REDD1 Is Involved in Amyloid ?-Induced Synaptic Dysfunction and Memory Impairment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
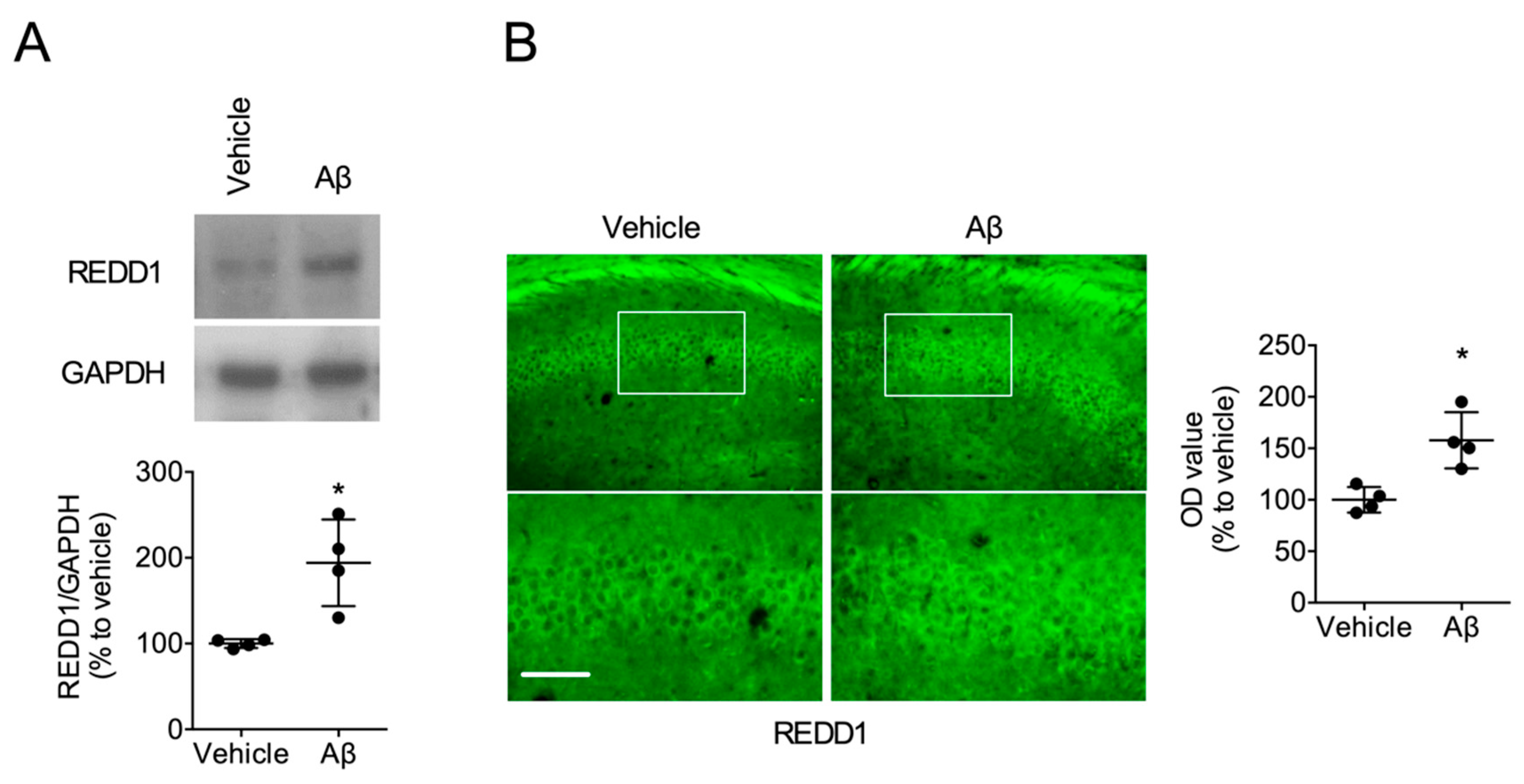
2.1. Aβ Increased REDD1 Levels in the Hippocampus
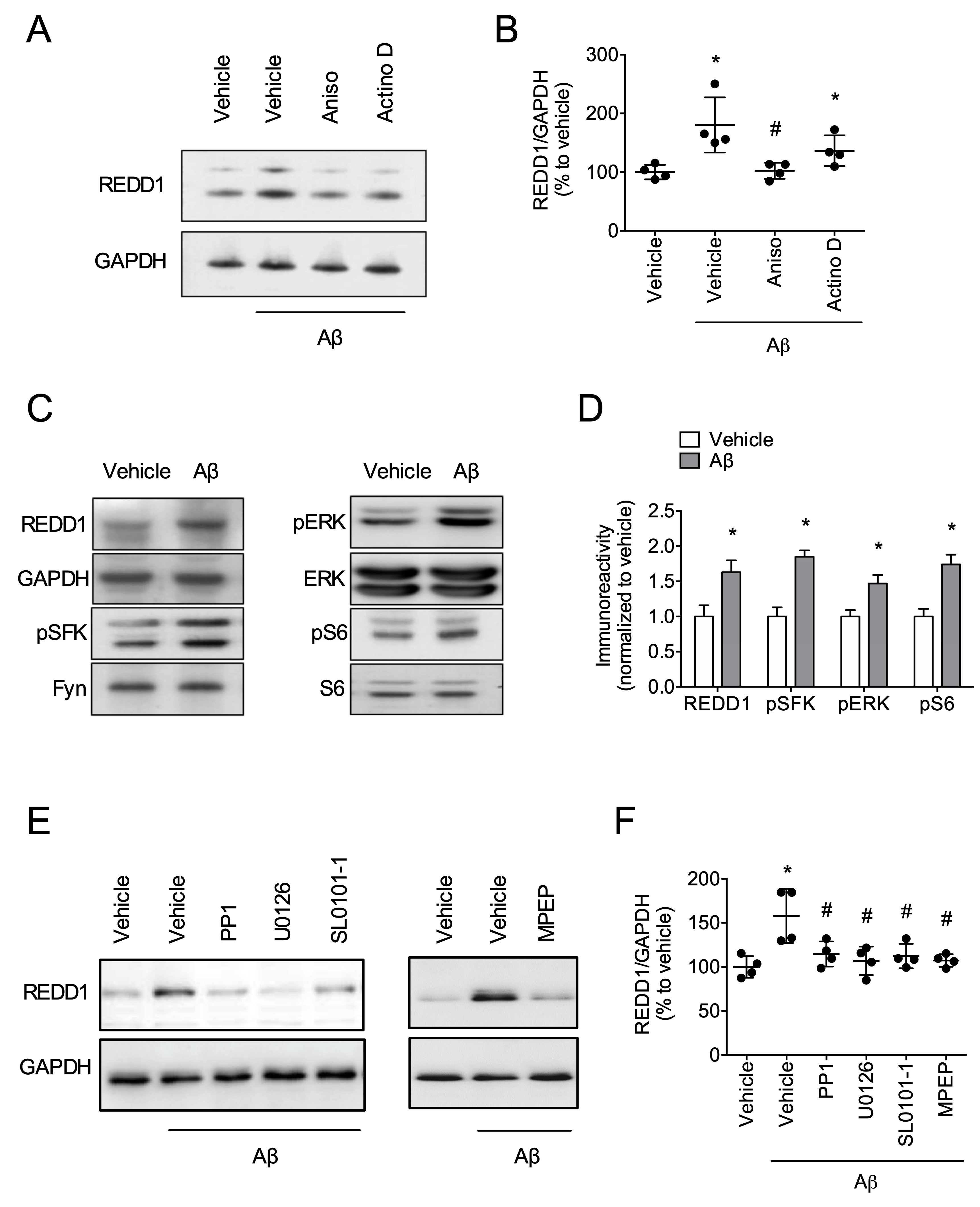
2.2. Fyn/ERK/S6 Signaling Is Involved in Aβ-Induced REDD1 Translation
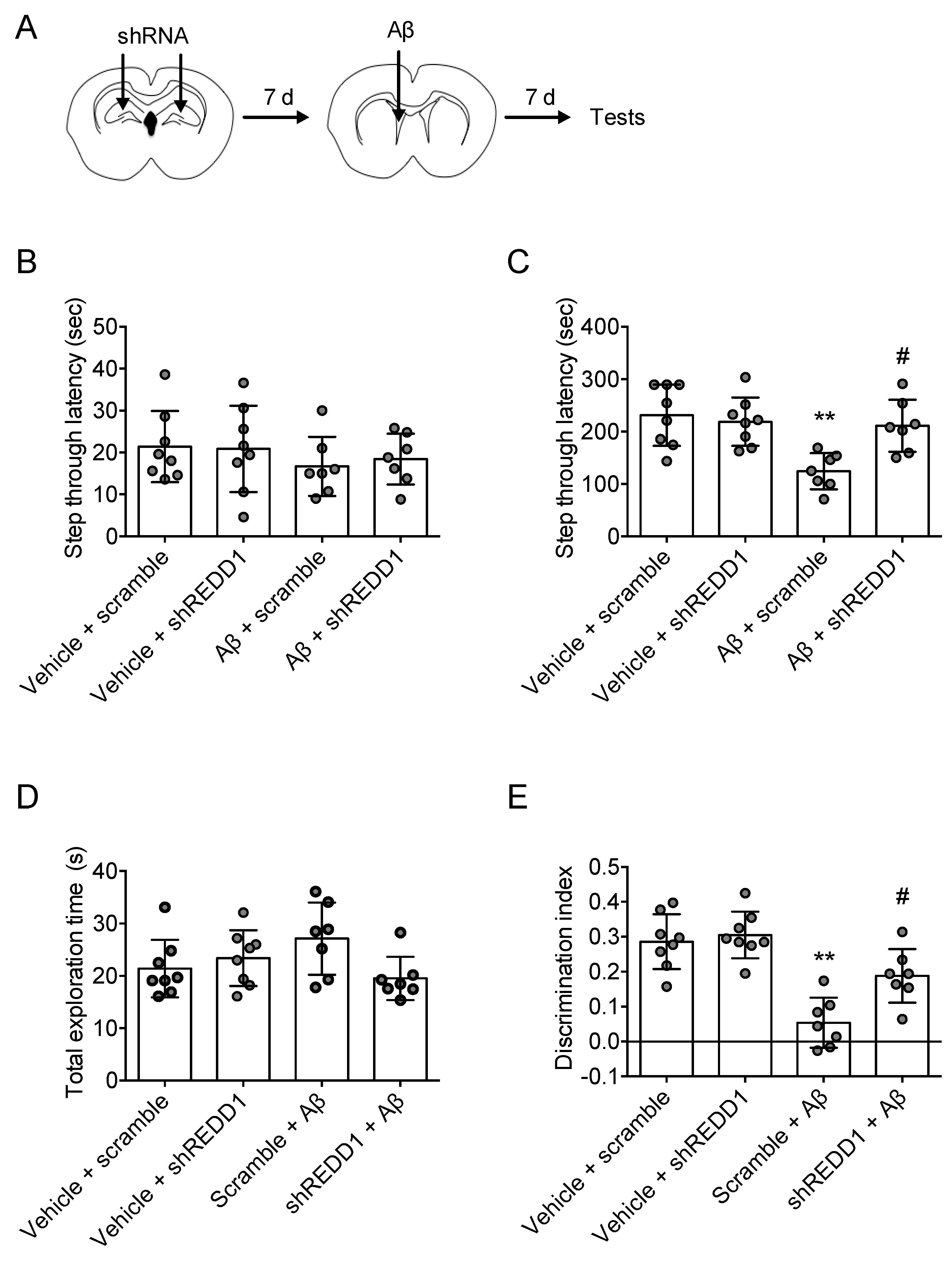
2.3. REDD1 Is Required for Aβ-Induced Synaptic Dysfunction
2.4. REDD1 Knockdown Rescued Aβ-Induced Memory Impairments
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. Aβ1–42 Preperation and Injection
4.4. REDD1 shRNA Injection
4.5. Immunohistochemistry for REDD1
4.6. Acute-Hippocampal-Slice Preparation
4.7. Western Blot
4.8. Electrophysiology
4.9. Passive-Avoidance Test
4.10. Object-Recognition Test
4.11. Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid β |
| REDD1 | Regulated in development and DNA damage response 1 |
| TSC | Tuberous sclerosis complex |
| ERK | Extracellular signal regulated kinase |
| SFK | Src family kinase |
| PBS | Phosphate-buffered saline |
| ACSF | Artificial cerebrospinal fluid |
| TTBS | Tween20/Tris-buffered saline |
| fEPSP | Field excitatory postsynaptic potential |
| HFS | High-frequency stimulation |
| LTP | Long-term potentiation |
References
- Weissberger, G.H.; Strong, J.V.; Stefanidis, K.B.; Summers, M.J.; Bondi, M.W.; Stricker, N.H. Diagnostic Accuracy of Memory Measures in Alzheimer’s Dementia and Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. Neuropsychol. Rev. 2017, 27, 354–388. [Google Scholar] [CrossRef] [PubMed]
- Celone, K.A.; Calhoun, V.D.; Dickerson, B.C.; Atri, A.; Chua, E.F.; Miller, S.L.; DePeau, K.; Rentz, D.M.; Selkoe, D.J.; Blacker, D.; et al. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: An independent component analysis. J. Neurosci. 2006, 26, 10222–10231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, D.; De Deyn, P.P. Animal models in the drug discovery pipeline for Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef] [Green Version]
- McGhee, N.K.; Jefferson, L.S.; Kimball, S.R. Elevated corticosterone associated with food deprivation upregulates expression in rat skeletal muscle of the mTORC1 repressor, REDD1. J. Nutr. 2009, 139, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Vadysirisack, D.D.; Baenke, F.; Ory, B.; Lei, K.; Ellisen, L.W. Feedback control of p53 translation by REDD1 and mTORC1 limits the p53-dependent DNA damage response. Mol. Cell. Biol. 2011, 31, 4356–4365. [Google Scholar] [CrossRef] [Green Version]
- Molitoris, J.K.; McColl, K.S.; Swerdlow, S.; Matsuyama, M.; Lam, M.; Finkel, T.H.; Matsuyama, S.; Distelhorst, C.W. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 2011, 286, 30181–30189. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.M.; Kubica, N.; Ellisen, L.W.; Jefferson, L.S.; Kimball, S.R. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J. Biol. Chem. 2006, 281, 39128–39134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.O.; Seo, S.K.; Woo, S.H.; Kim, Y.S.; Hong, S.E.; Yi, J.Y.; Noh, W.C.; Kim, E.K.; Lee, J.K.; Hong, S.I.; et al. Redd1 inhibits the invasiveness of non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2011, 407, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworski, J.; Sheng, M. The growing role of mTOR in neuronal development and plasticity. Mol. Neurobiol. 2006, 34, 205–219. [Google Scholar] [CrossRef]
- Malagelada, C.; Ryu, E.J.; Biswas, S.C.; Jackson-Lewis, V.; Greene, L.A. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci. 2006, 26, 9996–10005. [Google Scholar] [CrossRef] [Green Version]
- Malagelada, C.; Jin, Z.H.; Greene, L.A. RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 2008, 28, 14363–14371. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chu, S.F.; Wang, S.S.; Jiang, Y.N.; Gao, Y.; Yang, P.F.; Ai, Q.D.; Chen, N.H. RTP801 is a critical factor in the neurodegeneration process of A53T alpha-synuclein in a mouse model of Parkinson’s disease under chronic restraint stress. Br. J. Pharmacol. 2018, 175, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.R.; Lee, S.R.; Chung, H.J.; Kim, S.; Baek, S.H.; Kim, J.H.; Kim, Y.S. Identification of amyloid beta-peptide responsive genes by cDNA microarray technology: Involvement of RTP801 in amyloid beta-peptide toxicity. Exp. Mol. Med. 2003, 35, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Ota, K.T.; Liu, R.J.; Voleti, B.; Maldonado-Aviles, J.G.; Duric, V.; Iwata, M.; Dutheil, S.; Duman, C.; Boikess, S.; Lewis, D.A.; et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat. Med. 2014, 20, 531–535. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Gotz, J. Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J. 2017, 36, 3120–3138. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.A.; Satterly, N.; Versteeg, G.A.; Frantz, D.; Wei, S.G.; Williams, N.; Schmolke, M.; Pena-Llopis, S.; Brugarolas, J.; Forst, C.V.; et al. Chemical inhibition of RNA viruses reveals REDD1 as a host defense factor. Nat. Chem. Biol. 2011, 7, 712–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skendros, P.; Chrysanthopoulou, A.; Rousset, F.; Kambas, K.; Arampatzioglou, A.; Mitsios, A.; Bocly, V.; Konstantinidis, T.; Pellet, P.; Angelidou, I.; et al. Regulated in development and DNA damage responses 1 (REDD1) links stress with IL-1 beta-mediated familial Mediterranean fever attack through autophagy-driven neutrophil extracellular traps. J. Allergy Clin. Immunol. 2017, 140, 1378–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.H.; Baek, S.J.; Heo, S.; Park, H.J.; Kwon, H.; Lee, S.; Jung, J.; Park, S.J.; Kim, B.C.; Lee, Y.C.; et al. Direct pharmacological Akt activation rescues Alzheimer’s disease like memory impairments and aberrant synaptic plasticity. Neuropharmacology 2018, 128, 282–292. [Google Scholar] [CrossRef]
- Park, J.A.; Lee, C.H. Time-Course Change of Redd1 Expressions in the Hippocampal CA1 Region Following Chronic Cerebral Hypoperfusion. Cell. Mol. Neurobiol. 2017, 37, 563–569. [Google Scholar] [CrossRef]
- Pastor, F.; Dumas, K.; Barthelemy, M.A.; Regazzetti, C.; Druelle, N.; Peraldi, P.; Cormont, M.; Tanti, J.F.; Giorgetti-Peraldi, S. Implication of REDD1 in the activation of inflammatory pathways. Sci. Rep. 2017, 7, 7023. [Google Scholar] [CrossRef]
- Kabir, Z.D.; Lee, A.S.; Burgdorf, C.E.; Fischer, D.K.; Rajadhyaksha, A.M.; Mok, E.; Rizzo, B.; Rice, R.C.; Singh, K.; Ota, K.T.; et al. Cacna1c in the Prefrontal Cortex Regulates Depression-Related Behaviors via REDD1. Neuropsychopharmacology 2017, 42, 2032–2042. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Dai, W.; Kutzler, L.; Lacko, H.A.; Jefferson, L.S.; Dennis, M.D.; Kimball, S.R. ATF4-Mediated Upregulation of REDD1 and Sestrin2 Suppresses mTORC1 Activity during Prolonged Leucine Deprivation. J. Nutr. 2020, 150, 1022–1030. [Google Scholar] [CrossRef]
- Hugon, J.; Mouton-Liger, F.; Dumurgier, J.; Paquet, C. PKR involvement in Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 83. [Google Scholar] [CrossRef]
- Paquet, C.; Mouton-Liger, F.; Meurs, E.F.; Mazot, P.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. The PKR activator PACT is induced by Abeta: Involvement in Alzheimer’s disease. Brain Pathol. 2012, 22, 219–229. [Google Scholar] [CrossRef]
- Dennis, M.D.; Coleman, C.S.; Berg, A.; Jefferson, L.S.; Kimball, S.R. REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci. Signal. 2014, 7, ra68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereyra, M.; Katche, C.; de Landeta, A.B.; Medina, J.H. mTORC1 controls long-term memory retrieval. Sci. Rep. 2018, 8, 8759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artinian, J.; Jordan, A.; Khlaifia, A.; Honore, E.; La Fontaine, A.; Racine, A.S.; Laplante, I.; Lacaille, J.C. Regulation of Hippocampal Memory by mTORC1 in Somatostatin Interneurons. J. Neurosci. 2019, 39, 8439–8456. [Google Scholar] [CrossRef]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Koscielny, A.; Malik, A.R.; Liszewska, E.; Zmorzynska, J.; Tempes, A.; Tarkowski, B.; Jaworski, J. Adaptor Complex 2 Controls Dendrite Morphology via mTOR-Dependent Expression of GluA2. Mol. Neurobiol. 2018, 55, 1590–1606. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Barbaro, M.F.; Baraban, S.C. A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci. Lett. 2006, 401, 35–39. [Google Scholar] [CrossRef]
- Bevilaqua, L.R.M.; Cammarota, M. PERK, mTORC1 and eEF2 interplay during long term potentiation: An Editorial for ‘Genetic removal of eIF2a kinase PERK in mice enables hippocampal L-LTP independent of mTORC1 activity’ on page 133. J. Neurochem. 2018, 146, 119–121. [Google Scholar] [CrossRef]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Caccamo, A.; De Pinto, V.; Messina, A.; Branca, C.; Oddo, S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J. Neurosci. 2014, 34, 7988–7998. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Hoeffer, C.A.; Capetillo-Zarate, E.; Yu, F.; Wong, H.; Lin, M.T.; Tampellini, D.; Klann, E.; Blitzer, R.D.; Gouras, G.K. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e12845. [Google Scholar] [CrossRef] [PubMed]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Huang, W.; Costa-Mattioli, M. Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci. 2014, 37, 17–38. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.T.; Li, J.; Wu, D.; Sudhof, T.C.; Chen, L. Synaptic retinoic acid receptor signaling mediates mTOR-dependent metaplasticity that controls hippocampal learning. Proc. Natl. Acad. Sci. USA 2019, 116, 7113–7122. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Popov, V.N.; Starkov, A.A. Crosstalk between the mTOR and Nrf2/ARE signaling pathways as a target in the improvement of long-term potentiation. Exp. Neurol. 2020, 328, 113285. [Google Scholar] [CrossRef]
- Grier, M.D.; West, K.L.; Kelm, N.D.; Fu, C.; Does, M.D.; Parker, B.; McBrier, E.; Lagrange, A.H.; Ess, K.C.; Carson, R.P. Loss of mTORC2 signaling in oligodendrocyte precursor cells delays myelination. PLoS ONE 2017, 12, e0188417. [Google Scholar] [CrossRef] [Green Version]
- McCabe, M.P.; Cullen, E.R.; Barrows, C.M.; Shore, A.N.; Tooke, K.I.; Laprade, K.A.; Stafford, J.M.; Weston, M.C. Genetic inactivation of mTORC1 or mTORC2 in neurons reveals distinct functions in glutamatergic synaptic transmission. Elife 2020, 9, e51440. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Park, H.J.; Kwon, H.; Lee, J.H.; Cho, E.; Lee, Y.C.; Moon, M.; Jun, M.; Kim, D.H.; Jung, J.W. beta-Amyrin Ameliorates Alzheimer’s Disease-Like Aberrant Synaptic Plasticity in the Mouse Hippocampus. Biomol. Ther. (Seoul) 2020, 28, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Franklin, K.; Paxinos, G. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, Compact; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, J.H.; Kwon, H.; Cho, E.; Jeon, J.; Lee, J.; Lee, Y.C.; Cho, J.H.; Jun, M.; Moon, M.; Ryu, J.H.; et al. REDD1 Is Involved in Amyloid ?-Induced Synaptic Dysfunction and Memory Impairment. Int. J. Mol. Sci. 2020, 21, 9482. https://doi.org/10.3390/ijms21249482
Yi JH, Kwon H, Cho E, Jeon J, Lee J, Lee YC, Cho JH, Jun M, Moon M, Ryu JH, et al. REDD1 Is Involved in Amyloid ?-Induced Synaptic Dysfunction and Memory Impairment. International Journal of Molecular Sciences. 2020; 21(24):9482. https://doi.org/10.3390/ijms21249482
Chicago/Turabian StyleYi, Jee Hyun, Huiyoung Kwon, Eunbi Cho, Jieun Jeon, Jeongwon Lee, Young Choon Lee, Jong Hyun Cho, Mira Jun, Minho Moon, Jong Hoon Ryu, and et al. 2020. "REDD1 Is Involved in Amyloid ?-Induced Synaptic Dysfunction and Memory Impairment" International Journal of Molecular Sciences 21, no. 24: 9482. https://doi.org/10.3390/ijms21249482
APA StyleYi, J. H., Kwon, H., Cho, E., Jeon, J., Lee, J., Lee, Y. C., Cho, J. H., Jun, M., Moon, M., Ryu, J. H., Kim, J.-S., Choi, J. W., Park, S. J., Lee, S., & Kim, D. H. (2020). REDD1 Is Involved in Amyloid ?-Induced Synaptic Dysfunction and Memory Impairment. International Journal of Molecular Sciences, 21(24), 9482. https://doi.org/10.3390/ijms21249482