Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction



{kind=link}
{kind=link}
Abstract
:1. Introduction
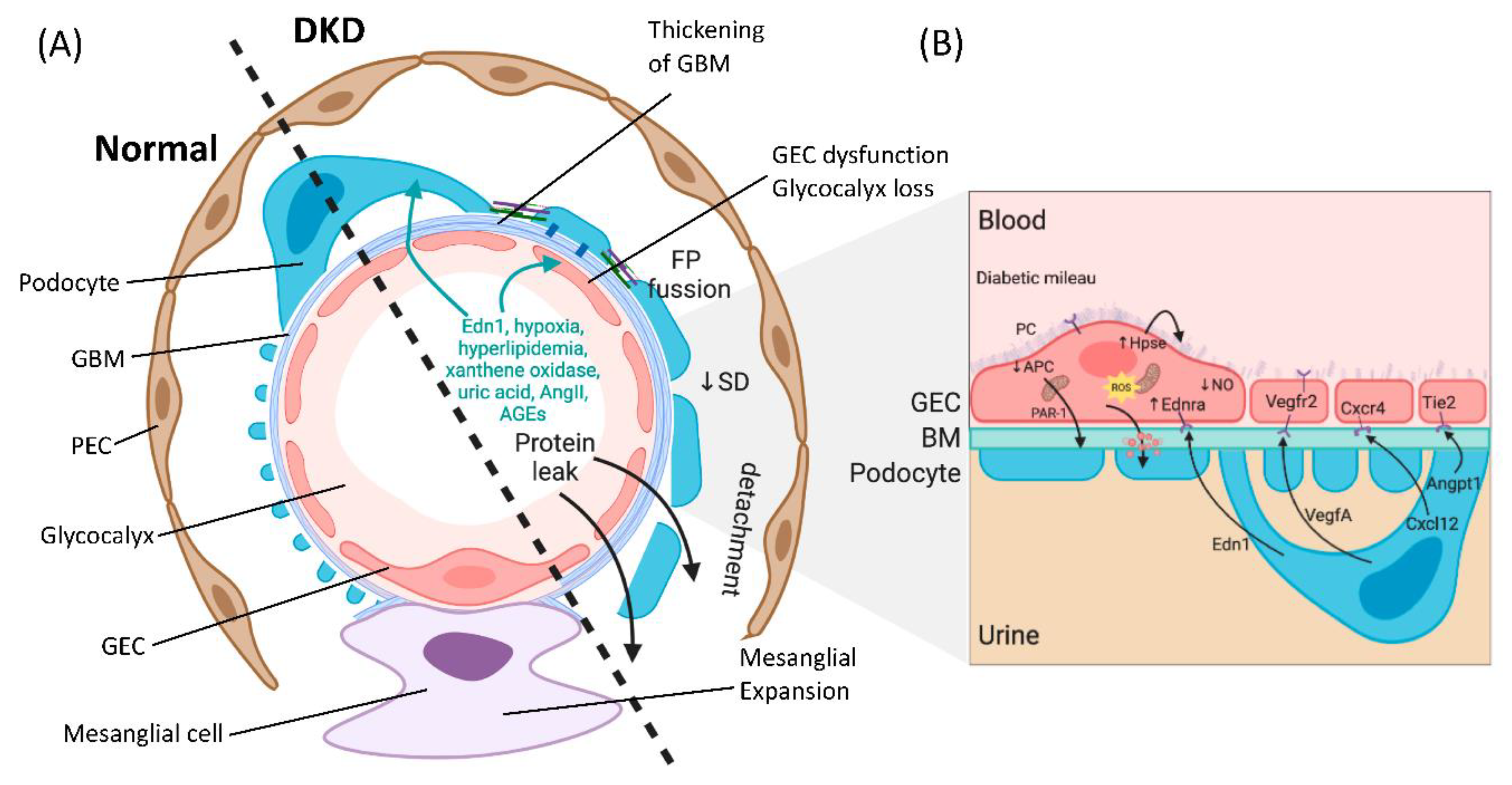
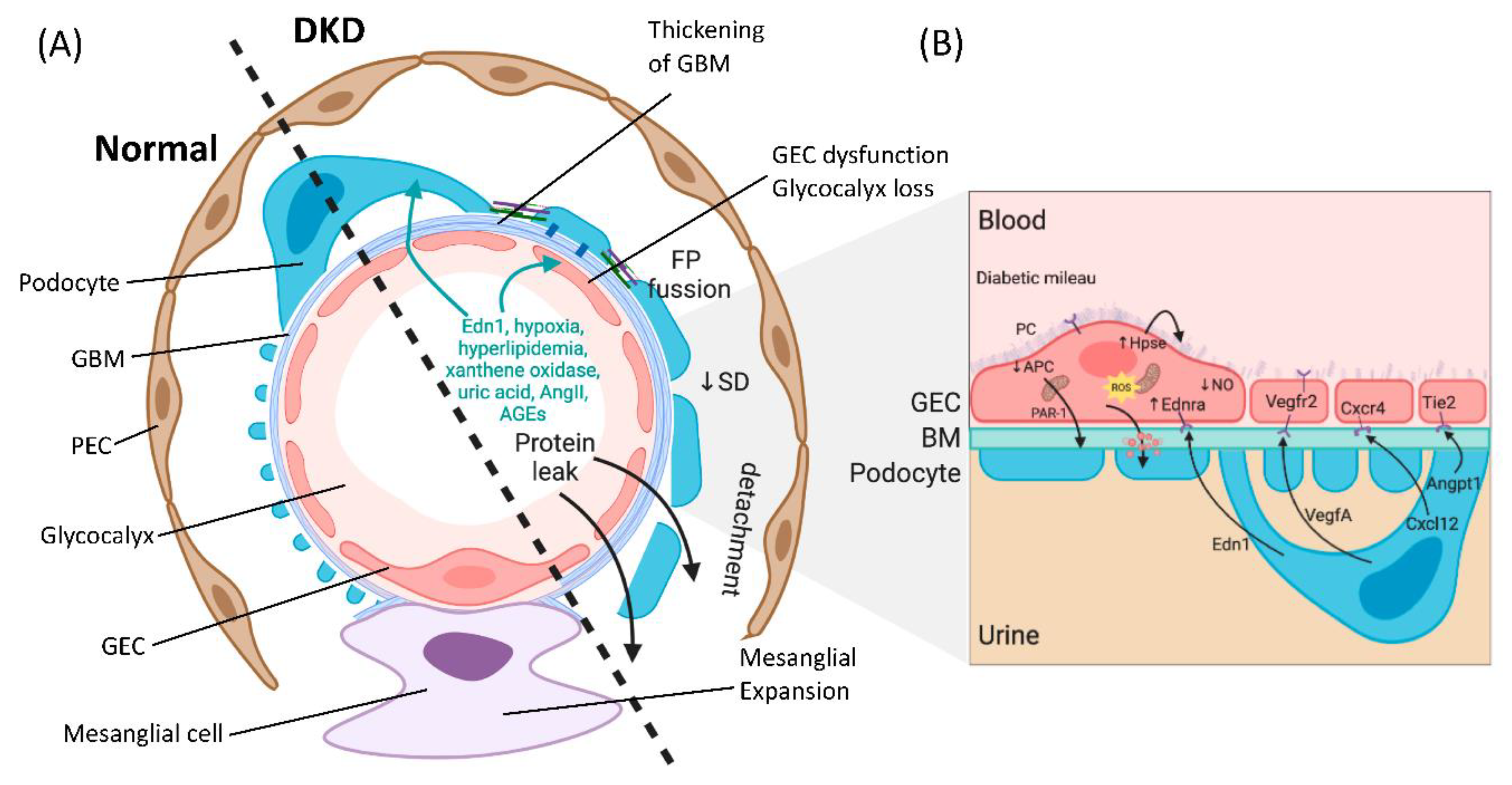
2. The Diabetic Milieu Affects Structure and Function of the Interconnected Glomerular Filtration Barrier
3. Crosstalk between GECs and Podocytes Is Essential for Filtration Barrier Function and Is Disturbed in DKD
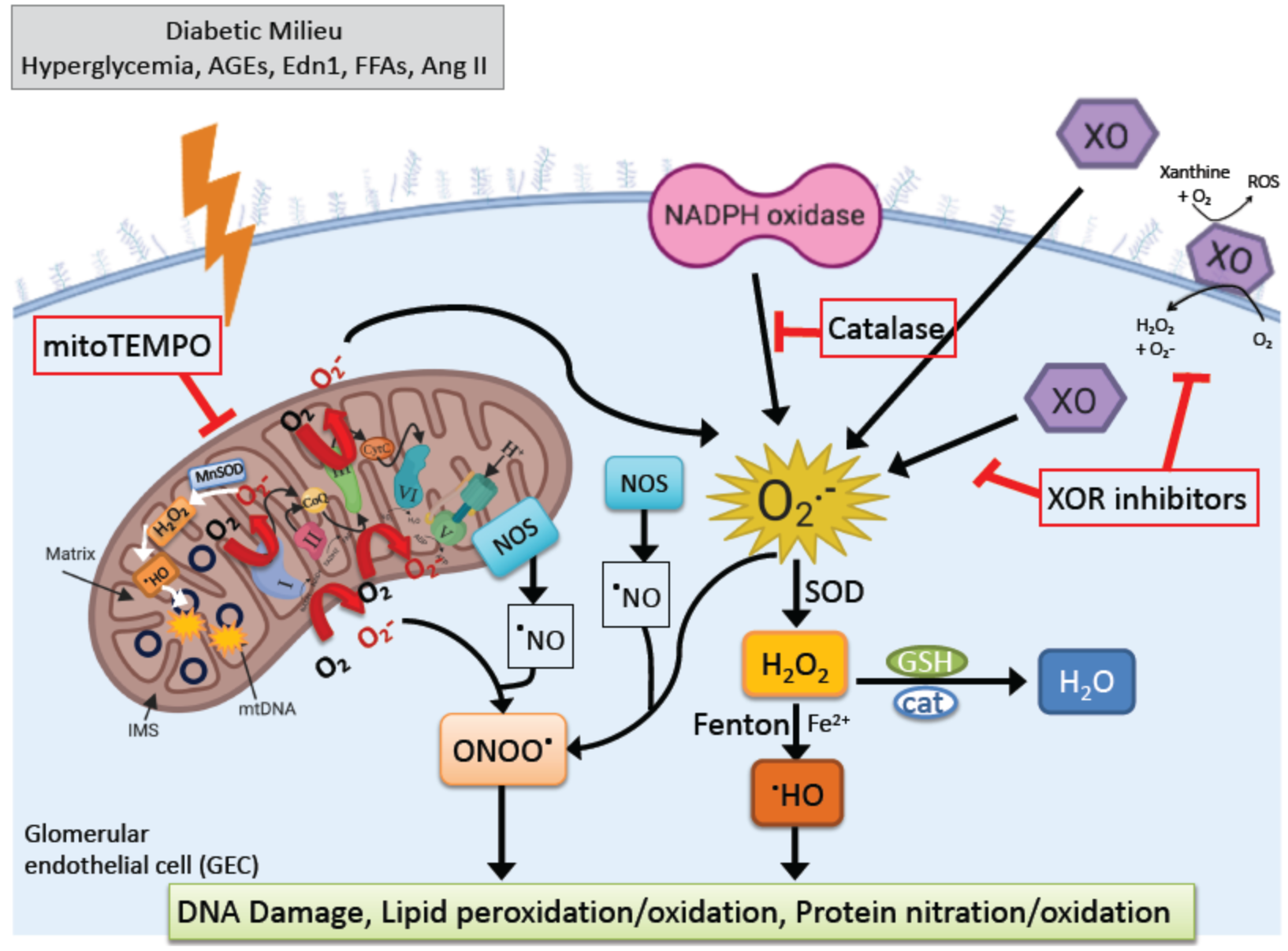
4. Oxidative Stress in DKD
4.1. Active Enzymatic ROS Generation in DKD
4.1.1. NADPH Oxidase (NOX)
4.1.2. Xanthine Oxidoreductase (XOR)
4.1.3. Mitochondrial ROS
4.2. ROS Interplay
4.3. Current Clinical Approaches for DKD and Their Effects on ROS
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette A1 |
| AGE | Advanced glycation end product |
| AMPK | AMP-activated protein kinase |
| Ang II | Angiotensin II |
| Angpt1 | Angiopoietin 1 |
| APC | Activated protein C |
| ATP | Adenosine triphosphate |
| CKD | Chronic kidney disease |
| CYP | Cytochrome P450 |
| DKD | Diabetic kidney disease |
| Ednra | Endothelin receptor A |
| Edn1 | Endothelin 1 |
| eNOS | Endothelial nitric oxide synthase |
| EPCR | Endothelial protein C receptor |
| ESL | Endothelial surface layer |
| ESRD | End-stage renal disease |
| FP | Foot process |
| FSGS | Focal segmental glomerulosclerosis |
| GBM | Glomerular basement membrane |
| GEC | Glomerular endothelial cell |
| GFR | Glomerular filtration rate |
| KLF2 | Krüppel-like factor 2 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| PAR-1 | Protease-activated receptor 1 |
| PEC | Parietal epithelial cell |
| PKC | Protein kinase C |
| RAAS | Renin angiotensin aldosterone system |
| ROS | Reactive oxygen species |
| SD | Slit diaphragm |
| SGLT2 | Sodium glucose cotransporter 2 |
| STZ | Streptozotocin |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TGFβ | Transforming growth factor β |
| UA | Uric acid |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR | Vascular endothelial growth factor receptor |
| XDH | Xanthine dehydrogenase |
| XO | Xanthine oxidase |
| XOR | Xanthine oxidoreductase |
References
- Chapter 1: Incidence, Prevalence, Patient Characteristics, and Treatment Modalities. Am. J. Kidney Dis. 2019, 73, S291–S332. [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.; Cavan, D.; Shaw, J.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.; Karuranga, S.; Malanda, B.; Saeedi, P.; Basit, A.; Besançon, S.; Bommer, C.; Esteghamati, A.; Ogurtsova, K.; Zhang, P.; et al. Global and regional estimates and projections of diabetes-related health expenditure: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2020, 162, 108072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2020. Diabetes Care 2019, 43, S135–S151. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.-J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar] [CrossRef]
- Caramori, M.L.; Parks, A.; Mauer, M. Renal Lesions Predict Progression of Diabetic Nephropathy in Type 1 Diabetes. J. Am. Soc. Nephrol. 2013, 24, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Drummond, K.N.; Kramer, M.S.; Suissa, S.; Lévy-Marchal, C.; Dell’Aniello, S.; Sinaiko, A.; Mauer, M. Effects of duration and age at onset of type 1 diabetes on preclinical manifestations of nephropathy. Diabetes 2003, 52, 1818–1824. [Google Scholar] [CrossRef]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Steffes, M.W.; Schmidt, D.; McCrery, R.; Basgen, J.M. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001, 59, 2104–2113. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.W.; Bennett, P.H.; Nelson, R.G. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 1999, 42, 1341–1344. [Google Scholar] [CrossRef] [Green Version]
- Pagtalunan, M.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dounousi, E.; Duni, A.; Leivaditis, K.; Vaios, V.; Eleftheriadis, T.; Liakopoulos, V. Improvements in the Management of Diabetic Nephropathy. Rev. Diabet. Stud. 2015, 12, 119–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katalin, S.; Susztak, K. Podocytes: The Weakest Link in Diabetic Kidney Disease? Curr. Diabetes Rep. 2016, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Stieger, N.; Worthmann, K.; Teng, B.; Engeli, S.; Das, A.M.; Haller, H.; Schiffer, M. Impact of high glucose and transforming growth factor–β on bioenergetic profiles in podocytes. Metab. Clin. Exp. 2012, 61, 1073–1086. [Google Scholar] [CrossRef]
- Kravets, I.; Mallipattu, S.K. The Role of Podocytes and Podocyte-Associated Biomarkers in Diagnosis and Treatment of Diabetic Kidney Disease. J. Endocr. Soc. 2020, 4, bvaa029. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Liu, Q.; Liu, B. Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef]
- Brosius, F.C.; Coward, R.J. Podocytes, Signaling Pathways, and Vascular Factors in Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2014, 21, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Mathieson, P.W. The podocyte as a target for therapies—New and old. Nat. Rev. Nephrol. 2011, 8, 52–56. [Google Scholar] [CrossRef]
- Zheng, X.; Soroush, F.; Long, J.; Hall, E.T.; Adishesha, P.K.; Bhattacharya, S.; Kiani, M.F.; Bhalla, V. Murine glomerular transcriptome links endothelial cell-specific molecule-1 deficiency with susceptibility to diabetic nephropathy. PLoS ONE 2017, 12, e0185250. [Google Scholar] [CrossRef]
- Sol, M.; Kamps, J.A.A.M.; Born, J.V.D.; Heuvel, M.C.V.D.; Van Der Vlag, J.; Krenning, G.; Hillebrands, J.-L. Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 2020, 11, 573557. [Google Scholar] [CrossRef]
- Kuwabara, A.; Satoh, M.; Tomita, N.; Sasaki, T.; Kashihara, N. Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia 2010, 53, 2056–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2016, 66, 763–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraldsson, B.; Nyström, J.; Deen, W.M. Properties of the Glomerular Barrier and Mechanisms of Proteinuria. Physiol. Rev. 2008, 88, 451–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jen, K.Y.; Laszik, Z.G. Endotheliopathies: Hemolytic Uremic Syndrome, Thrombotic Thrombocytopenic Purpura, and Preeclampsia. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 2767–2787. [Google Scholar] [CrossRef]
- Weinbaum, S.; Cancel, L.M.; Fu, B.M.; Tarbell, J.M. The Glycocalyx and Its Role in Vascular Physiology and Vascular Related Diseases. Cardiovasc. Eng. Technol. 2020, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Vink, H.; Hiramatsu, O.; Kajita, T.; Shigeto, F.; Spaan, J.A.E.; Kajiya, F. Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am. J. Physiol. Circ. Physiol. 2003, 285, H722–H726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florian, J.A.; Kosky, J.R.; Ainslie, K.; Pang, Z.; Dull, R.O.; Tarbell, J.M. Heparan Sulfate Proteoglycan Is a Mechanosensor on Endothelial Cells. Circ. Res. 2003, 93, e136–e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarbell, J.M.; Ebong, E.E. The Endothelial Glycocalyx: A Mechano-Sensor and -Transducer. Sci. Signal. 2008, 1, pt8. [Google Scholar] [CrossRef]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Popov, D. Endothelial cell dysfunction in hyperglycemia: Phenotypic change, intracellular signaling modification, ultrastructural alteration, and potential clinical outcomes. Int. J. Diabetes Mellit. 2010, 2, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Pearsall, E.A.; Cheng, R.; Matsuzaki, S.; Zhou, K.; Ding, L.; Ahn, B.; Kinter, M.; Humphries, K.M.; Quiambao, A.B.; Farjo, R.A.; et al. Neuroprotective effects of PPARα in retinopathy of type 1 diabetes. PLoS ONE 2019, 14, e0208399. [Google Scholar] [CrossRef] [Green Version]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.S. Role of nitric oxide in diabetic nephropathy. Semin. Nephrol. 2004, 24, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Sato, W.; Sautin, Y.Y.; Glushakova, O.; Croker, B.; Atkinson, M.A.; Tisher, C.C.; Johnson, R.J. Uncoupling of Vascular Endothelial Growth Factor with Nitric Oxide as a Mechanism for Diabetic Vasculopathy. J. Am. Soc. Nephrol. 2006, 17, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaimes, E.A.; Hua, P.; Tian, R.-X.; Raij, L. Human glomerular endothelium: Interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am. J. Physiol. Physiol. 2010, 298, F125–F132. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Zhao, H.J.; Wang, S.; Cheng, H.; Zhang, M.-Z.; Takahashi, T.; Fogo, A.B.; Breyer, M.D.; Harris, R.C. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J. Am. Soc. Nephrol. 2006, 17, 2664–2669. [Google Scholar] [CrossRef]
- Nakagawa, T.; Sato, W.; Glushakova, O.; Heinig, M.; Clarke, T.; Campbell-Thompson, M.; Yuzawa, Y.; Atkinson, M.A.; Johnson, R.J.; Croker, B. Diabetic Endothelial Nitric Oxide Synthase Knockout Mice Develop Advanced Diabetic Nephropathy. J. Am. Soc. Nephrol. 2007, 18, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF Inhibition and Renal Thrombotic Microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef]
- Feliers, D.; Chen, X.; Akis, N.; Ghosh-Choudhury, G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Sivaskandarajah, G.A.; Jeansson, M.; Maezawa, Y.; Eremina, V.; Baelde, H.J.; Quaggin, S.E. Vegfa Protects the Glomerular Microvasculature in Diabetes. Diabetes 2012, 61, 2958–2966. [Google Scholar] [CrossRef] [Green Version]
- De Vriese, A.S.; Tilton, R.G.; Elger, M.; Stephan, C.C.; Kriz, W.; Lameire, N.H. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J. Am. Soc. Nephrol. 2001, 12, 993–1000. [Google Scholar] [PubMed]
- Sung, S.H.; Ziyadeh, F.N.; Wang, A.; Pyagay, P.E.; Kanwar, Y.S.; Chen, S. Blockade of Vascular Endothelial Growth Factor Signaling Ameliorates Diabetic Albuminuria in Mice. J. Am. Soc. Nephrol. 2006, 17, 3093–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veron, D.; Reidy, K.J.; Bertuccio, C.; Teichman, J.; Villegas, G.; Jimenez, J.; Shen, W.; Kopp, J.B.; Thomas, D.B.; Tufro, A. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010, 77, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satchell, S.C.; Harper, S.J.; Tooke, J.; Kerjaschki, N.; Saleem, M.; Mathieson, P.W. Human podocytes express angiopoietin 1, a potential regulator of glomerular vascular endothelial growth factor. J. Am. Soc. Nephrol. 2002, 13, 544–550. [Google Scholar] [PubMed]
- Jeansson, M.; Gawlik, A.; Anderson, G.; Li, C.; Kerjaschki, D.; Henkelman, M.; Quaggin, S.E. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Investig. 2011, 121, 2278–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daehn, I.; Casalena, G.; Zhang, T.; Shi, S.; Fenninger, F.; Barasch, N.; Yu, L.; D’Agati, V.; Schlondorff, D.; Kriz, W.; et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 2014, 124, 1608–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebefors, K.; Wiener, R.J.; Yu, L.; Azeloglu, E.U.; Yi, Z.; Jia, F.; Zhang, W.; Baron, M.H.; He, J.C.; Haraldsson, B.; et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int. 2019, 96, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boels, M.G.S.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; Van Der Vlag, J.; Koster, A.J.; Van Zonneveld, A.J.; Van Faassen, E.; Gröne, H.-J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef] [Green Version]
- Garsen, M.; Lenoir, O.; Rops, A.L.; Dijkman, H.B.; Willemsen, B.; Van Kuppevelt, T.H.; Rabelink, T.J.; Berden, J.H.; Tharaux, P.-L.; Van Der Vlag, J. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J. Am. Soc. Nephrol. 2016, 27, 3545–3551. [Google Scholar] [CrossRef] [Green Version]
- Isermann, B.; Vinnikov, I.A.; Madhusudhan, T.; Herzog, S.; Kashif, M.; Blautzik, J.; Corat, M.A.; Zeier, M.; Blessing, E.; Oh, J.; et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat. Med. 2007, 13, 1349–1358. [Google Scholar] [CrossRef]
- Slater, S.C.; Ramnath, R.D.; Uttridge, K.; Saleem, M.A.; Cahill, P.; Mathieson, P.W.; Welsh, G.I.; Satchell, S.C. Chronic exposure to laminar shear stress induces Kruppel-like factor 2 in glomerular endothelial cells and modulates interactions with co-cultured podocytes. Int. J. Biochem. Cell Biol. 2012, 44, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Chen, H.; Wei, C.; Zhang, W.; Li, Z.; Jain, M.K.; Chuang, P.Y.; Chen, H.; Wang, Y.; Mallipattu, S.K.; et al. Reduced Krüppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int. 2015, 87, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, H.; Chen, A.; Cai, H.; Fu, J.; Salem, F.; Li, Y.; He, J.C.; Schlondorff, D.; Lee, K. Podocyte and endothelial-specific elimination of BAMBI identifies differential transforming growth factor-β pathways contributing to diabetic glomerulopathy. Kidney Int. 2020, 98, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, M.; Najafian, B.; Kim, Y.; Caramori, M.L.; Mauer, M. Podocyte Detachment and Reduced Glomerular Capillary Endothelial Fenestration in Human Type 1 Diabetic Nephropathy. Diabetes 2007, 56, 2155–2160. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-M.; Gao, Y.-B.; Cui, F.-Q.; Zhang, N. Exosomes from high glucose-treated glomerular endothelial cells activate mesangial cells to promote renal fibrosis. Biol. Open 2016, 5, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K.A. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gutterman, D.D. Regulation of Endothelial Function by Mitochondrial Reactive Oxygen Species. Antioxid. Redox Signal. 2011, 15, 1517–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy 2. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Alicigüzel, Y.; Ozen, I.; Aslan, M.; Karayalcin, U. Activities of xanthine oxidoreductase and antioxidant enzymes in different tissues of diabetic rats. J. Lab. Clin. Med. 2003, 142, 172–177. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nat. Cell Biol. 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Kakehi, T.; Yabe-Nishimura, C. NOX enzymes and diabetic complications. Semin. Immunopathol. 2008, 30, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Fórró, L.; Schlegel, W.; Krause, K.-H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free. Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free. Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Cao, Z.; Xu, X.; Meir, E.G.; Lambeth, J. Homologs of gp91 phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Bánfi, B.; Molnár, G.; Maturana, A.D.; Steger, K.; Hegedûs, B.; Demaurex, N.; Krause, K.-H. A Ca2+-activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holterman, C.E.; Thibodeau, J.-F.; Towaij, C.; Gutsol, A.; Montezano, A.C.; Parks, R.J.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R. Nephropathy and Elevated BP in Mice with Podocyte-Specific NADPH Oxidase 5 Expression. J. Am. Soc. Nephrol. 2013, 25, 784–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østergaard, J.A.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Targeting oxidative stress and anti-oxidant defence in diabetic kidney disease. J. Nephrol. 2020, 33, 1–13. [Google Scholar] [CrossRef]
- Block, K.; Eid, A.; Griendling, K.K.; Lee, D.-Y.; Wittrant, Y.; Gorin, Y. Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: Role in mesangial cell hypertrophy and fibronectin expression. J. Biol. Chem. 2008, 283, 24061–24076. [Google Scholar] [CrossRef] [Green Version]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of Podocyte Injury in Diabetes: Role of Cytochrome P450 and NADPH Oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef] [Green Version]
- Rajaram, R.D.; Dissard, R.; Faivre, A.; Ino, F.; Delitsikou, V.; Jaquet, V.; Cagarelli, T.; Lindenmeyer, M.; Jansen-Duerr, P.; Cohen, C.; et al. Tubular NOX4 expression decreases in chronic kidney disease but does not modify fibrosis evolution. Redox Biol. 2019, 26, 101234. [Google Scholar] [CrossRef]
- Schröder, R.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 Is a Protective Reactive Oxygen Species Generating Vascular NADPH Oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.-P.; Chang, S.-Y.; Liao, M.-C.; Lo, C.-S.; Chenier, I.; Luo, H.; Chiasson, J.-L.; Ingelfinger, J.R.; Chan, J.S.D.; Zhang, S.-L. Hedgehog Interacting Protein Promotes Fibrosis and Apoptosis in Glomerular Endothelial Cells in Murine Diabetes. Sci. Rep. 2018, 8, 5958. [Google Scholar] [CrossRef]
- Nagasu, H.; Satoh, M.; Kiyokage, E.; Kidokoro, K.; Toida, K.; Channon, K.M.; Kanwar, Y.S.; Sasaki, T.; Kashihara, N. Activation of endothelial NAD(P)H oxidase accelerates early glomerular injury in diabetic mice. Lab. Investig. 2016, 96, 25–36. [Google Scholar] [CrossRef]
- Breyer, M.D.; Bottinger, E.; Brosius, F.C.; Coffman, T.M.; Harris, R.C.; Heilig, C.W.; Sharma, K. Mouse Models of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2004, 16, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 2019, 62, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Cavaglieri, R.C.; Khazim, K.; Lee, D.-Y.; Bruno, F.; Thakur, S.; Fanti, P.; Szyndralewiez, C.; Barnes, J.L.; Block, K.; et al. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am. J. Physiol. Physiol. 2015, 308, F1276–F1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic Targeting or Pharmacologic Inhibition of NADPH Oxidase Nox4 Provides Renoprotection in Long-Term Diabetic Nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genkyotex Announces Top-Line Results of Phase 2 Clinical Program. Available online: https://www.businesswire.com/news/home/20150909005080/en/Genkyotex-Announces-Top-Line-Results-Phase-2-Clinical (accessed on 12 November 2020).
- Genkyotex Provides New Clinical Data from the PBC Phase 2 Trial Providing Further Evidence of the Anti-Fibrotic Activity of Setanaxib. Available online: https://www.businesswire.com/news/home/20200617005839/en/Genkyotex-provides-new-clinical-data-from-the-PBC-Phase-2-trial-providing-further-evidence-of-the-anti-fibrotic-activity-of-setanaxib (accessed on 12 November 2020).
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef]
- Safety, Tolerability and Renal Effects of APX-115 in Subjects with Type 2 Diabetes and Nephropathy. Available online: https://clinicaltrials.gov/ct2/show/NCT04534439 (accessed on 12 November 2020).
- Nishino, T.; Okamoto, K. Mechanistic insights into xanthine oxidoreductase from development studies of candidate drugs to treat hyperuricemia and gout. JBIC J. Biol. Inorg. Chem. 2015, 20, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Dissanayake, L.V.; Spires, D.R.; Palygin, O.; Staruschenko, A. Effects of uric acid dysregulation on the kidney. Am. J. Physiol. Physiol. 2020, 318, F1252–F1257. [Google Scholar] [CrossRef]
- Washio, K.; Kusunoki, Y.; Murase, T.; Nakamura, T.; Osugi, K.; Ohigashi, M.; Sukenaga, T.; Ochi, F.; Matsuo, T.; Katsuno, T.; et al. Xanthine oxidoreductase activity is correlated with insulin resistance and subclinical inflammation in young humans. Metab. Clin. Exp. 2017, 70, 51–56. [Google Scholar] [CrossRef]
- Miric, D.J.; Kisic, B.; Filipovic-Danic, S.; Grbic, R.; Dragojevic, I.; Miric, M.B.; Puhalo-Sladoje, D. Xanthine Oxidase Activity in Type 2 Diabetes Mellitus Patients with and without Diabetic Peripheral Neuropathy. J. Diabetes Res. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Boban, M.; Kocic, G.; Radenkovic, S.; Pavlovic, R.; Cvetkovic, T.; Deljanin-Ilic, M.; Ilic, S.; Bobana, M.D.; Djindjic, B.; Stojanovic, D.; et al. Circulating purine compounds, uric acid, and xanthine oxidase/dehydrogenase relationship in essential hypertension and end stage renal disease. Ren. Fail. 2014, 36, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, D.; Maahs, D.M.; Hovind, P.; Nakagawa, T. Uric Acid as a Mediator of Diabetic Nephropathy. Semin. Nephrol. 2011, 31, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanbay, M.; Yilmaz, M.I.; Sonmez, A.; Turgut, F.; Sağlam, M.; Cakir, E.; Yenicesu, M.; Covic, A.; Jalal, D.; Johnson, R.J. Serum uric acid level and endothelial dysfunction in patients with nondiabetic chronic kidney disease. Am. J. Nephrol. 2011, 33, 298–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patschan, D.; Patschan, S.; Gobe, G.G.; Chintala, S.; Goligorsky, M.S. Uric Acid Heralds Ischemic Tissue Injury to Mobilize Endothelial Progenitor Cells. J. Am. Soc. Nephrol. 2007, 18, 1516–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Lozada, L.G.; Lanaspa, M.A.; Cristóbal-García, M.; García-Arroyo, F.; Soto, V.; Cruz-Robles, D.; Nakagawa, T.; Yu, M.; Kang, D.-H.; Johnson, R.J. Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron 2013, 121, e71–e78. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.-A.; Sánchez-Lozada, L.G.; Johnson, R.J.; Kang, D.-H. Oxidative stress with an activation of the renin–angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef]
- Zharikov, S.; Krotova, K.; Hu, H.; Baylis, C.; Johnson, R.J.; Block, E.R.; Patel, J. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am. J. Physiol. Physiol. 2008, 295, C1183–C1190. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Bátkai, S.; Mukhopadhyay, B.; Patel, V.; Haskó, G.; Szabó, C.; Mabley, J.G.; Liaudet, L.; Pacher, P. Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J. Cell. Mol. Med. 2009, 13, 2330–2341. [Google Scholar] [CrossRef]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.; Cui, S.; Wang, L.; Fu, B.; Ding, R.; Yang, J.; Chen, X.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 51, 402–410. [Google Scholar] [CrossRef]
- Ficociello, L.H.; Rosolowsky, E.T.; Niewczas, M.A.; Maselli, N.J.; Weinberg, J.; Aschengrau, A.; Eckfeldt, J.H.; Stanton, R.C.; Galecki, A.T.; Doria, A.; et al. High-Normal Serum Uric Acid Increases Risk of Early Progressive Renal Function Loss in Type 1 Diabetes. Diabetes Care 2010, 33, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovind, P.; Rossing, P.; Tarnow, L.; Johnson, R.J.; Parving, H.-H. Serum Uric Acid as a Predictor for Development of Diabetic Nephropathy in Type 1 Diabetes: An Inception Cohort Study. Diabetes 2009, 58, 1668–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doria, A.; Galecki, A.T.; Spino, C.; Pop-Busui, R.; Cherney, D.Z.; Lingvay, I.; Parsa, A.; Rossing, P.; Sigal, R.J.; Afkarian, M.; et al. Serum Urate Lowering with Allopurinol and Kidney Function in Type 1 Diabetes. N. Engl. J. Med. 2020, 382, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; De Zoysa, J.R.; et al. Effects of Allopurinol on the Progression of Chronic Kidney Disease. N. Engl. J. Med. 2020, 382, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Galbusera, C.; Orth, P.; Fedida, D.; Spector, T. Superoxide radical production by allopurinol and xanthine oxidase. Biochem. Pharmacol. 2006, 71, 1747–1752. [Google Scholar] [CrossRef]
- Haberland, A.; Luther, H.; Schimke, I. Does allopurinol prevent superoxide radical production by xanthine oxidase (XOD)? Inflamm. Res. 1991, 32, 96–97. [Google Scholar] [CrossRef]
- Massey, V.; Komai, H.; Palmer, G.; Elion, G.B. On the mechanism of inactivation of xanthine oxidase by allopurinol and other pyrazolo[3,4-d]pyrimidines. J. Biol. Chem. 1970, 245, 2837–2844. [Google Scholar]
- Takano, Y.; Hase-Aoki, K.; Horiuchi, H.; Zhao, L.; Kasahara, Y.; Kondo, S.; Becker, M.A. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005, 76, 1835–1847. [Google Scholar] [CrossRef]
- Lee, H.-J.; Jeong, K.H.; Kim, Y.G.; Moon, J.Y.; Lee, S.H.; Ihm, C.G.; Sung, J.Y.; Lee, T.W. Febuxostat Ameliorates Diabetic Renal Injury in a Streptozotocin-Induced Diabetic Rat Model. Am. J. Nephrol. 2014, 40, 56–63. [Google Scholar] [CrossRef]
- Sircar, D.; Chatterjee, S.; Waikhom, R.; Golay, V.; Raychaudhury, A.; Chatterjee, S.; Pandey, R. Efficacy of Febuxostat for Slowing the GFR Decline in Patients With CKD and Asymptomatic Hyperuricemia: A 6-Month, Double-Blind, Randomized, Placebo-Controlled Trial. Am. J. Kidney Dis. 2015, 66, 945–950. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Hosoya, T.; Uchida, S.; Inaba, M.; Makino, H.; Maruyama, S.; Ito, S.; Yamamoto, T.; Tomino, Y.; Ohno, I.; et al. Febuxostat Therapy for Patients With Stage 3 CKD and Asymptomatic Hyperuricemia: A Randomized Trial. Am. J. Kidney Dis. 2018, 72, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niaudet, P. Mitochondrial disorders and the kidney. Arch. Dis. Child. 1998, 78, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; DiMauro, S.; Hirano, M. A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.T.; Naini, A.; DiMauro, S.; Hirano, M. Leigh Syndrome with Nephropathy and CoQ10 Deficiency Due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef] [Green Version]
- Mã¼Ller-Deile, J.; Schiffer, M.; Müller-Deile, J. The Podocyte Power-Plant Disaster and Its Contribution to Glomerulopathy. Front. Endocrinol. 2014, 5, 209. [Google Scholar] [CrossRef] [Green Version]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Özel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551–1566.e5. [Google Scholar] [CrossRef] [Green Version]
- Culic, O.; Gruwel, M.L.; Schrader, J. Energy turnover of vascular endothelial cells. Am. J. Physiol. Physiol. 1997, 273, C205–C213. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, J.-H.; Gomez, I.G.; Wada, Y.; Roach, A.; Mahad, D.; Duffield, J.S. Deletion of the Mitochondrial Complex-IV Cofactor Heme A:Farnesyltransferase Causes Focal Segmental Glomerulosclerosis and Interferon Response. Am. J. Pathol. 2018, 188, 2745–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency Destabilizes the Coenzyme Q Complex, Which Is Rescued by 2,4-Dihydroxybenzoic Acid Treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Yucel, N.; Kim, B.; Arany, Z. Local Mitochondrial ATP Production Regulates Endothelial Fatty Acid Uptake and Transport. Cell Metab. 2020, 32, 309–319.e7. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Kroemer, G. Life’s smile, death’s grin: Vital functions of apoptosis-executing proteins. Curr. Opin. Cell Biol. 2004, 16, 639–646. [Google Scholar] [CrossRef]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Tahara, E.B.; Navarete, F.D.T.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free. Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef]
- Ricci, J.-E.; Gottlieb, R.A.; Green, D.R. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daehn, I.S. Glomerular Endothelial Cell Stress and Cross-Talk with Podocytes in Early Diabetic Kidney Disease. Front. Med. 2018, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wei, C.; Lee, K.; Zhang, W.; He, W.; Chuang, P.; Liu, J.; He, J.C. Comparison of Glomerular and Podocyte mRNA Profiles in Streptozotocin-Induced Diabetes. J. Am. Soc. Nephrol. 2015, 27, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Ramnath, R.D.; Foster, R.R.; Wylie, E.C.; Fridén, V.; Dasgupta, I.; Haraldsson, B.; Welsh, G.I.; Mathieson, P.W.; Satchell, S.C. Reactive Oxygen Species Modulate the Barrier Function of the Human Glomerular Endothelial Glycocalyx. PLoS ONE 2013, 8, e55852. [Google Scholar] [CrossRef] [Green Version]
- Casalena, G.; Yu, L.; Gil, R.; Rodriguez, S.; Sosa, S.; Janssen, W.G.M.; Azeloglu, E.U.; Leventhal, J.S.; Daehn, I.S. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun. Signal. 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol. Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Luk, A.O.Y.; Tam, C.H.T.; Xie, F.; Carstensen, B.; Lau, E.S.H.; Lim, C.K.P.; Lee, H.M.; Ng, A.C.W.; Ng, M.C.Y.; et al. Progression of diabetic kidney disease and trajectory of kidney function decline in Chinese patients with Type 2 diabetes. Kidney Int. 2019, 95, 178–187. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, Z.; Liu, Q.; Quan, H.; Cheng, X. Effects of coenzyme Q10 intervention on diabetic kidney disease. Medicine 2019, 98, e15850. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.K.; Billings, I.F.T.; Claessens, A.J.; Roshanravan, B.; Linke, L.; Sundell, M.B.; Ahmad, S.; Shao, B.; Shen, D.; Ikizler, T.A.; et al. Coenzyme Q10 dose-escalation study in hemodialysis patients: Safety, tolerability, and effect on oxidative stress. BMC Nephrol. 2015, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.S.; Flemming, N.B.; Gallo, L.A.; Fotheringham, A.K.; McCarthy, D.A.; Zhuang, A.; Tang, P.H.; Borg, D.J.; Shaw, H.; Harvie, B.; et al. Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Baigent, C.; Lennon, R. Should We Increase GFR with Bardoxolone in Alport Syndrome? J. Am. Soc. Nephrol. 2018, 29, 357–359. [Google Scholar] [CrossRef]
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial Redox Signaling: Interaction of Mitochondrial Reactive Oxygen Species with Other Sources of Oxidative Stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Peraçoli, M.T.S.; Bannwart, C.F.; Cristofalo, R.; Borges, V.T.M.; Costa, R.A.A.; Witkin, S.S.; Peraçoli, J.C. Increased Reactive Oxygen Species and Tumor Necrosis Factor-Alpha Production by Monocytes are Associated with Elevated Levels of Uric Acid in Pre-Eclamptic Women. Am. J. Reprod. Immunol. 2011, 66, 460–467. [Google Scholar] [CrossRef]
- D’Ischia, M.; Napolitano, A.; Manini, P.; Panzella, L. Secondary Targets of Nitrite-Derived Reactive Nitrogen Species: Nitrosation/Nitration Pathways, Antioxidant Defense Mechanisms and Toxicological Implications. Chem. Res. Toxicol. 2011, 24, 2071–2092. [Google Scholar] [CrossRef]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Gladden, J.D.; Zelickson, B.R.; Wei, C.-C.; Ulasova, E.; Zheng, J.; Ahmed, M.I.; Chen, Y.; Bamman, M.; Ballinger, S.; Darley-Usmar, V.; et al. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free. Radic. Biol. Med. 2011, 51, 1975–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.; Gerstein, H.C. Effects of Vitamin E on Cardiovascular and Microvascular Outcomes in High-Risk Patients With Diabetes: Results of the HOPE Study and MICRO-HOPE Substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Heerspink, H.J.L.; et al. Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerspink, H.J.L.; Parving, H.-H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.-F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Fernandez-Prado, R.; Górriz, J.L.; Martinez-Castelao, A.; Navarro-González, J.F.; Porrini, E.; Soler, M.J.; Ortiz, A. Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation and Study of Diabetic Nephropathy with Atrasentan: What was learned about the treatment of diabetic kidney disease with canagliflozin and atrasentan? Clin. Kidney J. 2019, 12, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Trachtman, H.; Nelson, P.; Adler, S.G.; Campbell, K.N.; Chaudhuri, A.; Derebail, V.K.; Gambaro, G.; Gesualdo, L.; Gipson, D.S.; Hogan, J.; et al. DUET: A Phase 2 Study Evaluating the Efficacy and Safety of Sparsentan in Patients with FSGS. J. Am. Soc. Nephrol. 2018, 29, 2745–2754. [Google Scholar] [CrossRef] [Green Version]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef] [Green Version]
- Pedigo, C.E.; Ducasa, G.M.; Leclercq, F.; Sloan, A.; Mitrofanova, A.; Hashmi, T.; Molina-David, J.; Ge, M.; Lassenius, M.I.; Forsblom, C.; et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J. Clin. Investig. 2016, 126, 3336–3350. [Google Scholar] [CrossRef] [Green Version]
- Takagi, S.; Li, J.; Takagaki, Y.; Kitada, M.; Nitta, K.; Takasu, T.; Kanasaki, K.; Koya, D. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J. Diabetes Investig. 2018, 9, 1025–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes, Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, T.; Maeno, S.; Maeda, Y.; Yamato, M.; Sonoda, N.; Ogawa, Y.; Wakisaka, M.; Inoguchi, T. Amelioration of diabetic nephropathy by SGLT2 inhibitors independent of its glucose-lowering effect: A possible role of SGLT2 in mesangial cells. Sci. Rep. 2019, 9, 4703. [Google Scholar] [CrossRef]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; Van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef]
- Li, K.; Sun, J.; Huang, N.; Ma, Y.; Han, F.; Liu, Y.; Hou, N.; Sun, X. Liraglutide improves obesity-induced renal injury by alleviating uncoupling of the glomerular VEGF–NO axis in obese mice. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1978–1984. [Google Scholar] [CrossRef]
- Siddiqi, F.S.; Advani, A. Endothelial-Podocyte Crosstalk: The Missing Link between Endothelial Dysfunction and Albuminuria in Diabetes. Diabetes 2013, 62, 3647–3655. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. https://doi.org/10.3390/ijms21249456
Lassén E, Daehn IS. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. International Journal of Molecular Sciences. 2020; 21(24):9456. https://doi.org/10.3390/ijms21249456
Chicago/Turabian StyleLassén, Emelie, and Ilse S. Daehn. 2020. "Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction" International Journal of Molecular Sciences 21, no. 24: 9456. https://doi.org/10.3390/ijms21249456
APA StyleLassén, E., & Daehn, I. S. (2020). Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. International Journal of Molecular Sciences, 21(24), 9456. https://doi.org/10.3390/ijms21249456