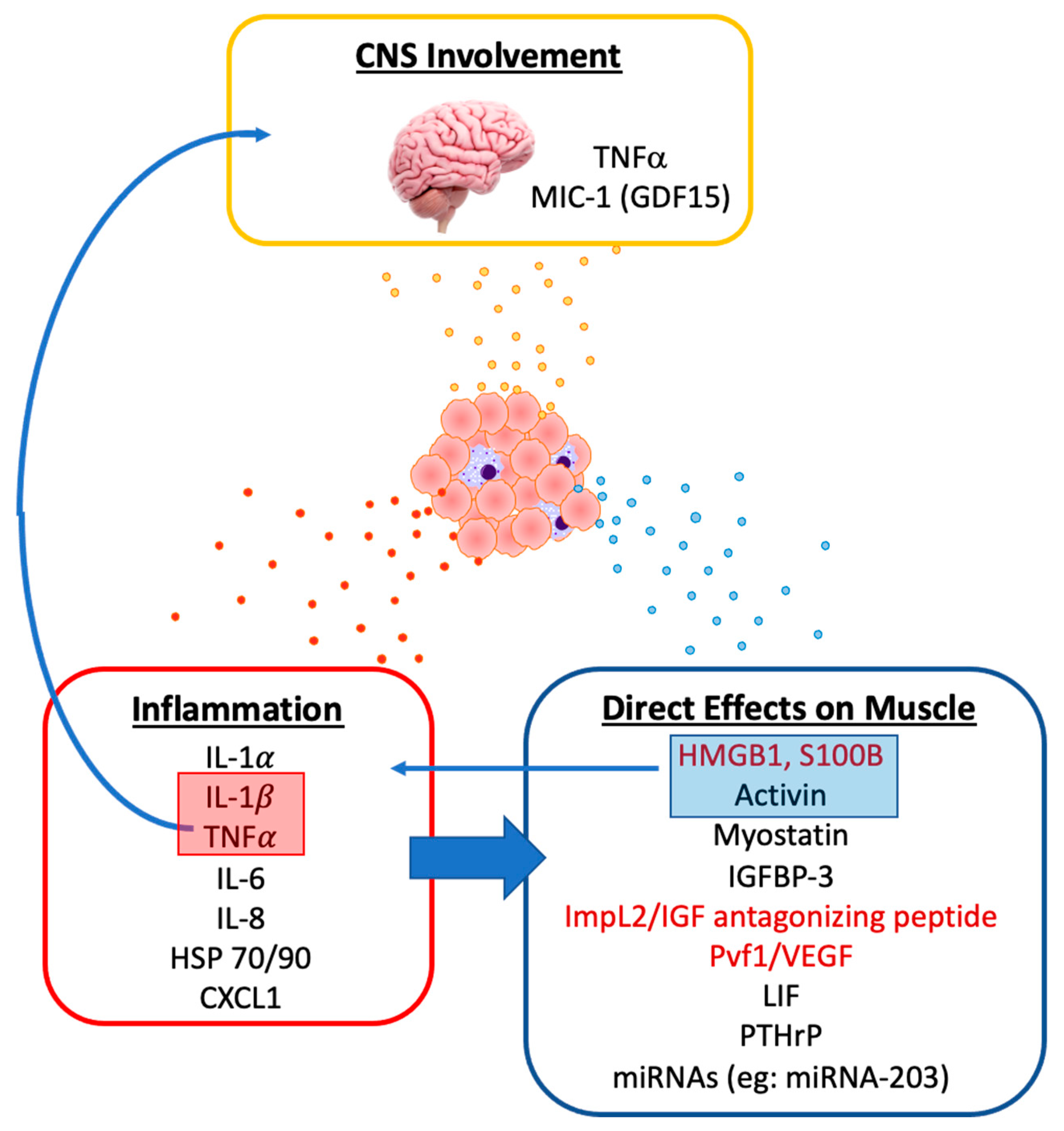
3. Tumor-Derived Mediators of Muscle Wasting
3.1. Evidence for the Role of Tumor on Muscle Wasting
3.2. Tumor-Derived Pro-Inflammatory Mediators
3.3. Tumor-Derived Mediators of Metabolism, Protein Catabolism and Myogenesis
3.4. Other Tumor-Derived Factors Associated with Muscle Wasting
4. Opportunities for Clinical Benefit
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fearon, K.C.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; Macdonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Prado, C.M.M.; Lieffers, J.R.; McCargar, L.J.; Reiman, T.; Sawyer, M.B.; Martin, L.; Baracos, V.E. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: A population-based study. Lancet Oncol. 2008, 9, 629–635. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Vagnildhaug, O.M.; Balstad, T.R.; Almberg, S.S.; Brunelli, C.; Knudsen, A.K.; Kaasa, S.; Thronaes, M.; Laird, B.; Solheim, T.S. A cross-sectional study examining the prevalence of cachexia and areas of unmet need in patients with cancer. Support. Care Cancer 2017, 26, 1871–1880. [Google Scholar] [CrossRef]
- Baracos, V.E.; Mazurak, V.; Bhullar, A.S. Cancer cachexia is defined by an ongoing loss of skeletal muscle mass. Ann. Palliat. Med. 2019, 8, 3–12. [Google Scholar] [CrossRef]
- Cornet, M.; Lim, C.; Salloum, C.; Lazzati, A.; Compagnon, P.; Pascal, G.; Azoulay, D. Prognostic value of sarcopenia in liver surgery. J. Visc. Surg. 2015, 152, 297–304. [Google Scholar] [CrossRef]
- Prado, C.M.M.; Baracos, V.E.; McCargar, L.J.; Reiman, T.; Mourtzakis, M.; Tonkin, K.; Mackey, J.R.; Koski, S.; Pituskin, E.; Sawyer, M.B. Sarcopenia as a Determinant of Chemotherapy Toxicity and Time to Tumor Progression in Metastatic Breast Cancer Patients Receiving Capecitabine Treatment. Clin. Cancer Res. 2009, 15, 2920–2926. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-M.; Dou, Q.-L.; Zeng, Y.; Yang, Y.; Cheng, A.S.; Zhang, W.-W. Sarcopenia as a predictor of mortality in women with breast cancer: A meta-analysis and systematic review. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F.R. Cancer-related inflammation. Nat. Cell Biol. 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J.; Busquets, S. Mediators of cachexia in cancer patients. Nutrition 2019, 66, 11–15. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Dolan, R.D.; McSorley, S.T.; Horgan, P.G.; Laird, B.; McMillan, D.C. The role of the systemic inflammatory response in predicting outcomes in patients with advanced inoperable cancer: Systematic review and meta-analysis. Crit. Rev. Oncol. 2017, 116, 134–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.; Chen, X.; He, W.; Wang, H.; Wang, Y.; Hu, P.; Rong, Y.; Fan, L.; Xia, L. Establishment of inflammation biomarkers-based nomograms to predict prognosis of advanced colorectal cancer patients based on real world data. PLoS ONE 2018, 13, e0208547. [Google Scholar] [CrossRef]
- Hamilton, T.D.; Leugner, D.; Kopciuk, K.; Dixon, E.; Sutherland, F.R.; Bathe, O.F. Identification of prognostic inflammatory factors in colorectal liver metastases. BMC Cancer 2014, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Miyata, H.; Sugimura, K.; Motoori, M.; Asukai, K.; Yanagimoto, Y.; Takahashi, Y.; Tomokuni, A.; Yamamoto, K.; Akita, H.; et al. Prognostic Significance of Sarcopenia and Systemic Inflammatory Response in Patients With Esophageal Cancer. Anticancer Res. 2018, 39, 449–458. [Google Scholar] [CrossRef]
- Nakayama, T.; Saito, K.; Kumagai, J.; Nakajima, Y.; Kijima, T.; Yoshida, S.; Kihara, K.; Fujii, Y. Higher Serum C-reactive Protein Level Represents the Immunosuppressive Tumor Microenvironment in Patients With Clear Cell Renal Cell Carcinoma. Clin. Genitourin. Cancer 2018, 16, e1151–e1158. [Google Scholar] [CrossRef]
- Deans, C.; Wigmore, S.J. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 265–269. [Google Scholar] [CrossRef]
- Heikkilä, K.; Ebrahim, S.; Lawlor, D.A. A systematic review of the association between circulating concentrations of C reactive protein and cancer. J. Epidemiol. Community Health 2007, 61, 824–833. [Google Scholar] [CrossRef]
- Graff, J.; Lalani, A.S.; Lee, S.; Curd, J.G.; Henner, W.D.; Ryan, C.W.; Venner, P.M.; Ruether, J.D.; Chi, K.N.; Beer, T.M.; et al. C-reactive protein as a prognostic marker for men with androgen-independent prostate cancer (AIPC): Results from the ASCENT trial. J. Clin. Oncol. 2007, 25, 5074. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ikeda, Y.; Korenaga, D.; Tanoue, K.; Hamatake, M.; Kawasaki, K.; Yamaoka, T.; Iwatani, Y.; Akazawa, K.; Takenaka, K. The impact of preoperative serum C-reactive protein on the prognosis of patients with hepatocellular carcinoma. Cancer 2005, 103, 1856–1864. [Google Scholar] [CrossRef]
- Hefler, L.A.; Concin, N.; Hofstetter, G.; Marth, C.; Mustea, A.; Sehouli, J.; Zeillinger, R.; Leipold, H.; Lass, H.; Grimm, C.; et al. Serum C-Reactive Protein as Independent Prognostic Variable in Patients with Ovarian Cancer. Clin. Cancer Res. 2008, 14, 710–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, M.; Natsugoe, S.; Ueno, S.; Baba, M.; Aikou, T. Significant Host- and Tumor-Related Factors for Predicting Prognosis in Patients With Esophageal Carcinoma. Ann. Surg. 2003, 238, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, N.B.; Glen, P.; McMillan, D.C.; McKay, C.J.; Foulis, A.K.; Carter, R.; Imrie, C.W. Systemic inflammatory response predicts outcome in patients undergoing resection for ductal adenocarcinoma head of pancreas. Br. J. Cancer 2004, 92, 21–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polterauer, S.; Grimm, C.; Tempfer, C.; Sliutz, G.; Speiser, P.; Reinthaller, A.; Hefler, L.A. C-reactive protein is a prognostic parameter in patients with cervical cancer. Gynecol. Oncol. 2007, 107, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Schneitter, A.; Hinterberger, S.; Seeber, J.; Reinthaller, A.; Hefler, L. Association of Elevated C-reactive Protein Levels With an Impaired Prognosis in Patients With Surgically Treated Endometrial Cancer. Obstet. Gynecol. 2007, 110, 1231–1236. [Google Scholar] [CrossRef]
- Bano, G.; Trevisan, C.; Carraro, S.; Solmi, M.; Luchini, C.; Stubbs, B.; Manzato, E.; Sergi, G.; Veronese, N. Inflammation and sarcopenia: A systematic review and meta-analysis. Maturitas 2017, 96, 10–15. [Google Scholar] [CrossRef]
- Okugawa, Y.; Toiyama, Y.; Yamamoto, A.; Shigemori, T.; Kitamura, A.; Ichikawa, T.; Ide, S.; Kitajima, T.; Fujikawa, H.; Yasuda, H.; et al. Close Relationship Between Immunological/Inflammatory Markers and Myopenia and Myosteatosis in Patients With Colorectal Cancer: A Propensity Score Matching Analysis. J. Parenter. Enter. Nutr. 2019, 43, 508–515. [Google Scholar] [CrossRef]
- Srdic, D.; Plestina, S.; Sverko-Peternac, A.; Nikolac, N.; Simundic, A.-M.; Samarzija, M. Cancer cachexia, sarcopenia and biochemical markers in patients with advanced non-small cell lung cancer—Chemotherapy toxicity and prognostic value. Support. Care Cancer 2016, 24, 4495–4502. [Google Scholar] [CrossRef]
- Volanakis, J.E. Human C-reactive protein: Expression, structure, and function. Mol. Immunol. 2001, 38, 189–197. [Google Scholar] [CrossRef]
- Scheede-Bergdahl, C.; Watt, H.L.; Trutschnigg, B.; Kilgour, R.D.; Haggarty, A.; Lucar, E.; Vigano, A. Is IL-6 the best pro-inflammatory biomarker of clinical outcomes of cancer cachexia? Clin. Nutr. 2012, 31, 85–88. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strassmann, G.; Fong, M.; Freter, C.E.; Windsor, S.; D’Alessandro, F.; Nordan, R.P. Suramin interferes with interleukin-6 receptor binding in vitro and inhibits colon-26-mediated experimental cancer cachexia in vivo. J. Clin. Investig. 1993, 92, 2152–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strassmann, G.; Fong, M.; Kenney, J.S.; Jacob, C.O. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J. Clin. Investig. 1992, 89, 1681–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, S.; Ouchi, K.F.; Mori, K.; Endo, M.; Matsumoto, T.; Eda, H.; Tanaka, Y.; Ishitsuka, H.; Tokita, H.; Yamaguchi, K. Involvement of human interleukin 6 in experimental cachexia induced by a human uterine cervical carcinoma xenograft. Clin. Cancer Res. 1995, 1, 1353–1358. [Google Scholar] [PubMed]
- Scott, H.R.; McMillan, D.C.; Crilly, A.; McArdle, C.S.; Milroy, R. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br. J. Cancer 1996, 73, 1560–1562. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.A.; Moses, A.G.; Maingay, J.; Sangster, K.; Fearon, K.C. Pro-inflammatory cytokine release by peripheral blood mononuclear cells from patients with advanced pancreatic cancer: Relationship to acute phase response and survival. Oncol. Rep. 2009, 21, 1091–1095. [Google Scholar] [CrossRef]
- Authier, F.J.; Chazaud, B.; Plonquet, A.; Eliezer-Vanerot, M.C.; Poron, F.; Belec, L.; Barlovatz-Meimon, G.; Gherardi, R.K. Differential expression of the IL-1 system components during in vitro myogenesis: Implication of IL-1beta in induction of myogenic cell apoptosis. Cell Death Differ. 1999, 6, 1012–1021. [Google Scholar] [CrossRef]
- Luo, G.; Hershko, D.D.; Robb, B.W.; Wray, C.J.; Hasselgren, P.O. IL-1beta stimulates IL-6 production in cultured skeletal muscle cells through activation of MAP kinase signaling pathway and NF-kappa B. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1249–R1254. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Wang, C.-J.; Chao, Y.-J.; Chen, H.-Y.; Wang, H.-C.; Tung, H.-L.; Lin, J.-T.; Shan, Y.-S. Elevated Serum Interleukin-8 Level Correlates with Cancer-Related Cachexia and Sarcopenia: An Indicator for Pancreatic Cancer Outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef] [Green Version]
- Bo, S.; Dianliang, Z.; Hongmei, Z.; XinXiang, W.; Yanbing, Z.; Xiaobo, L. Association of Interleukin-8 Gene Polymorphism With Cachexia From Patients With Gastric Cancer. J. Interf. Cytokine Res. 2010, 30, 9–14. [Google Scholar] [CrossRef]
- Callaway, C.S.; Delitto, A.E.; Patel, R.; Nosacka, R.L.; D’Lugos, A.C.; Delitto, D.; Deyhle, M.R.; Trevino, J.G.; Judge, S.M.; Judge, A.R. IL-8 Released from Human Pancreatic Cancer and Tumor-Associated Stromal Cells Signals through a CXCR2-ERK1/2 Axis to Induce Muscle Atrophy. Cancers 2019, 11, 1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milewska, M.; Domoradzki, T.; Majewska, A.; Blaszczyk, M.; Gajewska, M.; Hulanicka, M.; Ciecierska, A.; Grzelkowska-Kowalczyk, K. Interleukin-8 enhances myocilin expression, Akt-FoxO3 signaling and myogenic differentiation in rat skeletal muscle cells. J. Cell Physiol. 2019, 234, 19675–19690. [Google Scholar] [CrossRef] [PubMed]
- Dogra, C.; Changotra, H.; Wedhas, N.; Qin, X.; Wergedal, J.E.; Kumar, A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007, 21, 1857–1869. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yu, K.; Shyh-Chang, N.; Li, G.; Jiang, L.; Yu, S.; Xu, L.; Liu, R.; Guo, Z.; Xie, H.; et al. Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J. Cachex Sarcopenia Muscle 2019, 10, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Welc, S.S.; Wehling-Henricks, M.; Tidball, J.G. Myeloid cell-derived tumor necrosis factor-alpha promotes sarcopenia and regulates muscle cell fusion with aging muscle fibers. Aging Cell 2018, 17, e12828. [Google Scholar] [CrossRef]
- Coletti, D.; Moresi, V.; Adamo, S.; Molinaro, M.; Sassoon, D.A. Tumor necrosis factor-α gene transfer induces cachexia and inhibits muscle regeneration. Genes 2005, 43, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Saeman, M.R.; De Libero, J.; Wolf, S.E. Skeletal Muscle Loss is Associated with TNF Mediated Insufficient Skeletal Myogenic Activation After Burn. Shock 2015, 44, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Talbert, E.E.; Lewis, H.L.; Farren, M.R.; Ramsey, M.L.; Chakedis, J.M.; Rajasekera, P.; Haverick, E.; Sarna, A.; Bloomston, M.; Pawlik, T.M.; et al. Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J. Cachex Sarcopenia Muscle 2018, 9, 358–368. [Google Scholar] [CrossRef]
- Braun, T.P.; Zhu, X.; Szumowski, M.; Scott, G.D.; Grossberg, A.J.; Levasseur, P.R.; Graham, K.; Khan, S.; Damaraju, S.; Colmers, W.F.; et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic–pituitary-adrenal axis. J. Exp. Med. 2011, 208, 2449–2463. [Google Scholar] [CrossRef]
- Falconer, J.S.; Fearon, K.C.H.; Plester, C.E.; Ross, J.A.; Carter, D.C. Cytokines, the Acute-Phase Response, and Resting Energy Expenditure in Cachectic Patients with Pancreatic Cancer. Ann. Surg. 1994, 219, 325–331. [Google Scholar] [CrossRef]
- Fredrix, E.W.; Soeters, P.B.; Wouters, E.F.; Deerenberg, I.M.; Von Meyenfeldt, M.F.; Saris, W.H. Effect of different tumor types on resting energy expenditure. Cancer Res. 1991, 51, 6138–6141. [Google Scholar] [PubMed]
- Peacock, J.L.; Inculet, R.I.; Corsey, R.; Ford, D.B.; Rumble, W.F.; Lawson, D.; Norton, J.A. Resting energy expenditure and body cell mass alterations in noncachectic patients with sarcomas. Surgery 1987, 102, 465–472. [Google Scholar]
- Purcell, S.A.; Elliott, S.A.; Baracos, V.E.; Chu, Q.S.C.; Prado, C.M. Key determinants of energy expenditure in cancer and implications for clinical practice. Eur. J. Clin. Nutr. 2016, 70, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Vanhorebeek, I.; Berghe, G.V.D. The Neuroendocrine Response to Critical Illness is a Dynamic Process. Crit. Care Clin. 2006, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.R.; McMillan, D.C.; Watson, W.S.; Milroy, R.; McArdle, C.S. Longitudinal study of resting energy expenditure, body cell mass and the inflammatory response in male patients with non-small cell lung cancer. Lung Cancer 2001, 32, 307–312. [Google Scholar] [CrossRef]
- Brekel, A.J.S.-V.D.; Dentener, M.A.; Schols, A.M.; Buurman, W.A.; Wouters, E.F. Increased resting energy expenditure and weight loss are related to a systemic inflammatory response in lung cancer patients. J. Clin. Oncol. 1995, 13, 2600–2605. [Google Scholar] [CrossRef]
- Wu, J.; Huang, C.; Xiao, H.; Tang, Q.; Cai, W. Weight loss and resting energy expenditure in male patients with newly diagnosed esophageal cancer. Nutrition 2013, 29, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Bartoccioni, E.; Michaelis, D.; Hohlfeld, R. Constitutive and cytokine-induced production of interleukin-6 by human myoblasts. Immunol. Lett. 1994, 42, 135–138. [Google Scholar] [CrossRef]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Hindi, S.M.; Mishra, V.; Bhatnagar, S.; Tajrishi, M.M.; Ogura, Y.; Yan, Z.; Burkly, L.C.; Zheng, T.S.; Kumar, A. Regulatory circuitry of TWEAK-Fn14 system and PGC-1α in skeletal muscle atrophy program. FASEB J. 2014, 28, 1398–1411. [Google Scholar] [CrossRef] [Green Version]
- De Castro, G.S.; Simoes, E.; Correia-Lima, J.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcântara, P.S.M.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef] [Green Version]
- White, J.P.; Baltgalvis, K.A.; Puppa, M.J.; Sato, S.; Baynes, J.W.; Carson, J.A. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R201–R211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padrão, A.I.; Oliveira, P.A.; Vitorino, R.; Colaço, B.; Pires, M.J.; Marquez, M.; Castellanos, E.; Neuparth, M.J.; Teixeira-Guedes, C.I.; Costa, C.; et al. Bladder cancer-induced skeletal muscle wasting: Disclosing the role of mitochondria plasticity. Int. J. Biochem. Cell Biol. 2013, 45, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachex Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [Green Version]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- García-Prat, L.; Perdiguero, E.; Alonso-Martín, S.; Dell’Orso, S.; Ravichandran, S.; Brooks, S.R.; Juan, A.H.; Campanario, S.; Jiang, K.; Hong, X.; et al. FoxO maintains a genuine muscle stem-cell quiescent state until geriatric age. Nat. Cell Biol. 2020, 22, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Yin, P.; Duan, C. IGFBP-5 regulates muscle cell differentiation by binding to IGF-II and switching on the IGF-II auto-regulation loop. J. Cell Biol. 2008, 182, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-Y.; Huang, Z.-L.; Yang, J.-H.; Xu, Y.-H.; Sun, J.-S.; Zheng, Q.; Wei, C.; Song, W.; Yuan, Z. Pancreatic cancer cell-derived IGFBP-3 contributes to muscle wasting. J. Exp. Clin. Cancer Res. 2016, 35, 46. [Google Scholar] [CrossRef] [Green Version]
- Salih, D.A.M.; Tripathi, G.; Holding, C.; Szestak, T.A.M.; Gonzalez, M.I.; Carter, E.J.; Cobb, L.J.; Eisemann, J.E.; Pell, J.M. Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 4314–4319. [Google Scholar] [CrossRef] [Green Version]
- Acharyya, S.; Ladner, K.J.; Nelsen, L.L.; Damrauer, J.; Reiser, P.J.; Swoap, S.; Guttridge, D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Investig. 2004, 114, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 Coordinately Activates Protein Degradation by the Autophagic/Lysosomal and Proteasomal Pathways in Atrophying Muscle Cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, V.; Mangner, N.; Gasch, A.; Krohne, C.; Gielen, S.; Hirner, S.; Thierse, H.-J.; Witt, C.C.; Linke, A.; Schuler, G.; et al. Induction of MuRF1 Is Essential for TNF-α-Induced Loss of Muscle Function in Mice. J. Mol. Biol. 2008, 384, 48–59. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef] [Green Version]
- Zimmers, T.A.; Davies, M.V.; Koniaris, L.G.; Haynes, P.A.; Esquela, A.F.; Tomkinson, K.N.; McPherron, A.C.; Wolfman, N.M.; Lee, S.-J. Induction of Cachexia in Mice by Systemically Administered Myostatin. Science 2002, 296, 1486–1488. [Google Scholar] [CrossRef] [Green Version]
- Sulyok, S.; Wankell, M.; Alzheimer, C.; Werner, S. Activin: An important regulator of wound repair, fibrosis, and neuroprotection. Mol. Cell. Endocrinol. 2004, 225, 127–132. [Google Scholar] [CrossRef]
- Chen, J.L.; Walton, K.L.; Winbanks, C.E.; Murphy, K.T.; Thomson, R.E.; Makanji, Y.; Qian, H.; Lynch, G.S.; Harrison, C.A.; Gregorevic, P. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014, 28, 1711–1723. [Google Scholar] [CrossRef]
- Sidis, Y.; Mukherjee, A.; Keutmann, H.; Delbaere, A.; Sadatsuki, M.; Schneyer, A. Biological Activity of Follistatin Isoforms and Follistatin-Like-3 Is Dependent on Differential Cell Surface Binding and Specificity for Activin, Myostatin, and Bone Morphogenetic Proteins. Endocrinology 2006, 147, 3586–3597. [Google Scholar] [CrossRef] [Green Version]
- Harada, K.; Shintani, Y.; Sakamoto, Y.; Wakatsuki, M.; Shitsukawa, K.; Saito, S. Serum immunoreactive activin A levels in normal subjects and patients with various diseases. J. Clin. Endocrinol. Metab. 1996, 81, 2125–2130. [Google Scholar] [PubMed] [Green Version]
- Leto, G.; Incorvaia, L.; Badalamenti, G.; Tumminello, F.M.; Gebbia, N.; Flandina, C.; Crescimanno, M.; Rini, G. Activin A circulating levels in patients with bone metastasis from breast or prostate cancer. Clin. Exp. Metastasis 2006, 23, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbart, A.; Scharf, G.M.; Duncker, D.; Widera, C.; Gottlieb, J.; Vogel, A.; Schmidt, S.; Brandes, G.; Heuft, H.-G.; Lichtinghagen, R.; et al. Highly Specific Detection of Myostatin Prodomain by an Immunoradiometric Sandwich Assay in Serum of Healthy Individuals and Patients. PLoS ONE 2013, 8, e80454. [Google Scholar] [CrossRef] [PubMed]
- Loumaye, A.; De Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; Van Maanen, A.; Tréfois, P.; Gruson, A.D.; Thissen, J.P. Role of Activin A and Myostatin in Human Cancer Cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef] [Green Version]
- Dogra, C.; Hall, S.L.; Wedhas, N.; Linkhart, T.A.; Kumar, A. Fibroblast growth factor inducible 14 (Fn14) is required for the expression of myogenic regulatory factors and differentiation of myoblasts into myotubes. Evidence for TWEAK-independent functions of Fn14 during myogenesis. J. Biol. Chem. 2007, 282, 15000–15010. [Google Scholar] [CrossRef] [Green Version]
- Dogra, C.; Changotra, H.; Mohan, S.; Kumar, A. Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J. Biol. Chem. 2006, 281, 10327–10336. [Google Scholar] [CrossRef] [Green Version]
- Van Rooij, E.; Liu, N.; Olson, E.N. MicroRNAs flex their muscles. Trends Genet. 2008, 24, 159–166. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef] [Green Version]
- Langen, R.C.J.; Van Der Velden, J.L.J.; Schols, A.M.W.J.; Kelders, M.C.J.M.; Wouters, E.F.M.; Janssen-Heininger, Y.M.W. Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J. 2004, 18, 227–237. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nat. Cell Biol. 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol. 2017, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- White, Z.; Terrill, J.; White, R.B.; McMahon, C.; Sheard, P.; Grounds, M.D.; Shavlakadze, T. Voluntary resistance wheel exercise from mid-life prevents sarcopenia and increases markers of mitochondrial function and autophagy in muscles of old male and female C57BL/6J mice. Skelet. Muscle 2016, 6, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- White, Z.; White, R.B.; McMahon, C.; Grounds, M.D.; Shavlakadze, T. High mTORC1 signaling is maintained, while protein degradation pathways are perturbed in old murine skeletal muscles in the fasted state. Int. J. Biochem. Cell Biol. 2016, 78, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic Degradation Contributes to Muscle Wasting in Cancer Cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argilés, J.M.; Costelli, P.; Zorzano, A. Autophagy Exacerbates Muscle Wasting in Cancer Cachexia and Impairs Mitochondrial Function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef]
- Pettersen, K.; Andersen, S.; Van Der Veen, A.; Nonstad, U.; Hatakeyama, S.; Lambert, C.; Lach-Trifilieff, E.; Moestue, S.; Kim, J.; Grønberg, B.H.; et al. Autocrine activin A signalling in ovarian cancer cells regulates secretion of interleukin 6, autophagy, and cachexia. J. Cachex Sarcopenia Muscle 2020, 11, 195–207. [Google Scholar] [CrossRef] [Green Version]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, K.; Andersen, S.; Degen, S.; Tadini, V.; Grosjean, J.; Hatakeyama, S.; Tesfahun, A.N.; Moestue, S.A.; Kim, J.; Nonstad, U.; et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017, 7, 2046. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616. [Google Scholar] [CrossRef] [Green Version]
- Eley, H.L.; Tisdale, M.J. Skeletal Muscle Atrophy, a Link between Depression of Protein Synthesis and Increase in Degradation. J. Biol. Chem. 2007, 282, 7087–7097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.-H.; Li, Y.; Du, J.; Mitch, W.E.; Rosenthal, N.; Delafontaine, P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Investig. 2005, 115, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Semprun-Prieto, L.; Sukhanov, S.; Delafontaine, P. IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am. J. Physiol. Circ. Physiol. 2010, 298, H1565–H1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabello-Verrugio, C.; Morales, M.G.; Rivera, J.C.; Cabrera, D.; Simon, F. Renin-Angiotensin System: An Old Player with Novel Functions in Skeletal Muscle. Med. Res. Rev. 2015, 35, 437–463. [Google Scholar] [CrossRef]
- Brink, M.; Price, S.R.; Chrast, J.; Bailey, J.L.; Anwar, A.; Mitch, W.E.; Delafontaine, P. Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology 2001, 142, 1489–1496. [Google Scholar] [CrossRef]
- Brink, M.; Wellen, J.; Delafontaine, P. Angiotensin II causes weight loss and decreases circulating insulin-like growth factor I in rats through a pressor-independent mechanism. J. Clin. Investig. 1996, 97, 2509–2516. [Google Scholar] [CrossRef] [Green Version]
- Delafontaine, P.; Yoshida, T. The Renin-Angiotensin System and the Biology of Skeletal Muscle: Mechanisms of Muscle Wasting in Chronic Disease States. Trans. Am. Clin. Clim. Assoc. 2016, 127, 245–258. [Google Scholar]
- Freire, P.P.; Fernandez, G.J.; De Moraes, D.; Cury, S.S.; Dal-Pai-Silva, M.; Reis, P.P.; Rogatto, S.R.; Carvalho, R.F. The expression landscape of cachexia-inducing factors in human cancers. J. Cachex Sarcopenia Muscle 2020, 11, 947–961. [Google Scholar] [CrossRef] [Green Version]
- Cury, S.S.; De Moraes, D.; Freire, P.P.; De Oliveira, G.; Marques, D.V.P.; Fernandez, G.J.; Dal-Pai-Silva, M.; Hasimoto, É.N.; Dos Reis, P.P.; Rogatto, S.R.; et al. Tumor Transcriptome Reveals High Expression of IL-8 in Non-Small Cell Lung Cancer Patients with Low Pectoralis Muscle Area and Reduced Survival. Cancers 2019, 11, 1251. [Google Scholar] [CrossRef] [Green Version]
- Capece, D.; Verzella, D.; Tessitore, A.; Alesse, E.; Capalbo, C.; Zazzeroni, F. Cancer secretome and inflammation: The bright and the dark sides of NF-kappaB. Semin. Cell Dev. Biol. 2018, 78, 51–61. [Google Scholar] [CrossRef]
- Johnston, A.J.; Murphy, K.T.; Jenkinson, L.; Laine, D.; Emmrich, K.; Faou, P.; Weston, R.; Jayatilleke, K.M.; Schloegel, J.; Talbo, G.H.; et al. Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 2015, 162, 1365–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019, 156, 722–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pin, F.; Barreto, R.; Kitase, Y.; Mitra, S.; Erne, C.E.; Novinger, L.J.; Zimmers, T.A.; Couch, M.E.; Bonewald, L.F.; Bonetto, A. Growth of ovarian cancer xenografts causes loss of muscle and bone mass: A new model for the study of cancer cachexia. J. Cachex Sarcopenia Muscle 2018, 9, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, K.A.; Cho, D.S.; Arneson-Wissink, P.C.; Samani, A.; Palines, P.; Yang, Y.; Doles, J.D. Tumor-derived cytokines impair myogenesis and alter the skeletal muscle immune microenvironment. Cytokine 2018, 107, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 2017, 17, 165–178. [Google Scholar] [CrossRef]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 Paracrine Network Links Cancer Chemoresistance and Metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Eck, M.; Schmaußer, B.; Scheller, K.; Brändlein, S.; Müller-Hermelink, H.K. Pleiotropic effects of CXC chemokines in gastric carcinoma: Differences in CXCL8 and CXCL1 expression between diffuse and intestinal types of gastric carcinoma. Clin. Exp. Immunol. 2003, 134, 508–515. [Google Scholar] [CrossRef]
- Lo, H.-M.; Shieh, J.-M.; Chen, C.-L.; Tsou, C.-J.; Wu, W.-B. Vascular Endothelial Growth Factor Induces CXCL1 Chemokine Release via JNK and PI-3K-Dependent Pathways in Human Lung Carcinoma Epithelial Cells. Int. J. Mol. Sci. 2013, 14, 10090–10106. [Google Scholar] [CrossRef] [Green Version]
- Miyake, M.; Hori, S.; Morizawa, Y.; Tatsumi, Y.; Nakai, Y.; Anai, S.; Torimoto, K.; Aoki, K.; Tanaka, N.; Shimada, K.; et al. CXCL1-Mediated Interaction of Cancer Cells with Tumor-Associated Macrophages and Cancer-Associated Fibroblasts Promotes Tumor Progression in Human Bladder Cancer. Neoplasia 2016, 18, 636–646. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Kir, S.; Hong, S.; Hu, Y.; Wang, X.; Binari, R.; Tang, H.-W.; Chung, V.; Banks, A.S.; Spiegelman, B.; et al. Tumor-Derived Ligands Trigger Tumor Growth and Host Wasting via Differential MEK Activation. Dev. Cell 2019, 48, 277–286.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Ohkawa, S.; Li, H.; Roberts-Wilson, T.K.; Price, S.R. FOXO3a mediates signaling crosstalk that coordinates ubiquitin and atrogin-1/MAFbx expression during glucocorticoid-induced skeletal muscle atrophy. FASEB J. 2010, 24, 2660–2669. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta Bioenerg. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Song, W.; Droujinine, I.A.; Hu, Y.; Asara, J.M.; Perrimon, N. Systemic Organ Wasting Induced by Localized Expression of the Secreted Insulin/IGF Antagonist ImpL2. Dev. Cell 2015, 33, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, D.N.; Kandarian, S.C.; Jackman, R.W. A Key Role for Leukemia Inhibitory Factor in C26 Cancer Cachexia. J. Biol. Chem. 2015, 290, 19976–19986. [Google Scholar] [CrossRef] [Green Version]
- Kandarian, S.C.; Nosacka, R.L.; Delitto, A.E.; Judge, A.R.; Judge, S.M.; Ganey, J.D.; Moreira, J.D.; Jackman, R.W. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachex Sarcopenia Muscle 2018, 9, 1109–1120. [Google Scholar] [CrossRef]
- Luo, Y.; Chihara, Y.; Fujimoto, K.; Sasahira, T.; Kuwada, M.; Fujiwara, R.; Fujii, K.; Ohmori, H.; Kuniyasu, H. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur. J. Cancer 2013, 49, 741–751. [Google Scholar] [CrossRef]
- Michetti, F.; D’Ambrosi, N.; Toesca, A.; Puglisi, M.A.; Serrano, A.; Marchese, E.; Corvino, V.; Geloso, M.C. The S100B story: From biomarker to active factor in neural injury. J. Neurochem. 2018, 148, 168–187. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Dormoy-Raclet, V.; Cammas, A.; Celona, B.; Lian, X.J.; Van Der Giessen, K.; Zivojnovic, M.; Brunelli, S.; Riuzzi, F.; Sorci, G.; Wilhelm, B.T.; et al. HuR and miR-1192 regulate myogenesis by modulating the translation of HMGB1 mRNA. Nat. Commun. 2013, 4, 2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riuzzi, F.; Beccafico, S.; Sagheddu, R.; Chiappalupi, S.; Giambanco, I.; Bereshchenko, O.; Riccardi, C.; Sorci, G.; Donato, R. Levels of S100B protein drive the reparative process in acute muscle injury and muscular dystrophy. Sci. Rep. 2017, 7, 12537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuniyasu, H.; Chihara, Y.; Takahashi, T. Co-expression of receptor for advanced glycation end products and the ligand amphoterin associates closely with metastasis of colorectal cancer. Oncol. Rep. 2003, 10, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Sasahira, T.; Kirita, T.; Bhawal, U.K.; Ohmori, H.; Kuniyasu, H.; Yamamoto, K.; Fujii, K. Receptor for advanced glycation end products (RAGE) is important in the prediction of recurrence in human oral squamous cell carcinoma. Histopathology 2007, 51, 166–172. [Google Scholar] [CrossRef]
- Jing, R.; Cui, M.; Wang, J.; Wang, H. Receptor for advanced glycation end products (RAGE) soluble form (sRAGE): A new biomarker for lung cancer. Neoplasma 2010, 57, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Harpio, R.; Einarsson, R. S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. Clin. Biochem. 2004, 37, 512–518. [Google Scholar] [CrossRef]
- Lee, H.; Song, M.; Shin, N.; Shin, C.H.; Min, B.S.; Kim, H.-S.; Yoo, J.S.; Kim, H. Diagnostic Significance of Serum HMGB1 in Colorectal Carcinomas. PLoS ONE 2012, 7, e34318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, R.-Q.; Wu, X.-J.; Ding, Y.; Li, C.; Yu, X.-J.; Zhang, X.; Pan, Z.-Z.; Wan, D.-S.; Zheng, L.; Zeng, Y.-X.; et al. Co-expression of nuclear and cytoplasmic HMGB1 is inversely associated with infiltration of CD45RO+ T cells and prognosis in patients with stage IIIB colon cancer. BMC Cancer 2010, 10, 496. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, M.; Zhou, L.; Feng, X.; Cheng, J.; Yü, Y.; Gong, Y.; Zhu, Y.; Li, C.-Y.; Tian, L.; et al. Increased HMGB1 and cleaved caspase-3 stimulate the proliferation of tumor cells and are correlated with the poor prognosis in colorectal cancer. J. Exp. Clin. Cancer Res. 2015, 34, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-H.; Chen, C.-M.; Chen, H.-Y.; Yang, P.-C. Pathway-based gene signatures predicting clinical outcome of lung adenocarcinoma. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Ohmori, H.; Fujii, K.; Moriwaka, Y.; Sasahira, T.; Kurihara, M.; Tatsumoto, N.; Sasaki, T.; Yamashita, Y.; Kuniyasu, H. HMGB1 attenuates anti-metastatic defence of the liver in colorectal cancer. Eur. J. Cancer 2010, 46, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachex Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Yoneda, J.; Ohmori, H.; Sasaki, T.; Shimbo, K.; Eto, S.; Kato, Y.; Miyano, H.; Kobayashi, T.; Sasahira, T.; et al. Cancer Usurps Skeletal Muscle as an Energy Repository. Cancer Res. 2013, 74, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nat. Cell Biol. 2014, 513, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Toiyama, Y.; Okugawa, Y.; Ide, S.; Imaoka, H.; Boland, C.R.; Goel, A. Circulating microRNA-203 predicts prognosis and metastasis in human colorectal cancer. Gut 2017, 66, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Okugawa, Y.; Toiyama, Y.; Hur, K.; Yamamoto, A.; Yin, C.; Ide, S.; Kitajima, T.; Fujikawa, H.; Yasuda, H.; Koike, Y.; et al. Circulating miR-203 derived from metastatic tissues promotes myopenia in colorectal cancer patients. J. Cachex Sarcopenia Muscle 2019, 10, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Horak, M.; Novák, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef]
- Borner, T.; Arnold, M.; Ruud, J.; Breit, S.N.; Langhans, W.; Lutz, T.A.; Blomqvist, A.; Riediger, T. Anorexia-cachexia syndrome in hepatoma tumour-bearing rats requires the area postrema but not vagal afferents and is paralleled by increased MIC-1/GDF15. J. Cachex Sarcopenia Muscle 2016, 8, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Bauskin, A.R.; Brown, D.A.; Kuffner, T.; Johnen, H.; Luo, X.W.; Hunter, M.; Breit, S.N. Role of Macrophage Inhibitory Cytokine-1 in Tumorigenesis and Diagnosis of Cancer: Figure 1. Cancer Res. 2006, 66, 4983–4986. [Google Scholar] [CrossRef] [Green Version]
- Welsh, J.B.; Sapinoso, L.M.; Kern, S.G.; Brown, D.A.; Liu, T.; Bauskin, A.R.; Ward, R.L.; Hawkins, N.J.; Quinn, D.I.; Russell, P.J.; et al. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc. Natl. Acad. Sci. USA 2003, 100, 3410–3415. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Jeon, J.H.; Lee, M.J. Docosahexaenoic Acid, a Potential Treatment for Sarcopenia, Modulates the Ubiquitin–Proteasome and the Autophagy–Lysosome Systems. Nutrients 2020, 12, 2597. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Tetsumoto, S.; Kijima, T.; Kida, H.; Kumagai, T.; Takahashi, R.; Otani, Y.; Inoue, K.; Kuhara, H.; Shimada, K.; et al. Favorable Responses to Tocilizumab in Two Patients With Cancer-Related Cachexia. J. Pain Symptom Manag. 2013, 46, e9–e13. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Takahashi, F.; Motojima, S.; Nakashima, K.; Kaneko, N.; Hoshi, K.; Takahashi, K. Possible Role for Tocilizumab, an Anti-Interleukin-6 Receptor Antibody, in Treating Cancer Cachexia. J. Clin. Oncol. 2013, 31, e69–e72. [Google Scholar] [CrossRef] [PubMed]
- Moryoussef, F.; Dhooge, M.; Volet, J.; Barbe, C.; Brezault, C.; Hoeffel, C.; Coriat, R.; Bouché, O. Reversible sarcopenia in patients with gastrointestinal stromal tumor treated with imatinib. J. Cachex Sarcopenia Muscle 2015, 6, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan-Jun, Y.; Yang, Q.; Yong-Long, H.; Li-Li, W.; Jie, L.; Jin-Lu, H.; Jin, L.; Peng-Guo, C.; Run, G.; Guo, C. Selumetinib Attenuates Skeletal Muscle Wasting in Murine Cachexia Model through ERK Inhibition and AKT Activation. Mol. Cancer Ther. 2016, 16, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado, C.M.M.; Bekaii-Saab, T.; Doyle, L.A.; Shrestha, S.; Ghosh, S.; Baracos, V.E.; Sawyer, M.B. Skeletal muscle anabolism is a side effect of therapy with the MEK inhibitor: Selumetinib in patients with cholangiocarcinoma. Br. J. Cancer 2012, 106, 1583–1586. [Google Scholar] [CrossRef] [Green Version]
- Busquets, S.; Toledo, M.; Orpí, M.; Massa, D.; Porta, M.; Capdevila, E.; Padilla, N.; Frailis, V.; López-Soriano, F.J.; Han, H.Q.; et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J. Cachex Sarcopenia Muscle 2012, 3, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Toledo, M.; Busquets, S.; Penna, F.; Zhou, X.; Marmonti, E.; Betancourt, A.; Massa, D.; López-Soriano, F.J.; Han, H.Q.; Argilés, J.M. Complete reversal of muscle wasting in experimental cancer cachexia: Additive effects of activin type II receptor inhibition and beta-2 agonist. Int. J. Cancer 2016, 138, 2021–2029. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Hanna, D.L.; Zhang, W.; Baba, H.; Lenz, H.-J. Molecular Pathways: Cachexia Signaling—A Targeted Approach to Cancer Treatment. Clin. Cancer Res. 2016, 22, 3999–4004. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.; Panikkar, R. Sarcopenia associated with chemotherapy and targeted agents for cancer therapy. Ann. Palliat. Med. 2019, 8, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Antoun, S.; Birdsell, L.; Sawyer, M.B.; Venner, P.; Escudier, B.; Baracos, V.E. Association of Skeletal Muscle Wasting With Treatment With Sorafenib in Patients With Advanced Renal Cell Carcinoma: Results From a Placebo-Controlled Study. J. Clin. Oncol. 2010, 28, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Shimokata, T.; Honda, K.; Kondoh, C.; Hayashi, N.; Yoshino, Y.; Sassa, N.; Nakano, Y.; Gotoh, M.; Ando, Y. Muscle wasting associated with the long-term use of mTOR inhibitors. Mol. Clin. Oncol. 2016, 5, 641–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprague, K.L.D.B.L.; Gramling, R.E.; Ades, P.A. Exercise interventions for individuals with advanced cancer: A systematic review. Prev. Med. 2017, 104, 124–132. [Google Scholar] [CrossRef]
- Galvão, D.A.; Taaffe, D.R.; Spry, N.; Joseph, D.; Newton, R.U. Combined Resistance and Aerobic Exercise Program Reverses Muscle Loss in Men Undergoing Androgen Suppression Therapy for Prostate Cancer Without Bone Metastases: A Randomized Controlled Trial. J. Clin. Oncol. 2010, 28, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Grande, A.J.; Silva, V.; Maddocks, M. Exercise for cancer cachexia in adults: Executive summary of a Cochrane Collaboration systematic review. J. Cachex Sarcopenia Muscle 2015, 6, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.J.; Mozer, M. Differentiating Sarcopenia and Cachexia Among Patients With Cancer. Nutr. Clin. Pract. 2017, 32, 30–39. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armstrong, V.S.; Fitzgerald, L.W.; Bathe, O.F. Cancer-Associated Muscle Wasting—Candidate Mechanisms and Molecular Pathways. Int. J. Mol. Sci. 2020, 21, 9268. https://doi.org/10.3390/ijms21239268
Armstrong VS, Fitzgerald LW, Bathe OF. Cancer-Associated Muscle Wasting—Candidate Mechanisms and Molecular Pathways. International Journal of Molecular Sciences. 2020; 21(23):9268. https://doi.org/10.3390/ijms21239268
Chicago/Turabian StyleArmstrong, Victoria S., Liam W. Fitzgerald, and Oliver F. Bathe. 2020. "Cancer-Associated Muscle Wasting—Candidate Mechanisms and Molecular Pathways" International Journal of Molecular Sciences 21, no. 23: 9268. https://doi.org/10.3390/ijms21239268
APA StyleArmstrong, V. S., Fitzgerald, L. W., & Bathe, O. F. (2020). Cancer-Associated Muscle Wasting—Candidate Mechanisms and Molecular Pathways. International Journal of Molecular Sciences, 21(23), 9268. https://doi.org/10.3390/ijms21239268