Carbamazepine Restores Neuronal Signaling, Protein Synthesis, and Cognitive Function in a Mouse Model of Fragile X Syndrome



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
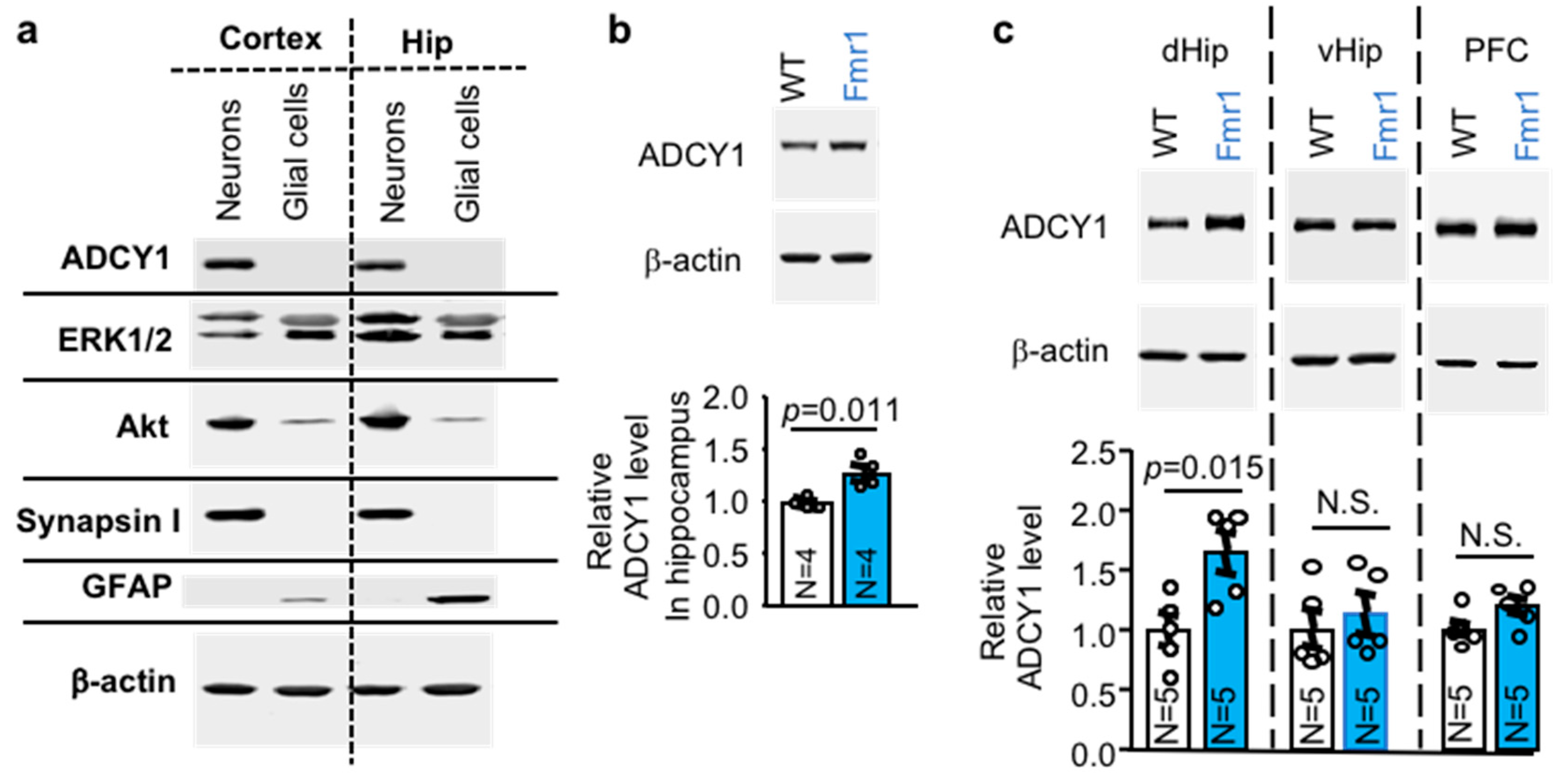
2.1. ADCY1 Is Expressed in Neurons and Elevated in the Dorsal Hippocampus of Fmr1 KO Mice
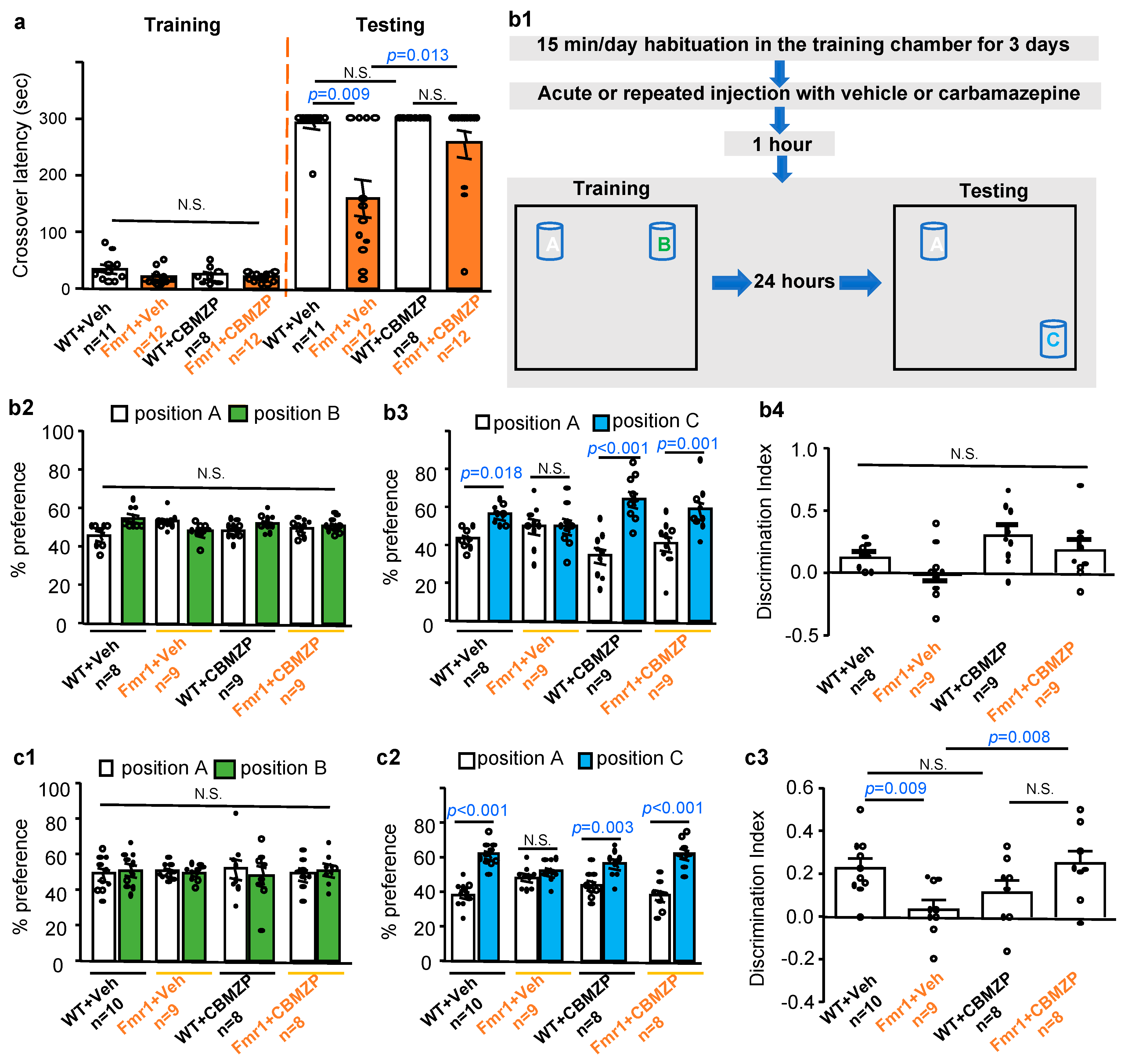
2.2. Carbamazepine Restores Hippocampus-Dependent Memory in Fmr1 KO Mice
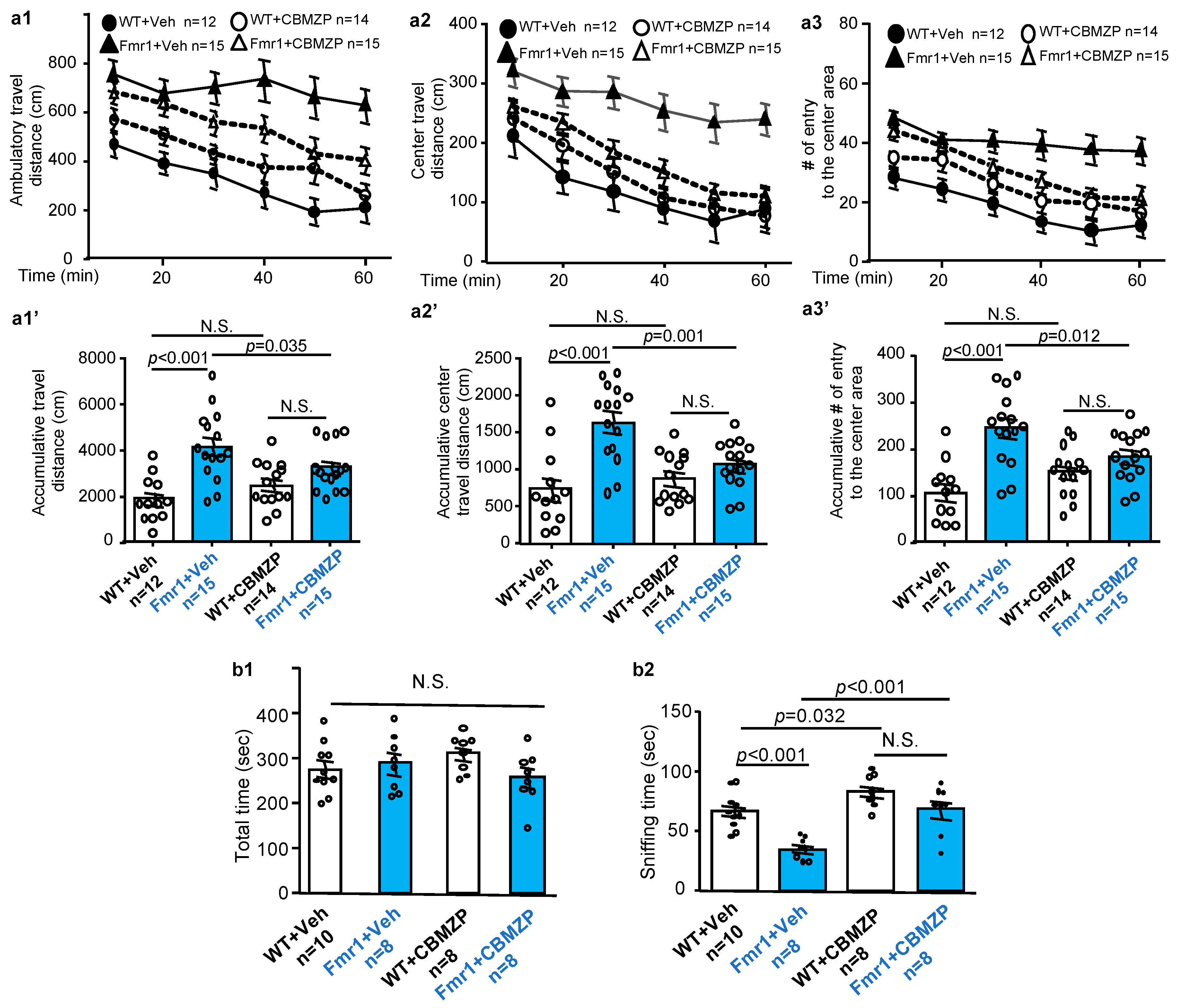
2.3. Effects of Carbamazepine on Non-Cognitive Behavioral Abnormalities in Fmr1 KO Mice
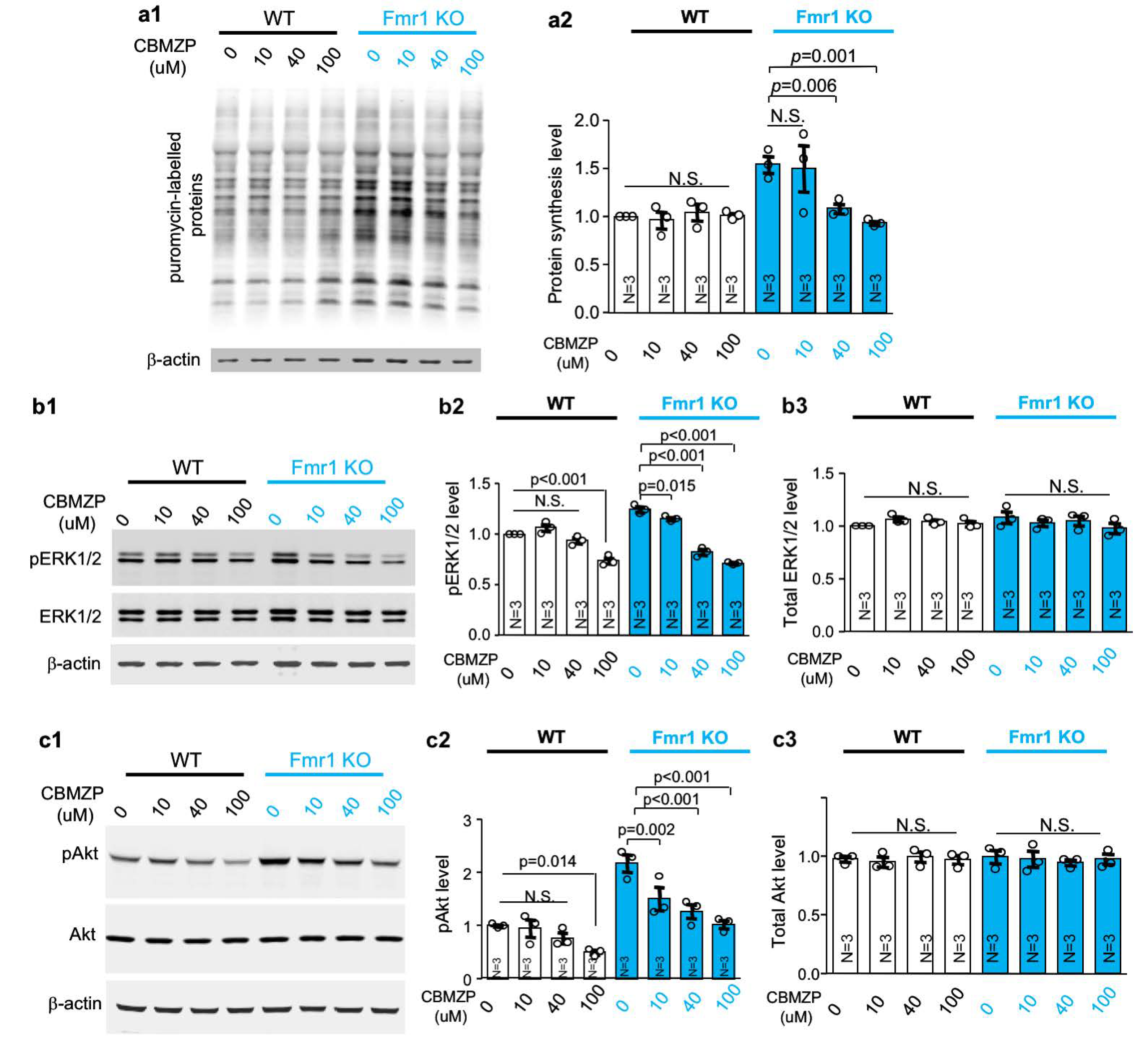
2.4. Carbamazepine Dampens the Elevated Protein Synthesis in Fmr1 KO Neurons
2.5. Carbamazepine Suppresses Both ERK½ and Akt Activity in Neurons
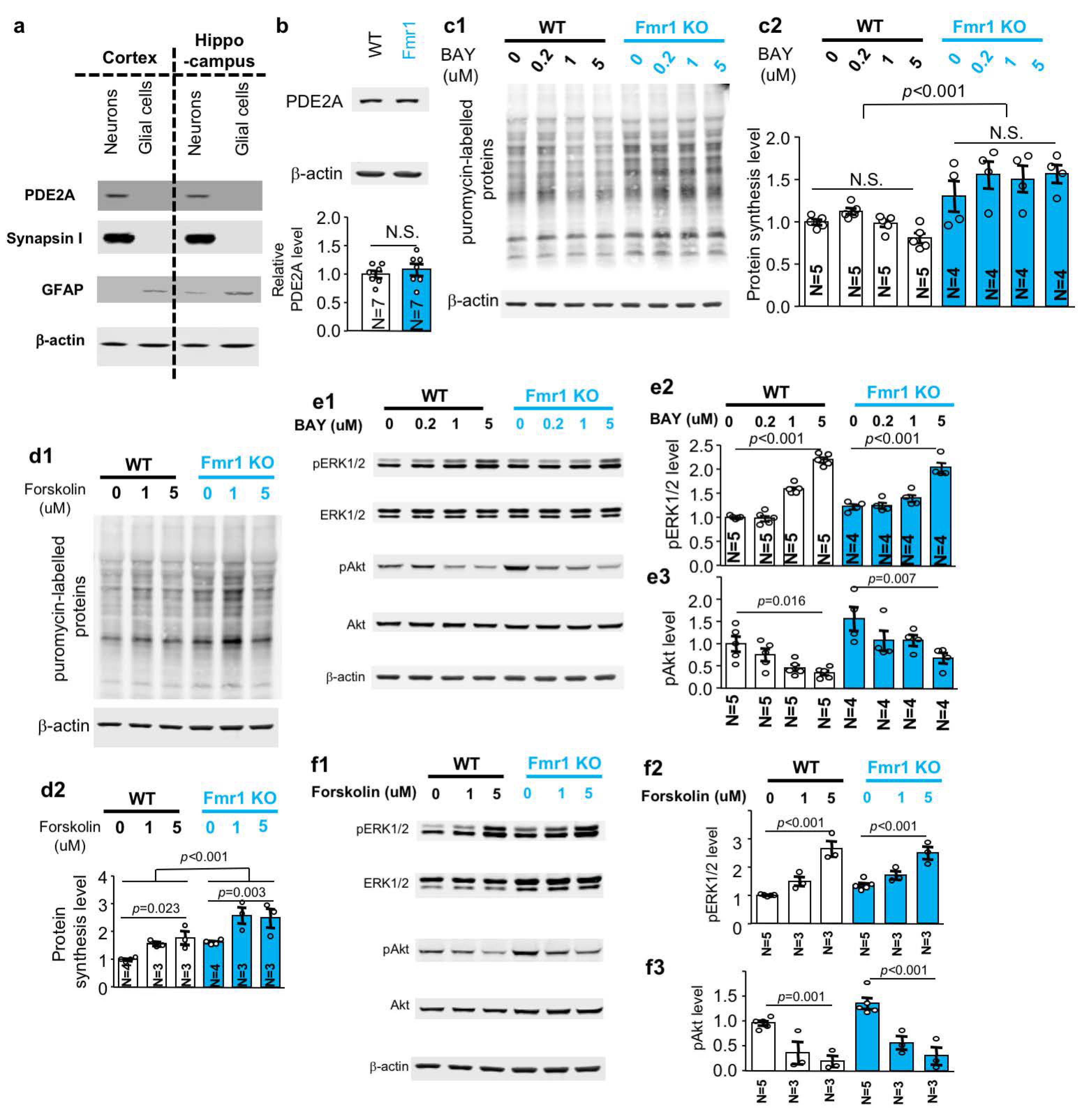
2.6. Pharmacological Inhibition of PDE2 and Activation of ADCY Do Not Correct the Elevated Protein Synthesis in FXS Mouse Neurons
2.7. Effects of PDE2 Inhibition and ADCY Activation on ERK½ and Akt Signaling
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. Western Blot Analysis
4.4. In Vivo Drug Administration
4.5. Behavioral Examination
4.6. Drug Effects on Protein Synthesis and Neuronal Signaling
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FXS | Fragile X syndrome |
| FMR1 | fragile X mental retardation 1 |
| FMRP | fragile X mental retardation protein |
| ADCY1 | type 1 adenylyl cyclase |
| KO | knockout |
| PDE | phosphodiesterase |
| ERK½ | extracellular signal-regulated kinase ½ |
| cAMP | cyclic AMP |
| WT | wild type |
| PI3K | Phosphoinositide 3 kinase |
| Akt | protein kinase B |
References
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu. Rev. Pathol. 2012, 7, 219–245. [Google Scholar] [CrossRef] [Green Version]
- Sethna, F.; Moon, C.; Wang, H. From FMRP function to potential therapies for fragile X syndrome. Neurochem. Res. 2014, 39, 1016–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.C.; Klann, E. The translation of translational control by FMRP: Therapeutic targets for FXS. Nat. Neurosci. 2013, 16, 1530–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.; Yao, X.; Pong, D.L.; Jeromin, A.; Bassell, G.J. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J. Neurosci. 2011, 31, 5693–5698. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Snape, M.; Klann, E.; Stone, J.G.; Singh, A.; Petersen, R.B.; Castellani, R.J.; Casadesus, G.; Smith, M.A.; Zhu, X. Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J. Neurochem. 2012, 121, 672–679. [Google Scholar] [CrossRef]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, D.; Caku, A.; Fradet, M.; Bouvier, P.; Dubé, J.; Corbin, F. Lovastatin corrects ERK pathway hyperactivation in fragile X syndrome: Potential of platelet’s signaling cascades as new outcome measures in clinical trials. Biomarkers 2016, 21, 497–508. [Google Scholar] [CrossRef]
- Ascano, M., Jr.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Sethna, F.; Feng, W.; Ding, Q.; Robison, A.J.; Feng, Y.; Wang, H. Enhanced expression of ADCY1 underlies aberrant neuronal signalling and behaviour in a syndromic autism model. Nat. Commun. 2017, 8, 14359. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ferguson, G.D.; Pineda, V.V.; Cundiff, P.E.; Storm, D.R. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat. Neurosci. 2004, 7, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cao, H.; Saraf, A.; Zweifel, L.S.; Storm, D.R. Overexpression of the type 1 adenylyl cyclase in the forebrain leads to deficits of behavioral inhibition. J. Neurosci. 2015, 35, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.; Bassell, G.J. Excess protein synthesis in FXS patient lymphoblastoid cells can be rescued with a p110beta-selective inhibitor. Mol. Med. 2012, 18, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Nakamoto, M.; Yao, X.; Chan, C.B.; Yim, S.Y.; Ye, K.; Warren, S.T.; Bassell, G.J. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 2010, 30, 10624–10638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, L.; Heldman, E.; Bersudsky, Y.; Vatner, S.F.; Ishikawa, Y.; Almog, O.; Belmaker, R.H.; Agam, G. Inhibition of specific adenylyl cyclase isoforms by lithium and carbamazepine, but not valproate, may be related to their antidepressant effect. Bipolar Disord. 2009, 11, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Maurin, T.; Lebrigand, K.; Castagnola, S.; Paquet, A.; Jarjat, M.; Popa, A.; Grossi, M.; Rage, F.; Bardoni, B. HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res. 2018, 46, 6344–6355. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Choi, E.J.; Wang, F.; Blazynski, C.; Storm, D.R. Type I calmodulin-sensitive adenylyl cyclase is neural specific. J. Neurochem. 1993, 60, 305–311. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, M. The role of Ca(2)(+)-stimulated adenylyl cyclases in bidirectional synaptic plasticity and brain function. Rev. Neurosci. 2012, 23, 67–78. [Google Scholar] [CrossRef]
- Ding, Q.; Sethna, F.; Wang, H. Behavioral analysis of male and female Fmr1 knockout mice on C57BL/6 background. Behav. Brain Res. 2014, 271, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Qin, M.; Kang, J.; Smith, C.B. A null mutation for Fmr1 in female mice: Effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience 2005, 135, 999–1009. [Google Scholar] [CrossRef]
- Assini, F.L.; Duzzioni, M.; Takahashi, R.N. Object location memory in mice: Pharmacological validation and further evidence of hippocampal CA1 participation. Behav. Brain Res. 2009, 204, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Malvaez, M.; Kramar, E.; Matheos, D.P.; Arrizon, A.; Cabrera, S.M.; Lynch, G.; Greene, R.W.; Wood, M.A. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology 2011, 36, 1545–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.T.; Zhao, Y.; Huang, Y.; Dorairaj, N.R.; Chandler, L.J.; O’Donnell, J.M. Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into the CA1 subregion of the rat hippocampus. Neuropsychopharmacology 2004, 29, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Wang, T.; Wan, H.; Han, L.; Qin, X.; Zhang, Y.; Wang, J.; Yu, C.; Berton, F.; Francesconi, W.; et al. Fmr1 deficiency promotes age-dependent alterations in the cortical synaptic proteome. Proc. Natl. Acad. Sci. USA 2015, 112, E4697–E4706. [Google Scholar] [CrossRef] [Green Version]
- Maurin, T.; Melancia, F.; Jarjat, M.; Castro, L.; Costa, L.; Delhaye, S.; Khayachi, A.; Castagnola, S.; Mota, E.; Di Giorgio, A.; et al. Involvement of Phosphodiesterase 2A Activity in the Pathophysiology of Fragile X Syndrome. Cereb. Cortex 2019, 29, 3241–3252. [Google Scholar] [CrossRef]
- Kim, H.; Lee, Y.; Park, J.Y.; Kim, J.E.; Kim, T.K.; Choi, J.; Lee, J.E.; Lee, E.H.; Kim, D.; Kim, K.S.; et al. Loss of Adenylyl Cyclase Type-5 in the Dorsal Striatum Produces Autistic-Like Behaviors. Mol. Neurobiol. 2017, 54, 7994–8008. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef]
- Sweatt, J.D. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 2004, 14, 311–317. [Google Scholar] [CrossRef]
- Sui, L.; Wang, J.; Li, B.M. Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn. Mem. 2008, 15, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Garelick, M.G.; Wang, H.; Li, V.; Athos, J.; Storm, D.R. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat. Neurosci. 2005, 8, 925–931. [Google Scholar] [CrossRef]
- Gross, C.; Raj, N.; Molinaro, G.; Allen, A.G.; Whyte, A.J.; Gibson, J.R.; Huber, K.M.; Gourley, S.L.; Bassell, G.J. Selective role of the catalytic PI3K subunit p110beta in impaired higher order cognition in fragile X syndrome. Cell Rep. 2015, 11, 681–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asiminas, A.; Jackson, A.D.; Louros, S.R.; Till, S.M.; Spano, T.; Dando, O.; Bear, M.F.; Chattarji, S.; Hardingham, G.E.; Osterweil, E.K.; et al. Sustained correction of associative learning deficits after brief, early treatment in a rat model of Fragile X Syndrome. Sci. Transl. Med. 2019, 11, eaao0498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.J.; Bhattacharyya, A.; Lahvis, G.P.; Yin, J.C.; Malter, J.; Davidson, R.J. The cyclic AMP phenotype of fragile X and autism. Neurosci. Biobehav. Rev. 2008, 32, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.J.; Davidson, R.J.; Elliott, J.L.; Lahvis, G.P.; Yin, J.C.; Bhattacharyya, A. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS ONE 2007, 2, e931. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Sklena, P. Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. Am. J. Med. Genet. 1993, 45, 81–87. [Google Scholar] [CrossRef]
- Conti, A.C.; Maas, J.W., Jr.; Muglia, L.M.; Dave, B.A.; Vogt, S.K.; Tran, T.T.; Rayhel, E.J.; Muglia, L.J. Distinct regional and subcellular localization of adenylyl cyclases type 1 and 8 in mouse brain. Neuroscience 2007, 146, 713–729. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; Cogram, P.; Deacon, R.M.; Rex, C.; Tranfaglia, M. Multiple Behavior Phenotypes of the Fragile-X Syndrome Mouse Model Respond to Chronic Inhibition of Phosphodiesterase-4D (PDE4D). Sci. Rep. 2017, 7, 14653. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, S.A.; Hagerman, R.J.; Ferri, R.; Bosco, P.; Bernardina, B.D.; Tassinari, C.A.; De Sarro, G.B.; Elia, M. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 1999, 40, 1092–1099. [Google Scholar] [CrossRef]
- Berry-Kravis, E. Epilepsy in fragile X syndrome. Dev. Med. Child. Neurol. 2002, 44, 724–728. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Kaufmann, W.E.; Ono, M.Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootsak, J.; et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Lin, D.S.; Zheng, F.; Sutton, M.A.; Wang, H. Intracellular calcium and calmodulin link brain-derived neurotrophic factor to p70S6 kinase phosphorylation and dendritic protein synthesis. J. Neurosci. Res. 2010, 88, 1420–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Oyarzabal, E.A.; Hong, J.S. Preparation of rodent primary cultures for neuron-glia, mixed glia, enriched microglia, and reconstituted cultures with microglia. Methods Mol. Biol. 2013, 1041, 231–240. [Google Scholar] [PubMed] [Green Version]
- Vogel-Ciernia, A.; Wood, M.A. Examining object location and object recognition memory in mice. Curr. Protoc. Neurosci. 2014, 69, 8–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moy, S.S.; Nadler, J.J.; Perez, A.; Barbaro, R.P.; Johns, J.M.; Magnuson, T.R.; Piven, J.; Crawley, J.N. Sociability and preference for social novelty in five inbred strains: An approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004, 3, 287–302. [Google Scholar] [CrossRef]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef]
- Yang, Y.M.; Arsenault, J.; Bah, A.; Krzeminski, M.; Fekete, A.; Chao, O.Y.; Pacey, L.K.; Wang, A.; Forman-Kay, J.; Hampson, D.R.; et al. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol. Psychiatry 2020, 25, 2017–2035. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Q.; Zhang, F.; Feng, Y.; Wang, H. Carbamazepine Restores Neuronal Signaling, Protein Synthesis, and Cognitive Function in a Mouse Model of Fragile X Syndrome. Int. J. Mol. Sci. 2020, 21, 9327. https://doi.org/10.3390/ijms21239327
Ding Q, Zhang F, Feng Y, Wang H. Carbamazepine Restores Neuronal Signaling, Protein Synthesis, and Cognitive Function in a Mouse Model of Fragile X Syndrome. International Journal of Molecular Sciences. 2020; 21(23):9327. https://doi.org/10.3390/ijms21239327
Chicago/Turabian StyleDing, Qi, Fan Zhang, Yue Feng, and Hongbing Wang. 2020. "Carbamazepine Restores Neuronal Signaling, Protein Synthesis, and Cognitive Function in a Mouse Model of Fragile X Syndrome" International Journal of Molecular Sciences 21, no. 23: 9327. https://doi.org/10.3390/ijms21239327
APA StyleDing, Q., Zhang, F., Feng, Y., & Wang, H. (2020). Carbamazepine Restores Neuronal Signaling, Protein Synthesis, and Cognitive Function in a Mouse Model of Fragile X Syndrome. International Journal of Molecular Sciences, 21(23), 9327. https://doi.org/10.3390/ijms21239327