The Role of Nitric Oxide in Cancer: Master Regulator or NOt?

 ,
,  ,
, 
Abstract
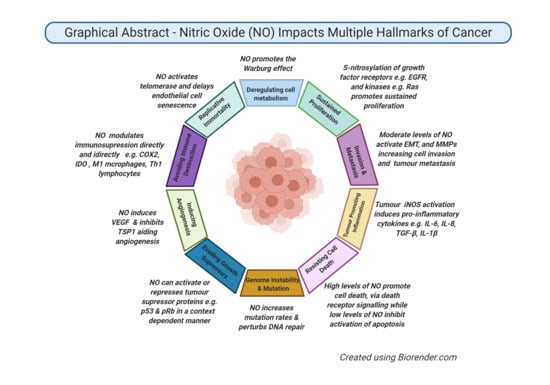
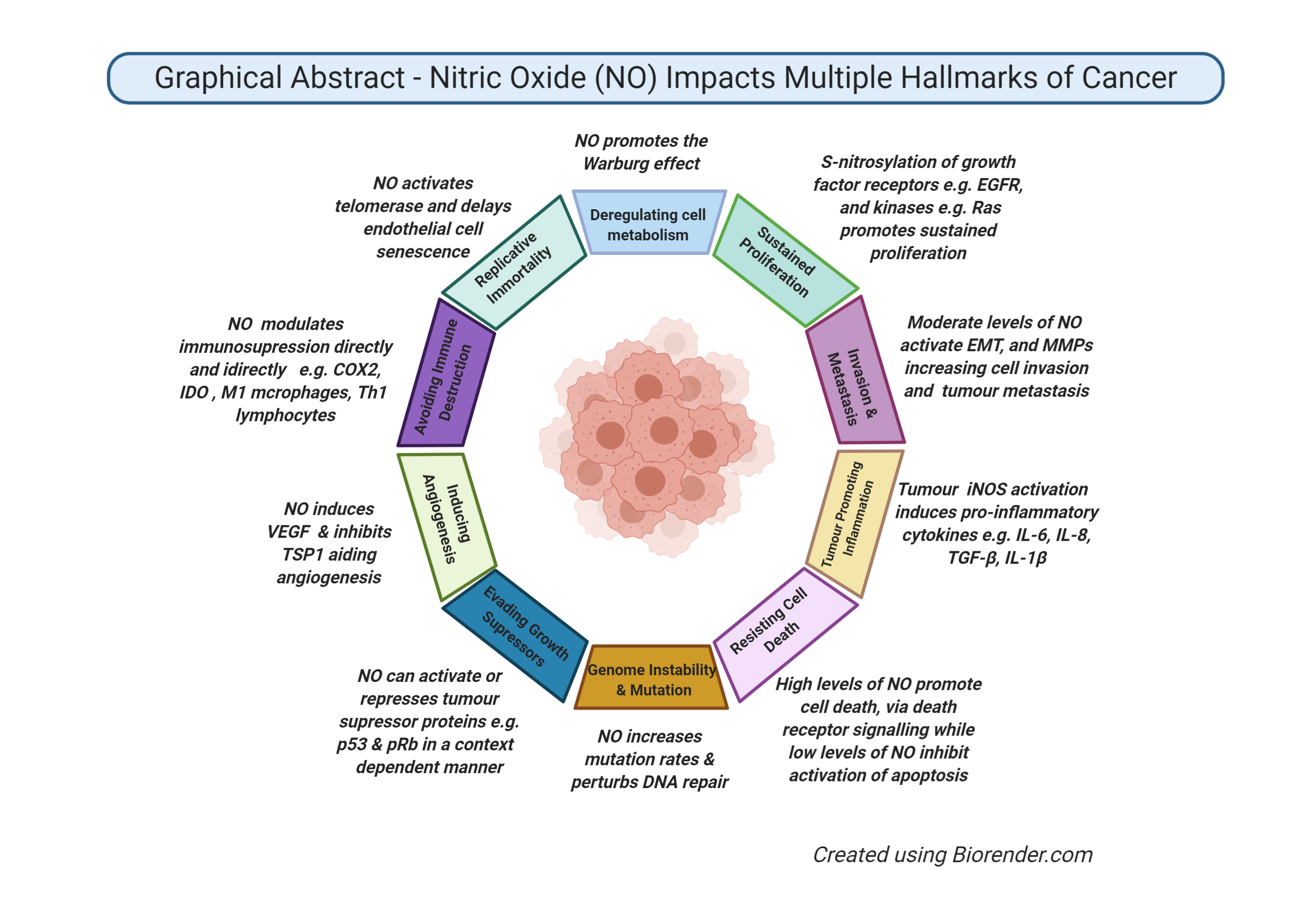{kind=link}
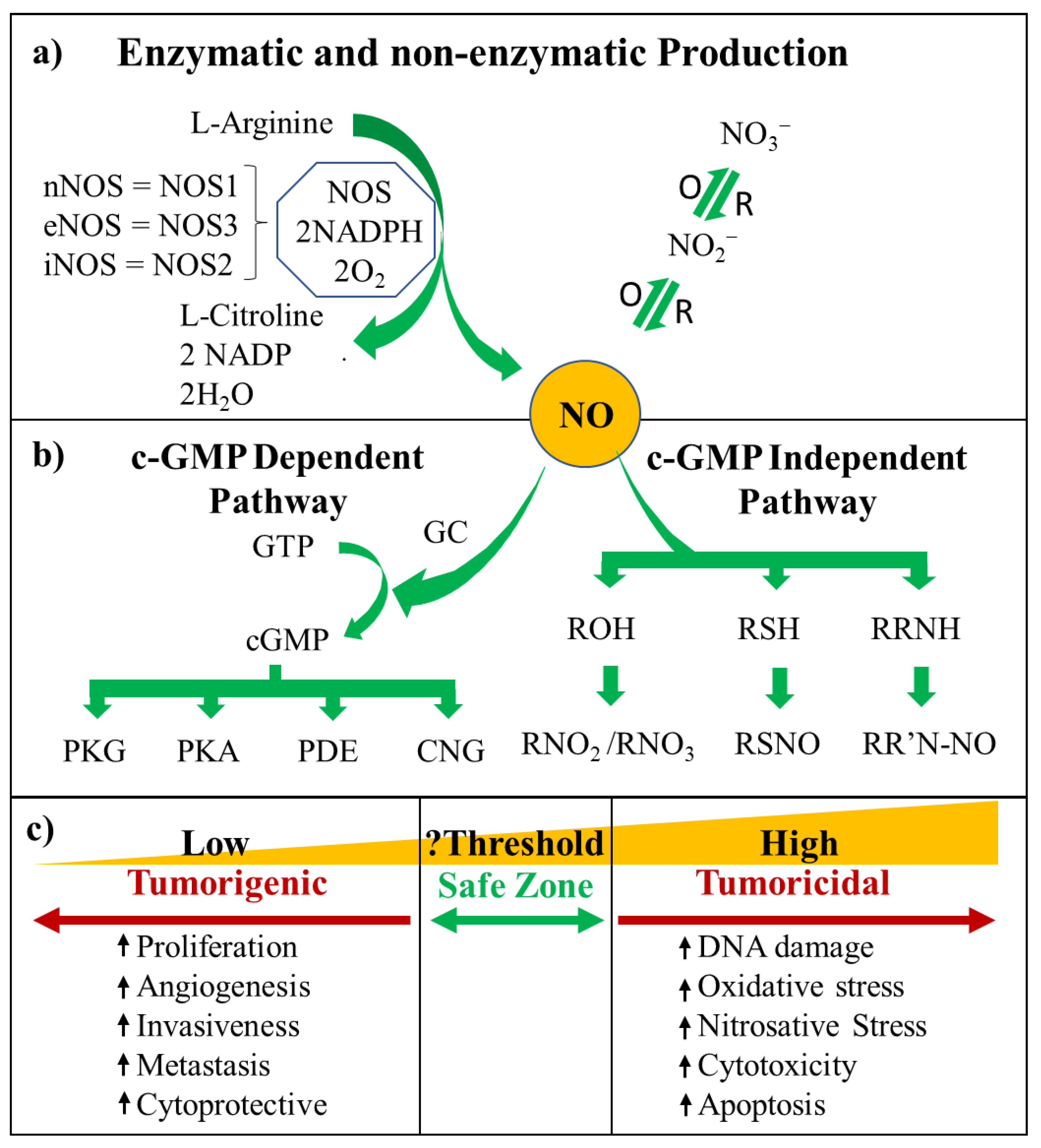{kind=link}
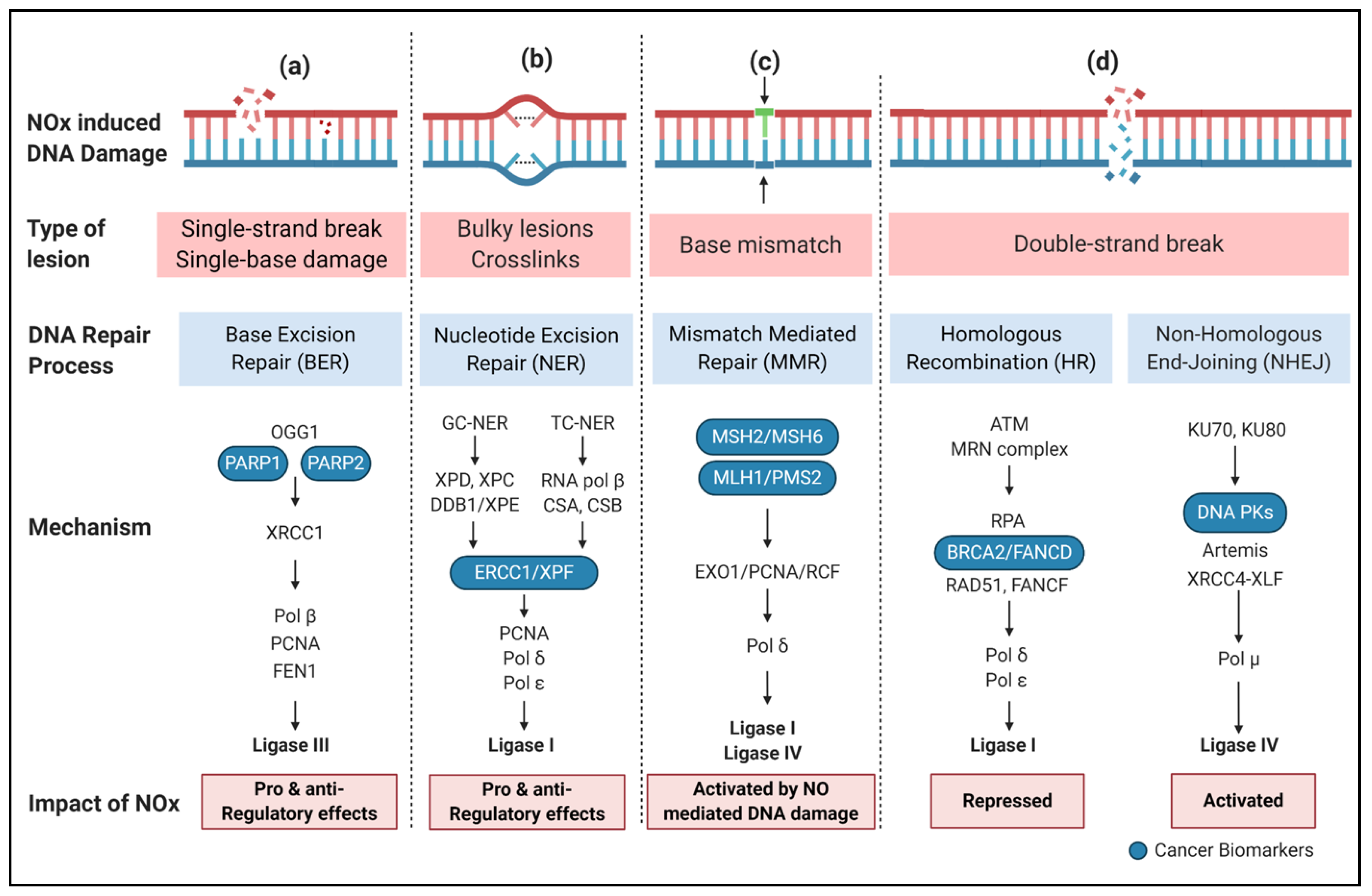{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- (a)
- Diffusion
- (b)
- Autoxidation
- (c)
- Reaction with superoxide to form peroxynitrite
2. NO Cell Signalling
2.1. cGMP-Dependent Pathway
2.2. cGMP-Independent Pathway
3. Role of NO in Cancer Biology
3.1. Genotoxicity and Mutagenesis
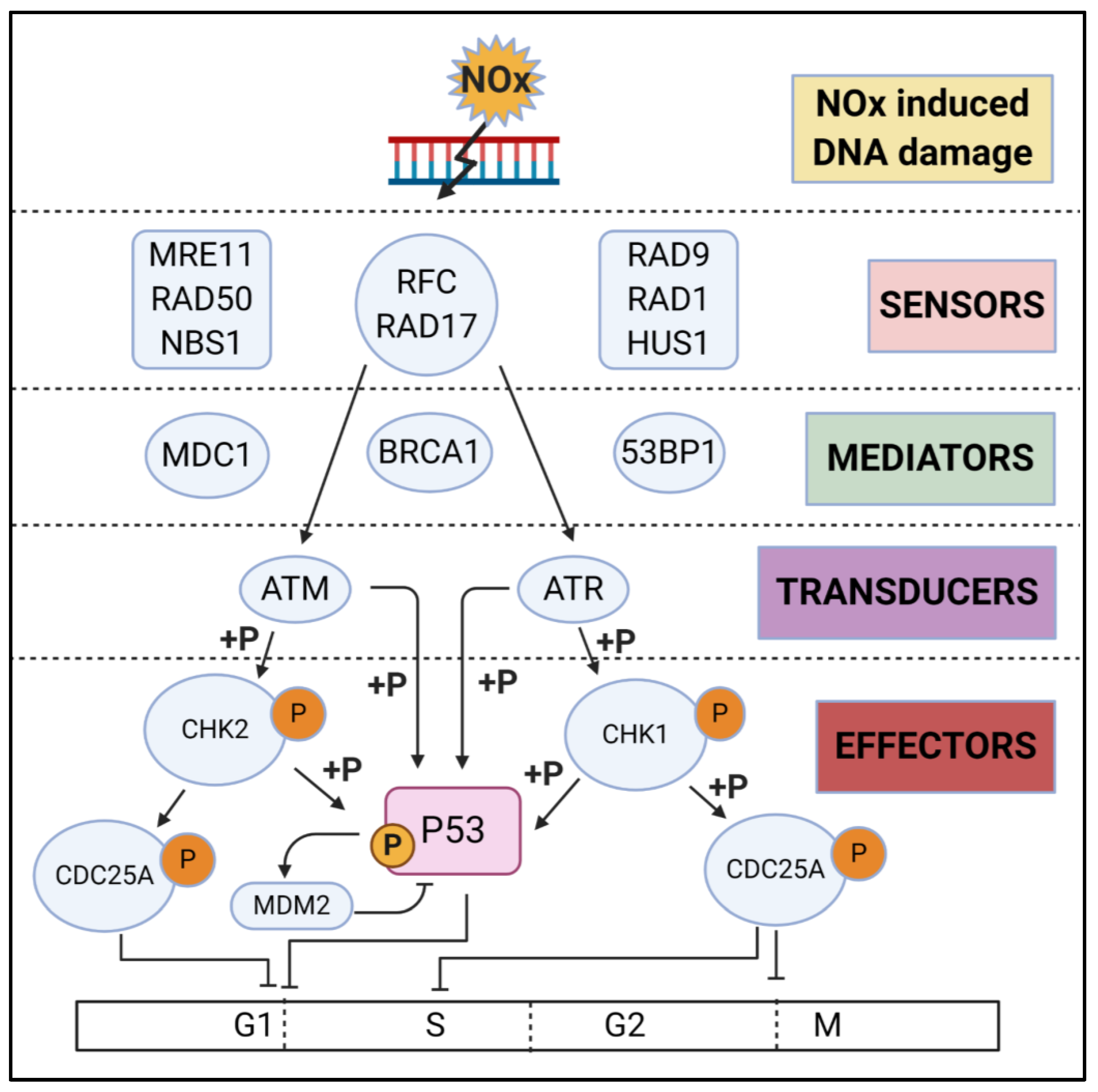
3.2. DNA Damage Repair (DDR)
3.3. Cell Cycle Arrest
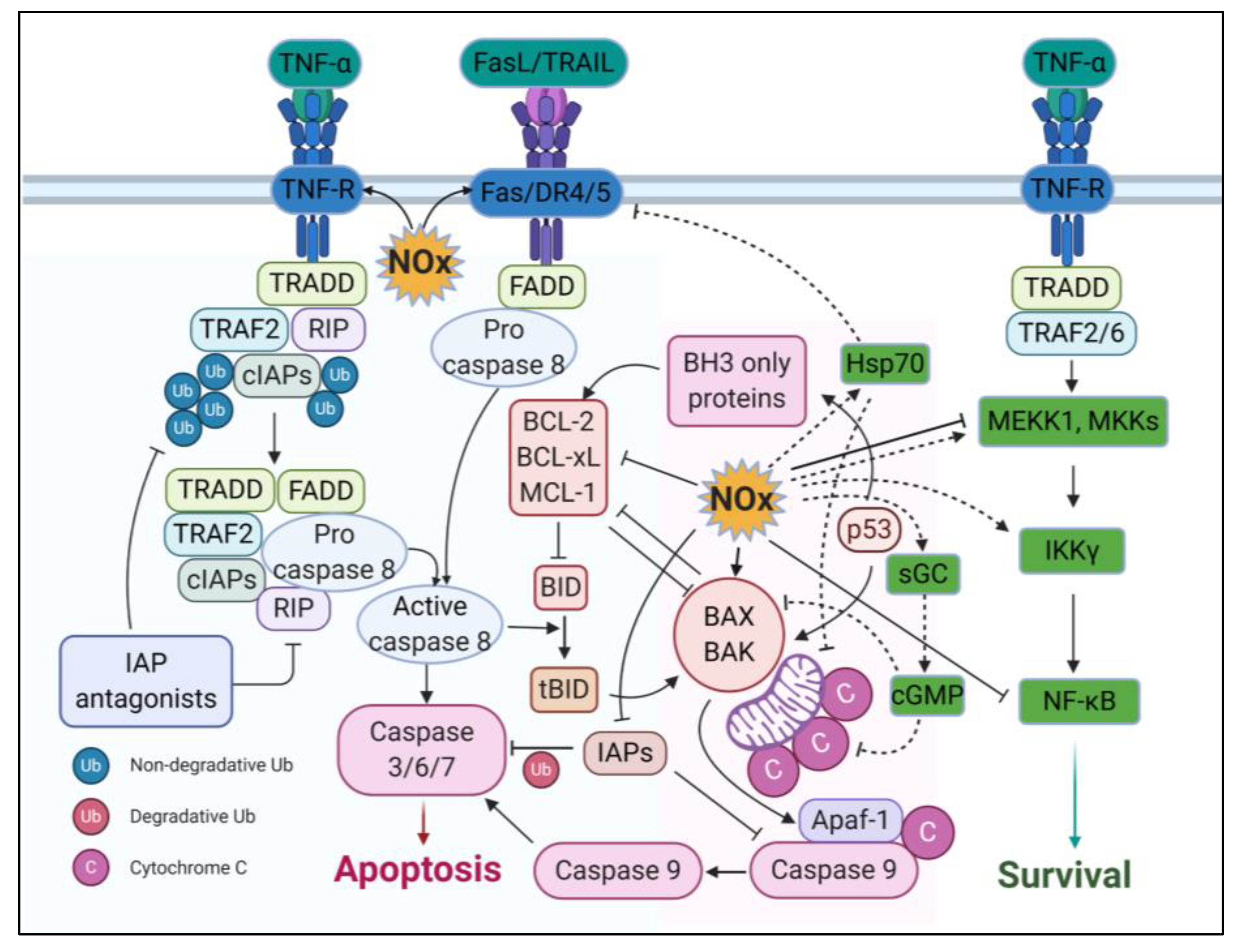
3.4. Apoptotic Effects
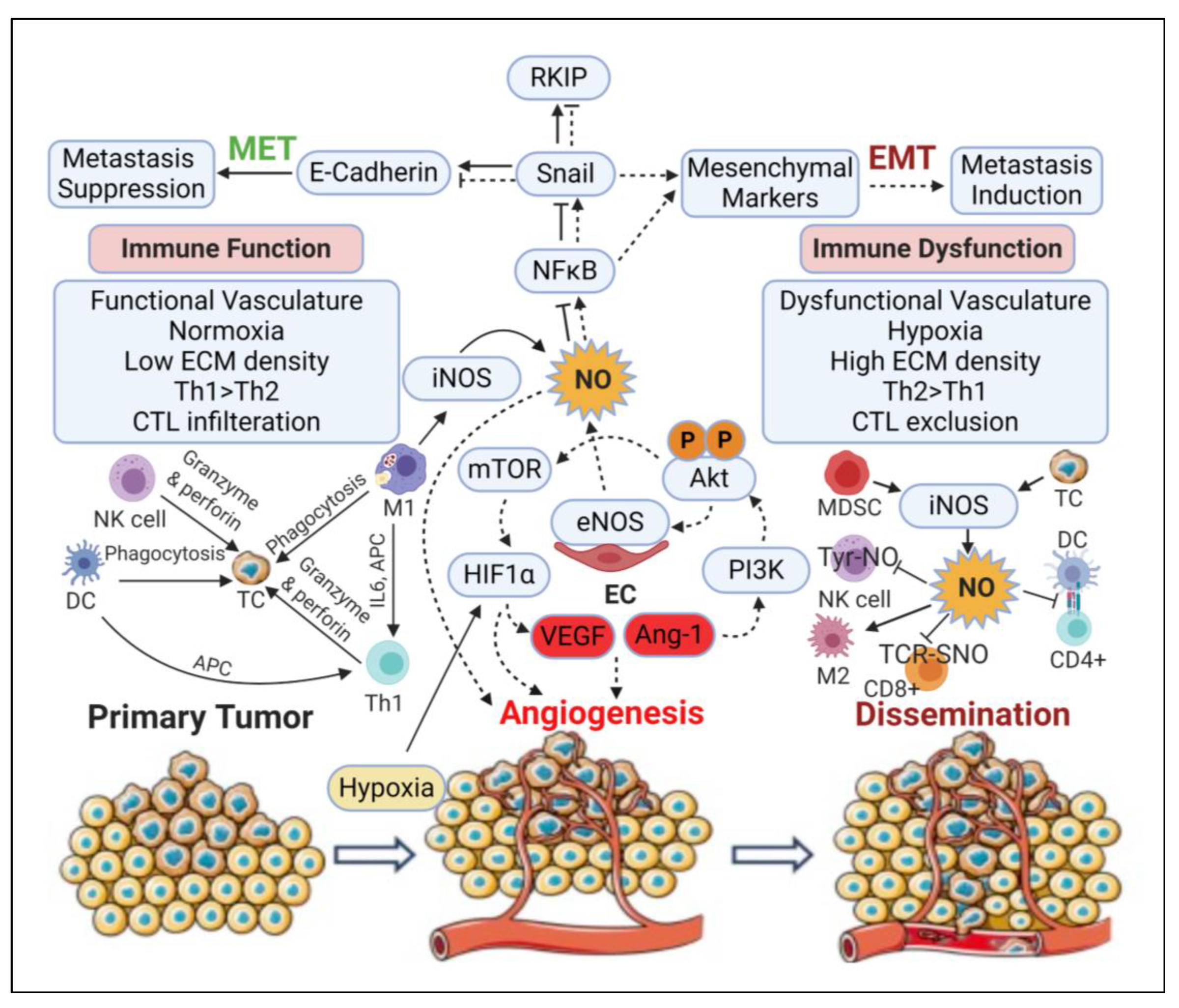
3.5. Angiogenic Effect
3.6. Epithelial-to-Mesenchymal Transition (EMT) and Metastatic Effects
3.7. Immunomodulatory Effects
4. NO-Mediated Strategies for Cancer Treatment
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NO | nitric Oxide |
| NOS | nitric oxide synthase |
| nNOS | neuronal nitric oxide synthase |
| eNOS | endothelial nitric oxide synthase |
| iNOS | inducible nitric oxide synthase |
| EDRF | endothelium-derived relaxing factor |
| NO2- | nitrate |
| NO3- | nitrite |
| NADPH | nicotinamide adenine dinucleotide phosphate NADPH |
| O2 | oxygen |
| N2O3 | nitrous anhydride |
| NO− | nitroxyl anion |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| cGMP | cyclic guanosine monophosphate |
| PDE | phosphodiesterase |
| GC | guanylate cyclase |
| GTP | guanosine triphosphate |
| PKG | protein kinase G |
| PKA | protein kinase A |
| CNG | cyclic nucleotide gated |
| VSMCs | vascular smooth muscle cells |
| ROH | alcohols |
| RSH | thiols |
| RR’NH | amines |
| RO-NO | nitrite/nitrate |
| RSNO | S-nitrosothiols |
| RR’N-NO | N-nitroso amines |
| NF-κB | nuclear factor kappa B |
| CREB | CRE-binding protein |
| MAPK | mitogen-activated protein kinases |
| PI3K | phosphatidylinositol-3 kinase |
| NTCP | Na+-taurocholate co-transporting polypeptide |
| GSH | glutathione |
| MDA | malondialdehyde |
| Fpg | protein formamidopyrimidine DNA glycosylase |
| DDR | DNA damage repair |
| NER | nucleotide excision repair |
| BER | base excision repair |
| MMR | mismatch repair |
| NHEJ | non-homologous end-joining |
| HDR | homology directed repair |
| FA | Fanconi anaemia |
| XP | Xeroderma pigmentosum |
| CS | Cockayne syndrome |
| TTD | trichothiodystrophy |
| AP | apurinic/apyrimidinic |
| Xan | xanthine |
| IDLs | insertion, deletion loops |
| PCNA | proliferating cell nuclear antigen |
| RFC | replication factor C |
| EXO1 | exonuclease |
| DSB | double strand breaks |
| Ku | Ku70-Ku80 heterodimer |
| DNA-PK | DNA-dependent protein kinase |
| SCID | severe combined immunodeficiency |
| MRN | MRE11-RAD50-NBS1 |
| DNA2 | dual endonuclease |
| D-loop | displacement loop |
| HR | homologous repair |
| DSBR | double strand break repair |
| SDSA | synthesis-dependent strand annealing |
| BIR | break induced replication |
| ATM | ataxia telangiectasa mutated |
| ATR | ATM and Rad3 related proteins |
| 53BP1 | p53 binding protein1 |
| TopBP1 | topoisomerase binding protein1 |
| MDC1 | mediator of DNA damage checkpoint1 |
| FADD | fas-associated death domain protein |
| TRADD | TNF-related death domain protein |
| PTP | permeability transition pore |
| AIF | apoptosis inducing factor |
| Apaf1 | adapter protein apoptotic protease activating factor |
| Hsp 70 | heat shock protein 70 |
| JNK | c-Jun N-terminal kinase |
| ERK1/2 | extracellular regulated kinases |
| VEGF | vascular endothelial growth factor |
| TSP1 | thrombospondin-1 |
| HIF-1α | hypoxia-inducible factor-1α |
| PGE2 | prostaglandin E2 |
| PGE1 | prostaglandin E1 |
| SNP | sodium nitroprusside |
| RKIP | Raf kinase inhibitor protein |
| NDRG1 | N-myc downstream-regulated gene 1 |
| Shh | sonic hedgehog |
| ECM | extracellular matrix |
| MMP | matrix metalloproteinase |
| TIMP | tissue inhibitor of metalloproteinase |
| PKC | protein Kinase C |
| ARG1 | arginase 1 |
| TILs | tumour-infiltrating lymphocytes |
| MDSCs | myeloid-derived suppressor cells |
| GTN | glyceryltrinitrate |
| ISDN | isosorbidedinitrate |
| RSNO | S-nitrosothiols |
| SNAP | S-nitroso-N-acetylpenicillamine |
| GSNO | S-nitrosoglutathione |
| SIN-1 | 3-morpholinosydnonimine |
| OONO- | peroxynitrite |
References
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Santolini, J.; Wang, Z.Q.; Wei, C.C.; Adak, S. Update on mechanism and catalytic regulation in the NO synthases. J. Biol. Chem. 2004, 279, 36167–36170. [Google Scholar] [CrossRef]
- Goligorsky, M.S.; Brodsky, S.V.; Noiri, E. NO bioavailability, endothelial dysfunction, and acute renal failure: New insights into pathophysiology. Semin. Nephrol. 2004, 24, 316–323. [Google Scholar] [CrossRef]
- Radomski, M.W. Nitric oxide: Biological mediator, modulator and effector. Ann. Med. 1995, 27, 321–329. [Google Scholar] [CrossRef]
- Kwon, N.S.; Stuehr, D.J.; Nathan, C.F. Inhibition of tumor cell ribonucleotide reductase by macrophage-derived nitric oxide. J. Exp. Med. 1991, 174, 761–767. [Google Scholar] [CrossRef]
- Roy, B.; Lepoivre, M.; Henry, Y.; Fontecave, M. Inhibition of ribonucleotide reductase by nitric oxide derived from thionitrites: Reversible modifications of both subunits. Biochemistry 1995, 34, 5411–5418. [Google Scholar] [CrossRef]
- Ford, P.C.; Wink, D.A.; Stanbury, D.M. Autoxidation kinetics of aqueous nitric oxide. FEBS Lett. 1993, 326, 1–3. [Google Scholar] [CrossRef]
- Uppu, R.M.; Squadrito, G.L.; Pryor, W.A. Acceleration of peroxynitrite oxidations by carbon dioxide. Arch. Biochem. Biophys. 1996, 327, 335–343. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Mayer, B.; Hemmens, B. Nitric Oxide: Chemical Puzzles Posed by a Biological Messenger. Angew. Chem. 1999, 38, 1714–1731. [Google Scholar] [CrossRef]
- Squadrito, G.L.; Pryor, W.A. Oxidative chemistry of nitric oxide: The roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic. Biol. Med. 1998, 25, 392–403. [Google Scholar] [CrossRef]
- Mocellin, S.; Bronte, V.; Nitti, D. Nitric oxide, a double edged sword in cancer biology: Searching for therapeutic opportunities. Med. Res. Rev. 2007, 27, 317–352. [Google Scholar] [CrossRef]
- Jenkins, D.C.; Charles, I.G.; Thomsen, L.L.; Moss, D.W.; Holmes, L.S.; Baylis, S.A.; Rhodes, P.; Westmore, K.; Emson, P.C.; Moncada, S. Roles of nitric oxide in tumor growth. Proc. Natl. Acad. Sci. USA 1995, 92, 4392–4396. [Google Scholar] [CrossRef]
- Vahora, H.; Khan, M.A.; Alalami, U.; Hussain, A. The Potential Role of Nitric Oxide in Halting Cancer Progression Through Chemoprevention. J. Cancer Prev. 2016, 21, 1–12. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Thomas, D.D.; Switzer, C.; Flores-Santana, W.; Isenberg, J.S.; Ambs, S.; Roberts, D.D.; Wink, D.A. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide 2008, 19, 73–76. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Patel, R.P.; McAndrew, J.; Sellak, H.; White, C.R.; Jo, H.; Freeman, B.A.; Darley-Usmar, V.M. Biological aspects of reactive nitrogen species. Biochim. Biophys. Acta 1999, 1411, 385–400. [Google Scholar] [CrossRef]
- Stamler, J.S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar] [CrossRef]
- Lau, K.S.; Grange, R.W.; Isotani, E.; Sarelius, I.H.; Kamm, K.E.; Huang, P.L.; Stull, J.T. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol. Genom. 2000, 2, 21–27. [Google Scholar] [CrossRef]
- Ghalayini, I.F. Nitric oxide-cyclic GMP pathway with some emphasis on cavernosal contractility. Int. J. Impot. Res. 2004, 16, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Muntane, J.; De la Rosa, A.J.; Marin, L.M.; Padillo, F.J. Nitric oxide and cell death in liver cancer cells. Mitochondrion 2013, 13, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, S.M.; Vaandrager, A.B.; Smolenski, A.; Walter, U.; De Jonge, H.R. Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem. Sci. 1997, 22, 307–312. [Google Scholar] [CrossRef]
- Degerman, E.; Belfrage, P.; Manganiello, V.C. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J. Biol. Chem. 1997, 272, 6823–6826. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D.; Milligan, G. Tailoring cAMP-signalling responses through isoform multiplicity. Trends Biochem. Sci. 1997, 22, 217–224. [Google Scholar] [CrossRef]
- Zagotta, W.N.; Siegelbaum, S.A. Structure and function of cyclic nucleotide-gated channels. Ann. Rev. Neurosci. 1996, 19, 235–263. [Google Scholar] [CrossRef]
- Warner, T.D.; Mitchell, J.A.; Sheng, H.; Murad, F. Effects of cyclic GMP on smooth muscle relaxation. Adv. Pharmacol. 1994, 26, 171–194. [Google Scholar] [CrossRef]
- Buechler, W.A.; Ivanova, K.; Wolfram, G.; Drummer, C.; Heim, J.M.; Gerzer, R. Soluble guanylyl cyclase and platelet function. Ann. N. Y. Acad. Sci. 1994, 714, 151–157. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Snyder, S.H. Nitric oxide: A neural messenger. Annu. Rev. Cell Dev. Biol. 1995, 11, 417–440. [Google Scholar] [CrossRef]
- Lehners, M.; Dobrowinski, H.; Feil, S.; Feil, R. cGMP Signaling and Vascular Smooth Muscle Cell Plasticity. J. Cardiovasc. Dev. Dis. 2018, 5, 20. [Google Scholar] [CrossRef]
- Jeremy, J.Y.; Rowe, D.; Emsley, A.M.; Newby, A.C. Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovasc. Res. 1999, 43, 580–594. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Somasundaram, V.; de Oliveira, G.A.; Kesarwala, A.; Heinecke, J.L.; Cheng, R.Y.; Glynn, S.A.; Ambs, S.; Wink, D.A.; Ridnour, L.A. Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression. Antioxid. Redox Signal. 2017, 26, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Krizaj, D.; Copenhagen, D.R. Calcium regulation in photoreceptors. Front. Biosci. J. Virtual Libr. 2002, 7, d2023–d2044. [Google Scholar] [CrossRef]
- Tricoire, L.; Vitalis, T. Neuronal nitric oxide synthase expressing neurons: A journey from birth to neuronal circuits. Front. Neural Circuits 2012, 6, 82. [Google Scholar] [CrossRef]
- Begara-Morales, J.C.; Sanchez-Calvo, B.; Chaki, M.; Valderrama, R.; Mata-Perez, C.; Padilla, M.N.; Corpas, F.J.; Barroso, J.B. Antioxidant Systems are Regulated by Nitric Oxide-Mediated Post-translational Modifications (NO-PTMs). Front. Plant Sci. 2016, 7, 152. [Google Scholar] [CrossRef]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. J. Biol. Inorg. Chem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef]
- Heck, D.E. *NO, RSNO, ONOO-, NO+, *NOO, NOx--dynamic regulation of oxidant scavenging, nitric oxide stores, and cyclic GMP-independent cell signaling. Antioxid. Redox Signal. 2001, 3, 249–260. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197. [Google Scholar] [CrossRef]
- Olson, S.Y.; Garban, H.J. Regulation of apoptosis-related genes by nitric oxide in cancer. Nitric Oxide 2008, 19, 170–176. [Google Scholar] [CrossRef]
- Sha, Y.; Marshall, H.E. S-nitrosylation in the regulation of gene transcription. Biochim. Biophys. Acta 2012, 1820, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, A.; Oh, E.; Taoka, A.; Sakurai, H.; Tsuchiya, T.; Tsuda, M. Rapid attenuation of AP-1 transcriptional factors associated with nitric oxide (NO)-mediated neuronal cell death. J. Biol. Chem. 1996, 271, 31061–31067. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stamler, J.S. S-nitrosothiols and the bioregulatory actions of nitrogen oxides through reactions with thiol groups. Curr. Top. Microbiol. Immunol. 1995, 196, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Hausladen, A. Oxidative modifications in nitrosative stress. Nat. Struct. Biol. 1998, 5, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Gitler, C.; Zarmi, B.; Kalef, E. General method to identify and enrich vicinal thiol proteins present in intact cells in the oxidized, disulfide state. Anal. Biochem. 1997, 252, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995, 369, 136–139. [Google Scholar] [CrossRef]
- Giulivi, C. Functional implications of nitric oxide produced by mitochondria in mitochondrial metabolism. Nat. Cell Biol. 1998, 332 Pt 3, 673–679. [Google Scholar] [CrossRef]
- Gonzalez, R.; Cruz, A.; Ferrin, G.; Lopez-Cillero, P.; Fernandez-Rodriguez, R.; Briceno, J.; Gomez, M.A.; Rufian, S.; Mata Mde, L.; Martinez-Ruiz, A.; et al. Nitric oxide mimics transcriptional and post-translational regulation during alpha-tocopherol cytoprotection against glycochenodeoxycholate-induced cell death in hepatocytes. J. Hepatol. 2011, 55, 133–144. [Google Scholar] [CrossRef]
- Xu, L.; Eu, J.P.; Meissner, G.; Stamler, J.S. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 1998, 279, 234–237. [Google Scholar] [CrossRef]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Identification of more than 100 structurally unique DNA-phosphate adducts formed during rat lung carcinogenesis by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 2018, 39, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Kovi, R.C.; Johnson, C.S.; Balbo, S.; Hecht, S.S.; O’Sullivan, M.G. Metastasis to the F344 Rat Pancreas from Lung Cancer Induced by 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone and Enantiomers of Its Metabolite 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol, Constituents of Tobacco Products. Toxicol. Pathol. 2018, 46, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef]
- Thomas, S.; Lowe, J.E.; Knowles, R.G.; Green, I.C.; Green, M.H. Factors affecting the DNA damaging activity of superoxide and nitric oxide. Mutat. Res. 1998, 402, 77–84. [Google Scholar] [CrossRef]
- Shapiro, R.; Dubelman, S.; Feinberg, A.M.; Crain, P.F.; McCloskey, J.A. Isolation and identification of cross-linked nucleosides from nitrous acid treated deoxyribonucleic acid. Am. Chem. Soc. 1977, 99, 302–303. [Google Scholar] [CrossRef]
- Liu, R.H.; Baldwin, B.; Tennant, B.C.; Hotchkiss, J.H. Elevated formation of nitrate and N-nitrosodimethylamine in woodchucks (Marmota monax) associated with chronic woodchuck hepatitis virus infection. Cancer Res. 1991, 51, 3925–3929. [Google Scholar]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; Felley-Bosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef]
- Wink, D.A.; Laval, J. The Fpg protein, a DNA repair enzyme, is inhibited by the biomediator nitric oxide in vitro and in vivo. Carcinogenesis 1994, 15, 2125–2129. [Google Scholar] [CrossRef]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch Biochem. Biophys. 1991, 288, 481–487. [Google Scholar] [CrossRef]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S.; et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Brunson, D.; Crespi, C.L.; Penman, B.W.; Wishnok, J.S.; Tannenbaum, S.R. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc. Natl. Acad. Sci. USA 1992, 89, 3030–3034. [Google Scholar] [CrossRef] [PubMed]
- deRojas-Walker, T.; Tamir, S.; Ji, H.; Wishnok, J.S.; Tannenbaum, S.R. Nitric oxide induces oxidative damage in addition to deamination in macrophage DNA. Chem. Res. Toxicol. 1995, 8, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.C.; Hollstein, M. Clinical implications of the p53 tumor-suppressor gene. N. Engl. J. Med. 1993, 329, 1318–1327. [Google Scholar] [CrossRef]
- Tornaletti, S.; Pfeifer, G.P. Complete and tissue-independent methylation of CpG sites in the p53 gene: Implications for mutations in human cancers. Oncogene 1995, 10, 1493–1499. [Google Scholar] [PubMed]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar]
- Routledge, M.N.; Wink, D.A.; Keefer, L.K.; Dipple, A. DNA sequence changes induced by two nitric oxide donor drugs in the supF assay. Chem. Res. Toxicol. 1994, 7, 628–632. [Google Scholar] [CrossRef]
- Marnett, L.J.; Burcham, P.C. Endogenous DNA adducts: Potential and paradox. Chem. Res. Toxicol. 1993, 6, 771–785. [Google Scholar] [CrossRef]
- Wu, D.; Hu, Q.; Zhu, D. An Update on Hydrogen Sulfide and Nitric Oxide Interactions in the Cardiovascular System. Oxidative Med. Cell. Longev. 2018, 2018, 4579140. [Google Scholar] [CrossRef]
- Baker, P.R.; Schopfer, F.J.; O’Donnell, V.B.; Freeman, B.A. Convergence of nitric oxide and lipid signaling: Anti-inflammatory nitro-fatty acids. Free Radic. Biol. Med. 2009, 46, 989–1003. [Google Scholar] [CrossRef]
- Hogg, N.; Kalyanaraman, B. Nitric oxide and low-density lipoprotein oxidation. Free. Radic. Res. 1998, 28, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, K.M.; Liochev, S.I.; Fridovich, I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. J. Biol. Chem. 1994, 269, 23471–23476. [Google Scholar] [PubMed]
- Yermilov, V.; Rubio, J.; Becchi, M.; Friesen, M.D.; Pignatelli, B.; Ohshima, H. Formation of 8-nitroguanine by the reaction of guanine with peroxynitrite in vitro. Carcinogenesis 1995, 16, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Juedes, M.J.; Wogan, G.N. Peroxynitrite-induced mutation spectra of pSP189 following replication in bacteria and in human cells. Mutat. Res. 1996, 349, 51–61. [Google Scholar] [CrossRef]
- Ramezanian, M.S.; Padmaja, S.; Koppenol, W.H. Nitration and hydroxylation of phenolic compounds by peroxynitrite. Chem. Res. Toxicol. 1996, 9, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Epe, B.; Ballmaier, D.; Roussyn, I.; Briviba, K.; Sies, H. DNA damage by peroxynitrite characterized with DNA repair enzymes. Nucleic Acids Res. 1996, 24, 4105–4110. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.C.; Heppner, G.H.; Paul, L.A.; Fulton, A.M. Macrophage-mediated induction of DNA strand breaks in target tumor cells. Cancer Res. 1989, 49, 6652–6657. [Google Scholar]
- Eastman, A.; Barry, M.A. The origins of DNA breaks: A consequence of DNA damage, DNA repair, or apoptosis? Cancer Investig. 1992, 10, 229–240. [Google Scholar] [CrossRef]
- Ross, W.E. DNA topoisomerases as targets for cancer therapy. Biochem. Pharmacol. 1985, 34, 4191–4195. [Google Scholar] [CrossRef]
- Broillet, M.C.; Firestein, S. Direct activation of the olfactory cyclic nucleotide-gated channel through modification of sulfhydryl groups by NO compounds. Neuron 1996, 16, 377–385. [Google Scholar] [CrossRef]
- Lander, H.M.; Ogiste, J.S.; Teng, K.K.; Novogrodsky, A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J. Biol. Chem. 1995, 270, 21195–21198. [Google Scholar] [CrossRef] [PubMed]
- Caselli, A.; Camici, G.; Manao, G.; Moneti, G.; Pazzagli, L.; Cappugi, G.; Ramponi, G. Nitric oxide causes inactivation of the low molecular weight phosphotyrosine protein phosphatase. J. Biol. Chem. 1994, 269, 24878–24882. [Google Scholar] [PubMed]
- Kim, S.F. The role of nitric oxide in prostaglandin biology; update. Nitric Oxide 2011, 25, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Messmer, U.K.; Brune, B. Nitric oxide (NO) in apoptotic versus necrotic RAW 264.7 macrophage cell death: The role of NO-donor exposure, NAD+ content, and p53 accumulation. Arch. Biochem. Biophys. 1996, 327, 1–10. [Google Scholar] [CrossRef]
- Muhl, H.; Sandau, K.; Brune, B.; Briner, V.A.; Pfeilschifter, J. Nitric oxide donors induce apoptosis in glomerular mesangial cells, epithelial cells and endothelial cells. Eur. J. Pharmacol. 1996, 317, 137–149. [Google Scholar] [CrossRef]
- Marnett, L.J. Generation of mutagens during arachidonic acid metabolism. Cancer Metastasis Rev. 1994, 13, 303–308. [Google Scholar] [CrossRef]
- Laval, F.; Wink, D.A. Inhibition by nitric oxide of the repair protein, O6-methylguanine-DNA-methyltransferase. Carcinogenesis 1994, 15, 443–447. [Google Scholar] [CrossRef]
- Mota, M.B.S.; Carvalho, M.A.; Monteiro, A.N.A.; Mesquita, R.D. DNA damage response and repair in perspective: Aedes aegypti, Drosophila melanogaster and Homo sapiens. Parasites Vectors 2019, 12, 533. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Diderich, K.; Alanazi, M.; Hoeijmakers, J.H. Premature aging and cancer in nucleotide excision repair-disorders. DNA Repair 2011, 10, 772–780. [Google Scholar] [CrossRef]
- Kennedy, R.D.; D’Andrea, A.D. DNA repair pathways in clinical practice: Lessons from pediatric cancer susceptibility syndromes. J. Clin. Oncol. 2006, 24, 3799–3808. [Google Scholar] [CrossRef]
- Rolig, R.L.; McKinnon, P.J. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000, 23, 417–424. [Google Scholar] [CrossRef]
- Akbari, M.; Krokan, H.E. Cytotoxicity and mutagenicity of endogenous DNA base lesions as potential cause of human aging. Mech. Ageing Dev. 2008, 129, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- de Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef]
- Lehmann, A.R. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003, 85, 1101–1111. [Google Scholar] [CrossRef]
- Bensenouci, S.; Louhibi, L.; De Verneuil, H.; Mahmoudi, K.; Saidi-Mehtar, N. Diagnosis of Xeroderma Pigmentosum Groups A and C by Detection of Two Prevalent Mutations in West Algerian Population: A Rapid Genotyping Tool for the Frequent XPC Mutation c.1643_1644delTG. BioMed Res. Int. 2016, 2016, 2180946. [Google Scholar] [CrossRef]
- Chien, Y.H.; Bau, D.T.; Jan, K.Y. Nitric oxide inhibits DNA-adduct excision in nucleotide excision repair. Free Radic. Biol. Med. 2004, 36, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Liu, G.H.; Huang, B.; Chen, C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res 2007, 35, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Parsons, J.L. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell. Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.A.; Bullock, H.A.; Xu, Y.; Tritt, R.; Zimmerman, E.; Ulbright, T.M.; Foster, R.S.; Einhorn, L.H.; Kelley, M.R. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001, 61, 2220–2225. [Google Scholar] [PubMed]
- Albertella, M.R.; Lau, A.; O’Connor, M.J. The overexpression of specialized DNA polymerases in cancer. DNA Repair 2005, 4, 583–593. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. Targeted therapy for cancer using PARP inhibitors. Curr. Opin. Pharmacol. 2008, 8, 363–369. [Google Scholar] [CrossRef]
- Liu, L.; Gerson, S.L. Therapeutic impact of methoxyamine: Blocking repair of abasic sites in the base excision repair pathway. Curr. Opin. Investig. Drugs 2004, 5, 623–627. [Google Scholar]
- Jaiswal, M.; LaRusso, N.F.; Shapiro, R.A.; Billiar, T.R.; Gores, G.J. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology 2001, 120, 190–199. [Google Scholar] [CrossRef]
- Nakano, T.; Katafuchi, A.; Shimizu, R.; Terato, H.; Suzuki, T.; Tauchi, H.; Makino, K.; Skorvaga, M.; Van Houten, B.; Ide, H. Repair activity of base and nucleotide excision repair enzymes for guanine lesions induced by nitrosative stress. Nucleic Acids Res. 2005, 33, 2181–2191. [Google Scholar] [CrossRef][Green Version]
- Mutamba, J.T.; Svilar, D.; Prasongtanakij, S.; Wang, X.H.; Lin, Y.C.; Dedon, P.C.; Sobol, R.W.; Engelward, B.P. XRCC1 and base excision repair balance in response to nitric oxide. DNA Repair (Amst) 2011, 10, 1282–1293. [Google Scholar] [CrossRef]
- Parrish, M.C.; Chaim, I.A.; Nagel, Z.D.; Tannenbaum, S.R.; Samson, L.D.; Engelward, B.P. Nitric oxide induced S-nitrosation causes base excision repair imbalance. DNA Repair (Amst) 2018, 68, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M., 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Guillotin, D.; Martin, S.A. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp. Cell Res. 2014, 329, 110–115. [Google Scholar] [CrossRef]
- Heinen, C.D. Mismatch repair defects and Lynch syndrome: The role of the basic scientist in the battle against cancer. DNA Repair (Amst.) 2016, 38, 127–134. [Google Scholar] [CrossRef]
- Belcheva, A.; Green, B.; Weiss, A.; Streutker, C.; Martin, A. Elevated incidence of polyp formation in APC(Min/(+))Msh2(-)/(-) mice is independent of nitric oxide-induced DNA mutations. PLoS ONE 2013, 8, e65204. [Google Scholar] [CrossRef]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef]
- Sun, J.; Lee, K.J.; Davis, A.J.; Chen, D.J. Human Ku70/80 protein blocks exonuclease 1-mediated DNA resection in the presence of human Mre11 or Mre11/Rad50 protein complex. J. Biol. Chem. 2012, 287, 4936–4945. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef]
- Jalal, S.; Earley, J.N.; Turchi, J.J. DNA repair: From genome maintenance to biomarker and therapeutic target. Clin. Cancer Res. 2011, 17, 6973–6984. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.E.; van der Burg, M.; IJspeert, H.; Carroll, P.; Wu, Q.; Ochi, T.; Leitch, A.; Miller, E.S.; Kysela, B.; Jawad, A.; et al. Mutations in the NHEJ component XRCC4 cause primordial dwarfism. Am. J. Hum. Genet. 2015, 96, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, L.; Smith, G.C.; Charles, L.G. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging anti-tumour agents. Nat. Cell Biol. 2000, 2, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Sharma, S.; Javadekar, S.M.; Pandey, M.; Srivastava, M.; Kumari, R.; Raghavan, S.C. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis. 2015, 6, e1697. [Google Scholar] [CrossRef]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef]
- Mazin, A.V.; Bornarth, C.J.; Solinger, J.A.; Heyer, W.D.; Kowalczykowski, S.C. Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell 2000, 6, 583–592. [Google Scholar] [CrossRef]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef]
- Yakovlev, V.A. Nitric oxide-dependent downregulation of BRCA1 expression promotes genetic instability. Cancer Res. 2013, 73, 706–715. [Google Scholar] [CrossRef]
- Mujoo, K.; Pandita, R.K.; Tiwari, A.; Charaka, V.; Chakraborty, S.; Singh, D.K.; Hambarde, S.; Hittelman, W.N.; Horikoshi, N.; Hunt, C.R.; et al. Differentiation of Human Induced Pluripotent or Embryonic Stem Cells Decreases the DNA Damage Repair by Homologous Recombination. Stem Cell Rep. 2017, 9, 1660–1674. [Google Scholar] [CrossRef]
- Aqil, M.; Elseth, K.M.; Vesper, B.J.; Deliu, Z.; Aydogan, B.; Xue, J.; Radosevich, J.A. Part I-mechanism of adaptation: High nitric oxide adapted A549 cells show enhanced DNA damage response and activation of antiapoptotic pathways. Tumour Biol. 2014, 35, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Venclovas, C.; Thelen, M.P. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 2000, 28, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.P.; Dutta, A. DNA replication in eukaryotic cells. Ann. Rev. bioChem. 2002, 71, 333–374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef]
- Shibata, A.; Barton, O.; Noon, A.T.; Dahm, K.; Deckbar, D.; Goodarzi, A.A.; Lobrich, M.; Jeggo, P.A. Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest. Mol. Cell. Biol. 2010, 30, 3371–3383. [Google Scholar] [CrossRef]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef]
- Dai, Y.; Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef]
- Buscemi, G.; Perego, P.; Carenini, N.; Nakanishi, M.; Chessa, L.; Chen, J.; Khanna, K.; Delia, D. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene 2004, 23, 7691–7700. [Google Scholar] [CrossRef]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuasen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid destruction of human Cdc25A in response to DNA damage. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Urist, M.; Prives, C. Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J. Biol. Chem. 2003, 278, 20480–20489. [Google Scholar] [CrossRef] [PubMed]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Petrini, J.H.; Williams, B.R.; Lukas, J.; Bartek, J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat. Genet. 2002, 30, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, Z.; Gunasekera, A.H.; Sowin, T.J.; Rosenberg, S.H.; Fesik, S.; Zhang, H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 2003, 278, 21767–21773. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Kim, S.T.; Xu, B.; Maser, R.S.; Lin, J.; Petrini, J.H.; Kastan, M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 2000, 404, 613–617. [Google Scholar] [CrossRef]
- Wilson, K.A.; Stern, D.F. NFBD1/MDC1, 53BP1 and BRCA1 have both redundant and unique roles in the ATM pathway. Cell Cycle 2008, 7, 3584–3594. [Google Scholar] [CrossRef][Green Version]
- Hofseth, L.J.; Saito, S.; Hussain, S.P.; Espey, M.G.; Miranda, K.M.; Araki, Y.; Jhappan, C.; Higashimoto, Y.; He, P.; Linke, S.P.; et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc. Natl. Acad. Sci. USA 2003, 100, 143–148. [Google Scholar] [CrossRef]
- Wang, X.; Zalcenstein, A.; Oren, M. Nitric oxide promotes p53 nuclear retention and sensitizes neuroblastoma cells to apoptosis by ionizing radiation. Cell Death Differ. 2003, 10, 468–476. [Google Scholar] [CrossRef]
- de Gooijer, M.C.; van den Top, A.; Bockaj, I.; Beijnen, J.H.; Wurdinger, T.; van Tellingen, O. The G2 checkpoint-a node-based molecular switch. FEBS Open Bio 2017, 7, 439–455. [Google Scholar] [CrossRef]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Thornton, T.M.; Rincon, M. Non-classical p38 map kinase functions: Cell cycle checkpoints and survival. Int. J. Biol. Sci. 2009, 5, 44–51. [Google Scholar] [CrossRef]
- Liu, Z.; Li, G.; Gou, Y.; Xiao, D.; Luo, G.; Saavedra, J.E.; Liu, J.; Wang, H. JS-K, a nitric oxide prodrug, induces DNA damage and apoptosis in HBV-positive hepatocellular carcinoma HepG2.2.15 cell. Biomed. Pharmacother. 2017, 92, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Kiziltepe, T.; Hideshima, T.; Ishitsuka, K.; Ocio, E.M.; Raje, N.; Catley, L.; Li, C.Q.; Trudel, L.J.; Yasui, H.; Vallet, S.; et al. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood 2007, 110, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Williams, J.L. Nitric oxide-donating aspirin induces G2/M phase cell cycle arrest in human cancer cells by regulating phase transition proteins. Int. J. Oncol. 2012, 41, 325–330. [Google Scholar] [CrossRef]
- Nakanishi, M.; Shimada, M.; Niida, H. Genetic instability in cancer cells by impaired cell cycle checkpoints. Cancer Sci. 2006, 97, 984–989. [Google Scholar] [CrossRef]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef]
- Grasselli, A.; Nanni, S.; Colussi, C.; Aiello, A.; Benvenuti, V.; Ragone, G.; Moretti, F.; Sacchi, A.; Bacchetti, S.; Gaetano, C.; et al. Estrogen receptor-alpha and endothelial nitric oxide synthase nuclear complex regulates transcription of human telomerase. Circ. Res. 2008, 103, 34–42. [Google Scholar] [CrossRef]
- Kim, P.K.; Zamora, R.; Petrosko, P.; Billiar, T.R. The regulatory role of nitric oxide in apoptosis. Int. Immunopharmacol. 2001, 1, 1421–1441. [Google Scholar] [CrossRef]
- Johlfs, M.G.; Fiscus, R.R. Protein kinase G type-Ialpha phosphorylates the apoptosis-regulating protein Bad at serine 155 and protects against apoptosis in N1E-115 cells. Neurochem. Int. 2010, 56, 546–553. [Google Scholar] [CrossRef]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef]
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134. [Google Scholar] [CrossRef]
- Blaise, G.A.; Gauvin, D.; Gangal, M.; Authier, S. Nitric oxide, cell signaling and cell death. Toxicology 2005, 208, 177–192. [Google Scholar] [CrossRef]
- Pilz, R.B.; Casteel, D.E. Regulation of gene expression by cyclic GMP. Circ. Res. 2003, 93, 1034–1046. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Garban, H.J.; Bonavida, B. Nitric oxide sensitizes ovarian tumor cells to Fas-induced apoptosis. Gynecol. Oncol. 1999, 73, 257–264. [Google Scholar] [CrossRef]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell 2011, 145, 92–103. [Google Scholar] [CrossRef]
- Brune, B. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ. 2003, 10, 864–869. [Google Scholar] [CrossRef]
- Chae, H.J.; So, H.S.; Chae, S.W.; Park, J.S.; Kim, M.S.; Oh, J.M.; Chung, Y.T.; Yang, S.H.; Jeong, E.T.; Kim, H.M.; et al. Sodium nitroprusside induces apoptosis of H9C2 cardiac muscle cells in a c-Jun N-terminal kinase-dependent manner. Int. Immunopharmacol. 2001, 1, 967–978. [Google Scholar] [CrossRef]
- Kim, S.J.; Ju, J.W.; Oh, C.D.; Yoon, Y.M.; Song, W.K.; Kim, J.H.; Yoo, Y.J.; Bang, O.S.; Kang, S.S.; Chun, J.S. ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J. Biol. Chem. 2002, 277, 1332–1339. [Google Scholar] [CrossRef]
- Edilova, M.I.; Abdul-Sater, A.A.; Watts, T.H. TRAF1 Signaling in Human Health and Disease. Front. Immunol. 2018, 9, 2969. [Google Scholar] [CrossRef]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef]
- Kazerounian, S.; Lawler, J. Integration of pro- and anti-angiogenic signals by endothelial cells. J. Cell Commun. Signal. 2018, 12, 171–179. [Google Scholar] [CrossRef]
- Morbidelli, L.; Donnini, S.; Ziche, M. Role of nitric oxide in the modulation of angiogenesis. Curr. Pharm. Des. 2003, 9, 521–530. [Google Scholar] [CrossRef]
- Wink, D.A.; Vodovotz, Y.; Laval, J.; Laval, F.; Dewhirst, M.W.; Mitchell, J.B. The multifaceted roles of nitric oxide in cancer. Carcinogenesis 1998, 19, 711–721. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Zhou, J.; Schmid, T.; Brune, B. HIF-1alpha and p53 as targets of NO in affecting cell proliferation, death and adaptation. Curr. Mol. Med. 2004, 4, 741–751. [Google Scholar] [CrossRef]
- Ying, L.; Hofseth, L.J. An emerging role for endothelial nitric oxide synthase in chronic inflammation and cancer. Cancer Res. 2007, 67, 1407–1410. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Chen, J.X.; Meyrick, B. Hypoxia increases Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery endothelium. Lab. Investig. 2004, 84, 182–190. [Google Scholar] [CrossRef]
- Brouet, A.; Sonveaux, P.; Dessy, C.; Balligand, J.L.; Feron, O. Hsp90 ensures the transition from the early Ca2+-dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J. Biol. Chem. 2001, 276, 32663–32669. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Ridnour, L.A.; Perruccio, E.M.; Espey, M.G.; Wink, D.A.; Roberts, D.D. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc. Natl. Acad. Sci. USA 2005, 102, 13141–13146. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Isenberg, J.S.; Espey, M.G.; Thomas, D.D.; Roberts, D.D.; Wink, D.A. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc. Natl. Acad. Sci. USA 2005, 102, 13147–13152. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, S.; Lee, S.J.; Kim, C.K.; Kim, Y.M.; Chung, H.T.; Lee, H.; Han, J.A.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Prostaglandin E2 stimulates angiogenesis by activating the nitric oxide/cGMP pathway in human umbilical vein endothelial cells. Exp. Mol. Med. 2005, 37, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Morbidelli, L.; Masini, E.; Amerini, S.; Granger, H.J.; Maggi, C.A.; Geppetti, P.; Ledda, F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J. Clin. Investig. 1994, 94, 2036–2044. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Gos, M.; Miloszewska, J.; Przybyszewska, M. Epithelial-mesenchymal transition in cancer progression. Postep. Biochem. 2009, 55, 121–128. [Google Scholar]
- Banyard, J.; Bielenberg, D.R. The role of EMT and MET in cancer dissemination. Connect. Tissue Res. 2015, 56, 403–413. [Google Scholar] [CrossRef]
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle 2010, 9, 4931–4940. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S. Inhibition of Epithelial-to-Mesenchymal Transition (EMT) in Cancer by Nitric Oxide: Pivotal Roles of Nitrosylation of NF-kappaB, YY1 and Snail. Forum Immunopathol. Dis. Ther. 2012, 3, 125–133. [Google Scholar] [CrossRef]
- Hickok, J.R.; Sahni, S.; Mikhed, Y.; Bonini, M.G.; Thomas, D.D. Nitric oxide suppresses tumor cell migration through N-Myc downstream-regulated gene-1 (NDRG1) expression: Role of chelatable iron. J. Biol. Chem. 2011, 286, 41413–41424. [Google Scholar] [CrossRef]
- Vyas-Read, S.; Shaul, P.W.; Yuhanna, I.S.; Willis, B.C. Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L212–L221. [Google Scholar] [CrossRef]
- Jespersen, C.; Doller, A.; Akool, E.S.; Bachmann, M.; Muller, R.; Gutwein, P.; Muhl, H.; Pfeilschifter, J.; Eberhardt, W. Molecular mechanisms of nitric oxide-dependent inhibition of TPA-induced matrix metalloproteinase-9 (MMP-9) in MCF-7 cells. J. Cell Physiol. 2009, 219, 276–287. [Google Scholar] [CrossRef]
- Gonzalez-Avila, G.; Sommer, B.; Garcia-Hernandez, A.A.; Ramos, C. Matrix Metalloproteinases’ Role in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 97–131. [Google Scholar] [CrossRef]
- Singh, T.; Chaudhary, S.C.; Kapur, P.; Weng, Z.; Elmets, C.A.; Kopelovich, L.; Athar, M. Nitric oxide donor exisulind is an effective inhibitor of murine photocarcinogenesis. Photochem. Photobiol. 2012, 88, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.M.; Stetler-Stevenson, W.G. The role of matrix metalloproteases and their inhibitors in tumour invasion, metastasis and angiogenesis. Eur. Respir. J. 1994, 7, 2062–2072. [Google Scholar]
- Akool, E.S.; Kleinert, H.; Hamada, F.M.; Abdelwahab, M.H.; Forstermann, U.; Pfeilschifter, J.; Eberhardt, W. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol. Cell. Biol. 2003, 23, 4901–4916. [Google Scholar] [CrossRef]
- Weiss, J.M.; Ridnour, L.A.; Back, T.; Hussain, S.P.; He, P.; Maciag, A.E.; Keefer, L.K.; Murphy, W.J.; Harris, C.C.; Wink, D.A.; et al. Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy. J. Exp. Med. 2010, 207, 2455–2467. [Google Scholar] [CrossRef]
- Le, X.; Wei, D.; Huang, S.; Lancaster, J.R., Jr.; Xie, K. Nitric oxide synthase II suppresses the growth and metastasis of human cancer regardless of its up-regulation of protumor factors. Proc. Natl. Acad. Sci. USA 2005, 102, 8758–8763. [Google Scholar] [CrossRef]
- El Hasasna, H.; Saleh, A.; Al Samri, H.; Athamneh, K.; Attoub, S.; Arafat, K.; Benhalilou, N.; Alyan, S.; Viallet, J.; Al Dhaheri, Y.; et al. Rhus coriaria suppresses angiogenesis, metastasis and tumor growth of breast cancer through inhibition of STAT3, NFkappaB and nitric oxide pathways. Sci. Rep. 2016, 6, 21144. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Supriatno; Kawaguchi, S.; Tomitaro, O.; Yoshida, H.; Sato, M. Overexpression of iNOS gene suppresses the tumorigenicity and metastasis of oral cancer cells. In Vivo 2004, 18, 449–455. [Google Scholar] [PubMed]
- Wei, D.; Richardson, E.L.; Zhu, K.; Wang, L.; Le, X.; He, Y.; Huang, S.; Xie, K. Direct demonstration of negative regulation of tumor growth and metastasis by host-inducible nitric oxide synthase. Cancer Res. 2003, 63, 3855–3859. [Google Scholar] [PubMed]
- Shi, Q.; Xiong, Q.; Wang, B.; Le, X.; Khan, N.A.; Xie, K. Influence of nitric oxide synthase II gene disruption on tumor growth and metastasis. Cancer Res. 2000, 60, 2579–2583. [Google Scholar] [PubMed]
- Irwin, C.; Roberts, W.; Naseem, K.M. Nitric oxide inhibits platelet adhesion to collagen through cGMP-dependent and independent mechanisms: The potential role for S-nitrosylation. Platelets 2009, 20, 478–486. [Google Scholar] [CrossRef]
- Switzer, C.H.; Glynn, S.A.; Cheng, R.Y.; Ridnour, L.A.; Green, J.E.; Ambs, S.; Wink, D.A. S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol. Cancer Res. MCR 2012, 10, 1203–1215. [Google Scholar] [CrossRef]
- Jadeski, L.C.; Lala, P.K. Nitric oxide synthase inhibition by N(G)-nitro-L-arginine methyl ester inhibits tumor-induced angiogenesis in mammary tumors. Am. J. Pathol. 1999, 155, 1381–1390. [Google Scholar] [CrossRef]
- Orucevic, A.; Bechberger, J.; Green, A.M.; Shapiro, R.A.; Billiar, T.R.; Lala, P.K. Nitric-oxide production by murine mammary adenocarcinoma cells promotes tumor-cell invasiveness. Int. J. Cancer 1999, 81, 889–896. [Google Scholar] [CrossRef]
- Siegert, A.; Rosenberg, C.; Schmitt, W.D.; Denkert, C.; Hauptmann, S. Nitric oxide of human colorectal adenocarcinoma cell lines promotes tumour cell invasion. Br. J. Cancer 2002, 86, 1310–1315. [Google Scholar] [CrossRef]
- Sun, M.H.; Han, X.C.; Jia, M.K.; Jiang, W.D.; Wang, M.; Zhang, H.; Han, G.; Jiang, Y. Expressions of inducible nitric oxide synthase and matrix metalloproteinase-9 and their effects on angiogenesis and progression of hepatocellular carcinoma. World J. Gastroenterol. 2005, 11, 5931–5937. [Google Scholar] [CrossRef]
- Veeravalli, K.K.; Rao, J.S. MMP-9 and uPAR regulated glioma cell migration. Cell Adhes. Migr. 2012, 6, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Kasic, T.; Gri, G.; Gallana, K.; Borsellino, G.; Marigo, I.; Battistini, L.; Iafrate, M.; Prayer-Galetti, T.; Pagano, F.; et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med. 2005, 201, 1257–1268. [Google Scholar] [CrossRef]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Gehad, A.E.; Lichtman, M.K.; Schmults, C.D.; Teague, J.E.; Calarese, A.W.; Jiang, Y.; Watanabe, R.; Clark, R.A. Nitric oxide-producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J. Investig. Dermatol. 2012, 132, 2642–2651. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef] [PubMed]
- Douguet, L.; Bod, L.; Lengagne, R.; Labarthe, L.; Kato, M.; Avril, M.F.; Prevost-Blondel, A. Nitric oxide synthase 2 is involved in the pro-tumorigenic potential of gammadelta17 T cells in melanoma. Oncoimmunology 2016, 5, e1208878. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.; Wang, J.; Vangundy, Z.; You, J.; Yildiz, V.; Yu, L.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4(+) T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7, 15424. [Google Scholar] [CrossRef]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.; McMichael, E.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor-Mediated Natural Killer Cell Function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef]
- Jayaraman, P.; Alfarano, M.G.; Svider, P.F.; Parikh, F.; Lu, G.; Kidwai, S.; Xiong, H.; Sikora, A.G. iNOS expression in CD4+ T cells limits Treg induction by repressing TGFbeta1: Combined iNOS inhibition and Treg depletion unmask endogenous antitumor immunity. Clin. Cancer Res. 2014, 20, 6439–6451. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Chen, X.; Huang, Y.; Li, W.; Li, J.; Cao, K.; Cao, G.; Zhang, L.; Li, F.; Roberts, A.I.; et al. Phylogenetic distinction of iNOS and IDO function in mesenchymal stem cell-mediated immunosuppression in mammalian species. Cell Death Differ. 2014, 21, 388–396. [Google Scholar] [CrossRef]
- Chinnadurai, R.; Sands, J.; Rajan, D.; Liu, X.; Arafat, D.; Das, R.; Anania, F.A.; Gibson, G.; Kisseleva, T.; Galipeau, J. Molecular Genetic and Immune Functional Responses Distinguish Bone Marrow Mesenchymal Stromal Cells from Hepatic Stellate Cells. Stem Cells 2019, 37, 1075–1082. [Google Scholar] [CrossRef]
- Hoos, M.D.; Vitek, M.P.; Ridnour, L.A.; Wilson, J.; Jansen, M.; Everhart, A.; Wink, D.A.; Colton, C.A. The impact of human and mouse differences in NOS2 gene expression on the brain’s redox and immune environment. Mol. Neurodegener. 2014, 9, 50. [Google Scholar] [CrossRef]
- Ryan, A.E.; Burke, A.J.; Giles, F.J.; Sullivan, F.J.; Glynn, S.A. Mechanisms of Nitric Oxide-Dependent Regulation of Tumor Invasion and Metastasis. In Nitric Oxide and Cancer: Pathogenesis and Therapy, 1st ed.; Bonavida, B., Ed.; Springer: Los Angeles, CA, USA, 2015; pp. 49–63. [Google Scholar]
- Ridnour, L.A.; Thomas, D.D.; Donzelli, S.; Espey, M.G.; Roberts, D.D.; Wink, D.A.; Isenberg, J.S. The biphasic nature of nitric oxide responses in tumor biology. Antioxid. Redox Signal. 2006, 8, 1329–1337. [Google Scholar] [CrossRef]
- Bonavida, B.; Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015, 6, 486–494. [Google Scholar] [CrossRef]
- Huang, Z.; Fu, J.; Zhang, Y. Nitric Oxide Donor-Based Cancer Therapy: Advances and Prospects. J. Med. Chem. 2017, 60, 7617–7635. [Google Scholar] [CrossRef]
- Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P.; Pantziarka, P. Repurposing Drugs in Oncology (ReDO)-nitroglycerin as an anti-cancer agent. Ecancermedicalscience 2015, 9, 568. [Google Scholar] [CrossRef] [PubMed]
- Huerta, S.; Chilka, S.; Bonavida, B. Nitric oxide donors: Novel cancer therapeutics (review). Int. J. Oncol. 2008, 33, 909–927. [Google Scholar] [CrossRef] [PubMed]
- Park, I.C.; Woo, S.H.; Park, M.J.; Lee, H.C.; Lee, S.J.; Hong, Y.J.; Lee, S.H.; Hong, S.I.; Rhee, C.H. Ionizing radiation and nitric oxide donor sensitize Fas-induced apoptosis via up-regulation of Fas in human cervical cancer cells. Oncol. Rep. 2003, 10, 629–633. [Google Scholar] [PubMed]
- Mitchell, J.B.; Wink, D.A.; DeGraff, W.; Gamson, J.; Keefer, L.K.; Krishna, M.C. Hypoxic mammalian cell radiosensitization by nitric oxide. Cancer Res. 1993, 53, 5845–5848. [Google Scholar] [PubMed]
- Wang, P.G.; Xian, M.; Tang, X.; Wu, X.; Wen, Z.; Cai, T.; Janczuk, A.J. Nitric oxide donors: Chemical activities and biological applications. Chem. Rev. 2002, 102, 1091–1134. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Kawanishi, S. Oxidative DNA damage induced by simultaneous generation of nitric oxide and superoxide. FEBS Lett. 1995, 371, 86–88. [Google Scholar] [CrossRef]
- Keefer, L.K. Progress toward clinical application of the nitric oxide-releasing diazeniumdiolates. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 585–607. [Google Scholar] [CrossRef]
- Thatcher, G.R.; Nicolescu, A.C.; Bennett, B.M.; Toader, V. Nitrates and NO release: Contemporary aspects in biological and medicinal chemistry. Free Radic. Biol. Med. 2004, 37, 1122–1143. [Google Scholar] [CrossRef]
- Glynn, S.A.; Boersma, B.J.; Dorsey, T.H.; Yi, M.; Yfantis, H.G.; Ridnour, L.A.; Martin, D.N.; Switzer, C.H.; Hudson, R.S.; Wink, D.A.; et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J. Clin. Investig. 2010, 120, 3843–3854. [Google Scholar] [CrossRef]
- Walsh, E.M.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of Triple Negative Breast Cancer and the Impact of Inducible Nitric Oxide Synthase on Tumor Biology and Patient Outcomes. Crit. Rev. Oncog. 2016, 21, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Switzer, C.H.; Lizardo, M.M.; Khanna, C.; Glynn, S.A.; Hussain, S.P.; Young, H.A.; Ambs, S.; et al. Tumor microenvironment-based feed-forward regulation of NOS2 in breast cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, 6323–6328. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Glynn, S.A.; Greer, M.; Somasundaram, V.; No, J.H.; Scheiblin, D.A.; Garrido, P.; Heinz, W.F.; Ryan, A.E.; Weiss, J.M.; et al. Coexpression of NOS2 and COX2 accelerates tumor growth and reduces survival in estrogen receptor-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13030–13035. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.; Gonzalez, D.D.; Liu, Z.B.; Li, X.; Wong, H.; Granados, S.; Ezzedine, N.E.; Sieglaff, D.H.; Ensor, J.E.; Miller, K.D.; et al. Role of RPL39 in Metaplastic Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw292. [Google Scholar] [CrossRef] [PubMed]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 2017, 8, 80568–80588. [Google Scholar] [CrossRef] [PubMed]
- Davila-Gonzalez, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; et al. Pharmacological Inhibition of NOS Activates ASK1/JNK Pathway Augmenting Docetaxel-Mediated Apoptosis in Triple-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Pershing, N.L.; Yang, C.F.J.; Xu, M.; Counter, C.M. Treatment with the nitric oxide synthase inhibitor L-NAME provides a survival advantage in a mouse model of Kras mutation-positive, non-small cell lung cancer. Oncotarget 2016, 7, 42385–42392. [Google Scholar] [CrossRef]
- Camp, E.R.; Yang, A.; Liu, W.; Fan, F.; Somcio, R.; Hicklin, D.J.; Ellis, L.M. Roles of nitric oxide synthase inhibition and vascular endothelial growth factor receptor-2 inhibition on vascular morphology and function in an in vivo model of pancreatic cancer. Clin. Cancer Res. 2006, 12, 2628–2633. [Google Scholar] [CrossRef]
- Lampson, B.L.; Kendall, S.D.; Ancrile, B.B.; Morrison, M.M.; Shealy, M.J.; Barrientos, K.S.; Crowe, M.S.; Kashatus, D.F.; White, R.R.; Gurley, S.B.; et al. Targeting eNOS in pancreatic cancer. Cancer Res. 2012, 72, 4472–4482. [Google Scholar] [CrossRef]
- Fujita, M.; Somasundaram, V.; Basudhar, D.; Cheng, R.Y.S.; Ridnour, L.A.; Higuchi, H.; Imadome, K.; No, J.H.; Bharadwaj, G.; Wink, D.A. Role of nitric oxide in pancreatic cancer cells exhibiting the invasive phenotype. Redox Biol. 2019, 22, 101158. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? Int. J. Mol. Sci. 2020, 21, 9393. https://doi.org/10.3390/ijms21249393
Khan FH, Dervan E, Bhattacharyya DD, McAuliffe JD, Miranda KM, Glynn SA. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? International Journal of Molecular Sciences. 2020; 21(24):9393. https://doi.org/10.3390/ijms21249393
Chicago/Turabian StyleKhan, Faizan H., Eoin Dervan, Dibyangana D. Bhattacharyya, Jake D. McAuliffe, Katrina M. Miranda, and Sharon A. Glynn. 2020. "The Role of Nitric Oxide in Cancer: Master Regulator or NOt?" International Journal of Molecular Sciences 21, no. 24: 9393. https://doi.org/10.3390/ijms21249393
APA StyleKhan, F. H., Dervan, E., Bhattacharyya, D. D., McAuliffe, J. D., Miranda, K. M., & Glynn, S. A. (2020). The Role of Nitric Oxide in Cancer: Master Regulator or NOt? International Journal of Molecular Sciences, 21(24), 9393. https://doi.org/10.3390/ijms21249393