Comprehensive Mutational and Phenotypic Characterization of New Metastatic Cutaneous Squamous Cell Carcinoma Cell Lines Reveal Novel Drug Susceptibilities

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Validation of Novel Cell Lines from Patients with Metastatic cSCC
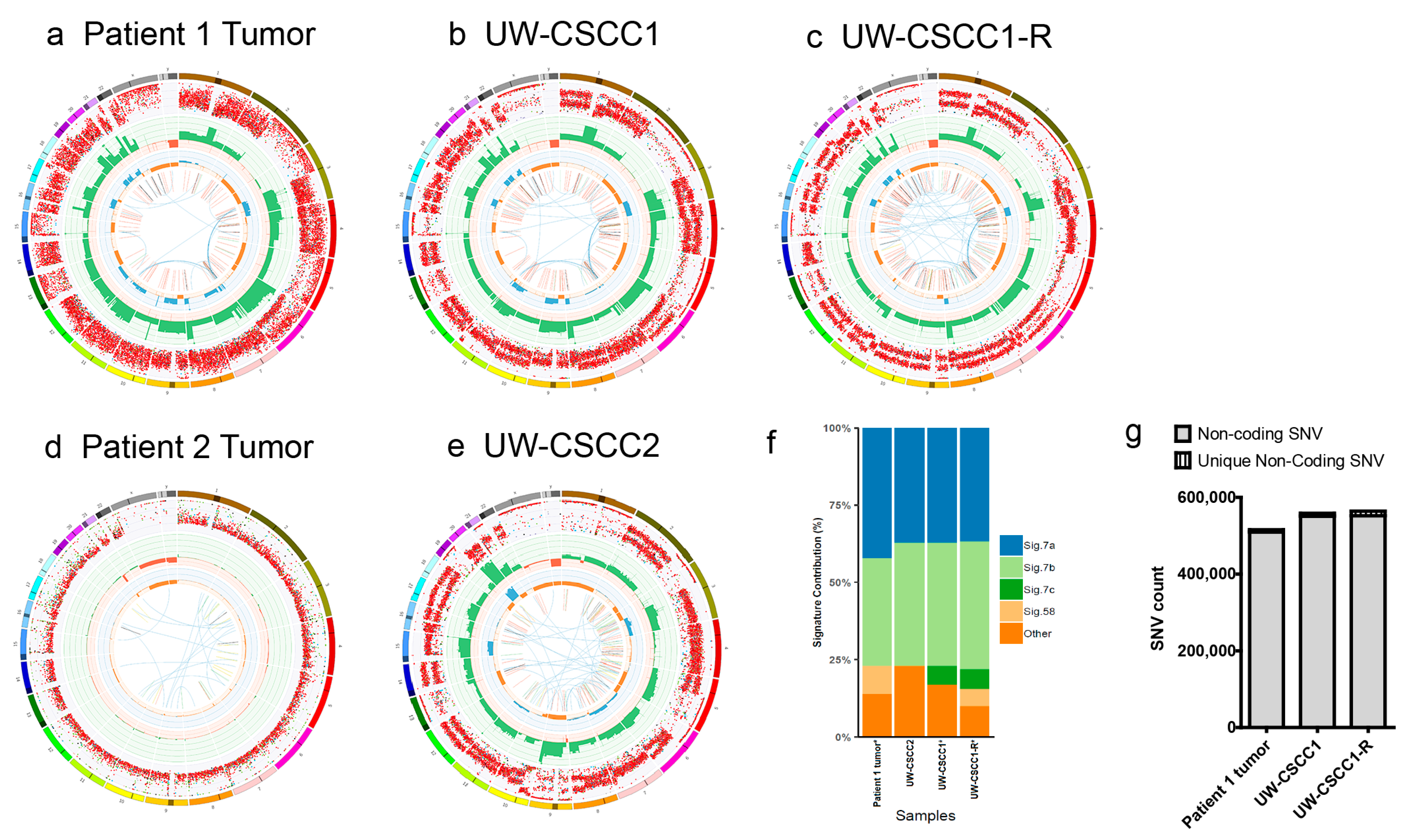
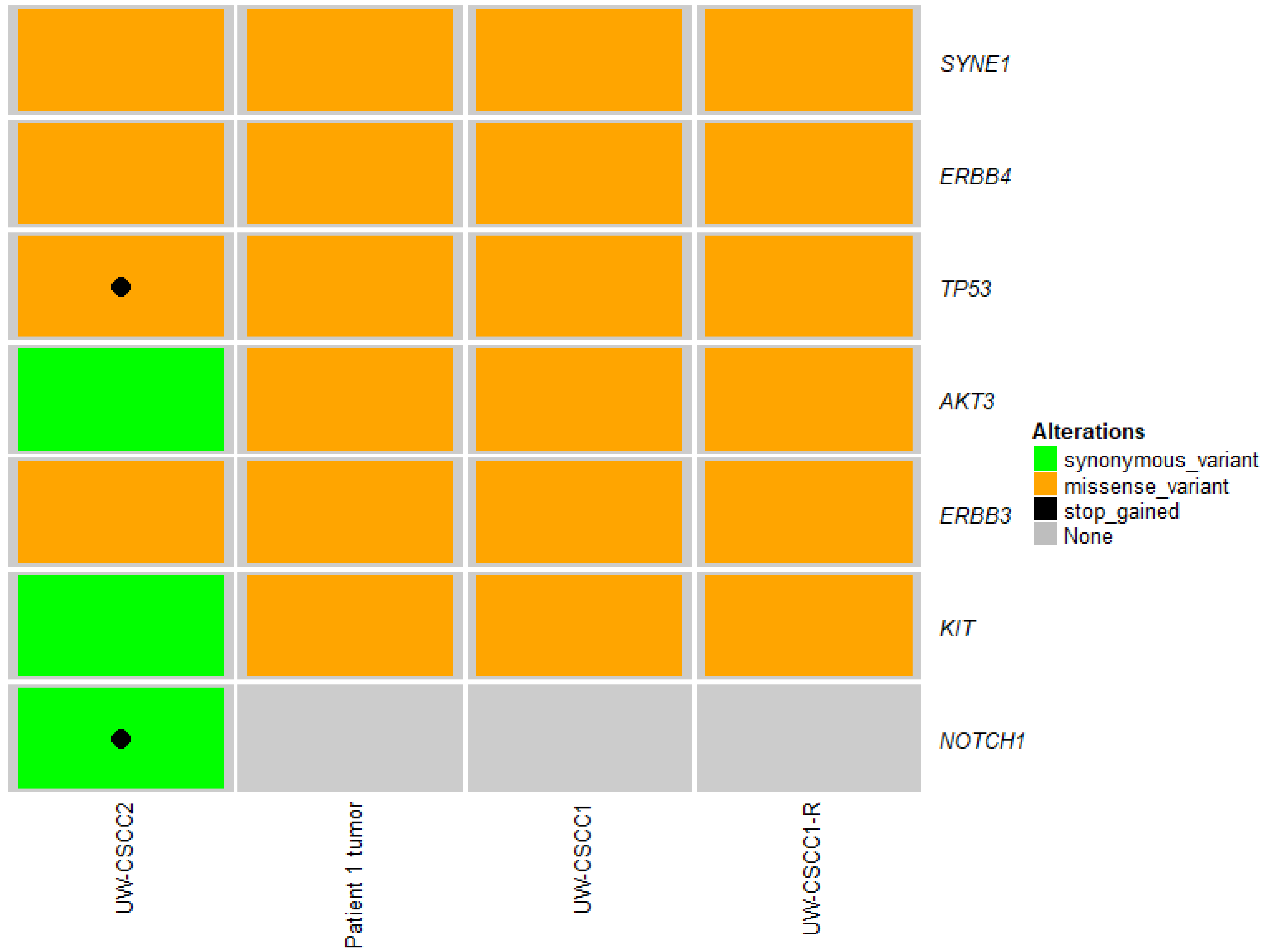
2.2. Genotypic Validation of Novel Cell Lines from Patients with Metastatic cSCC
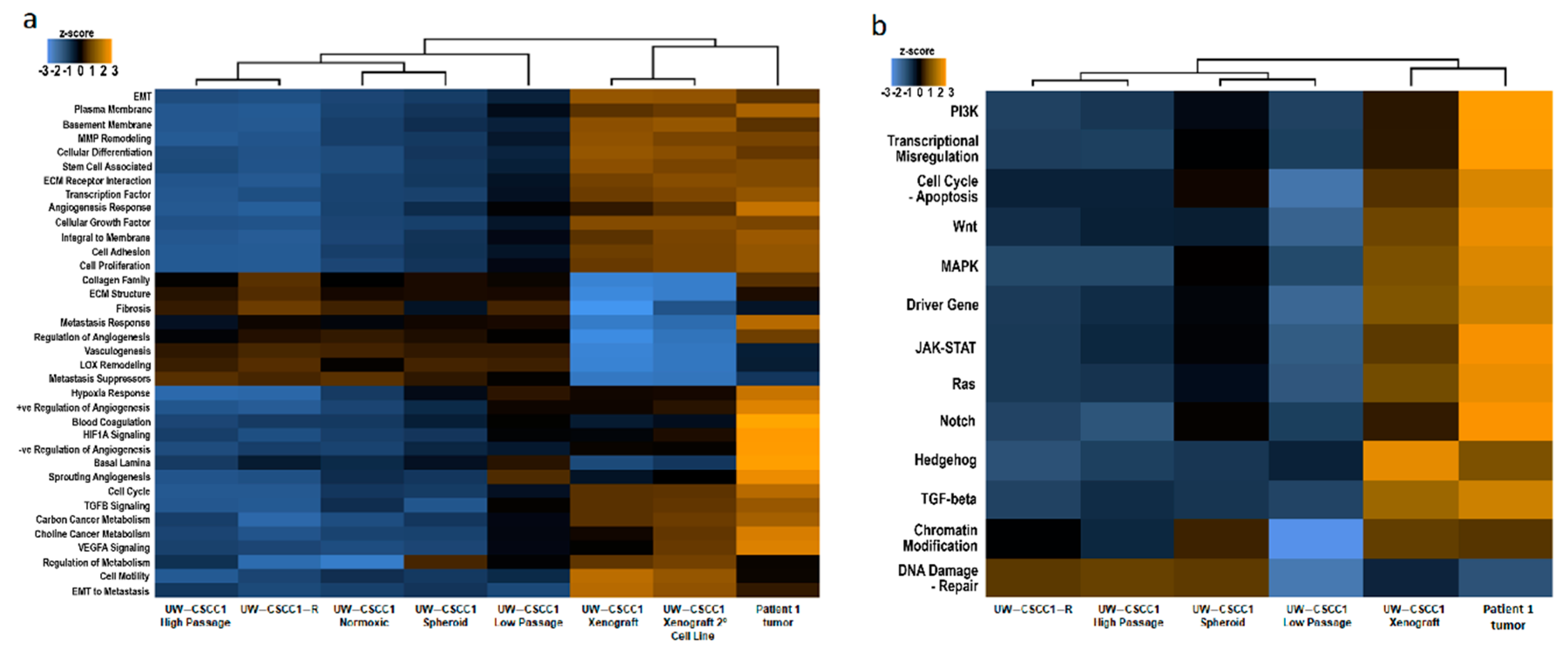
2.3. Effect of In Vitro Culture on Cancer Gene Expression Patterns
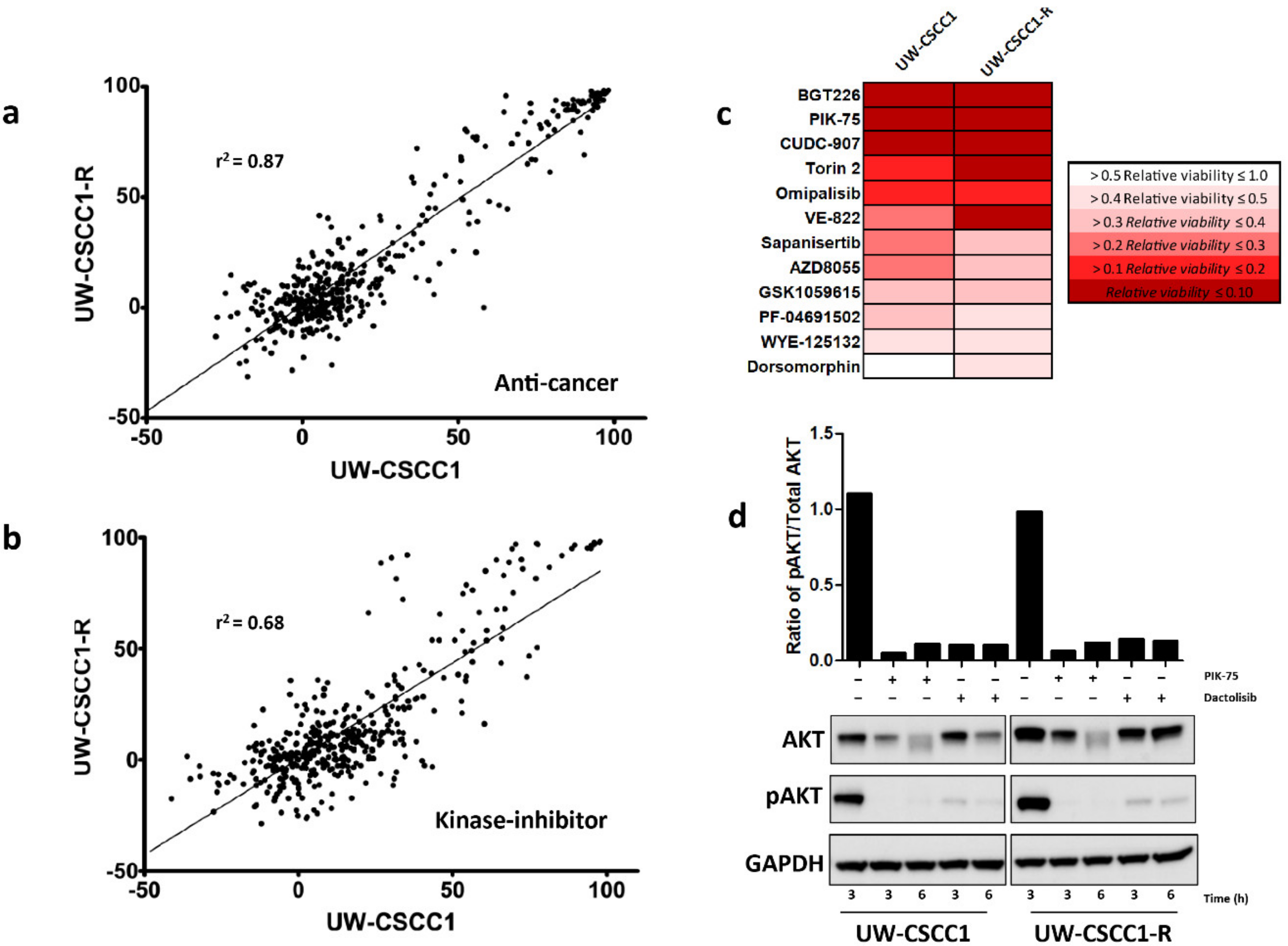
2.4. High-Throughput and Secondary Drug Screens Reveal Novel Therapeutic Targets in Metastatic cSCC Cell Lines
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics and Specimen Selection
4.2. Cell Culture Development and Maintenance
4.3. Generation of Mouse Xenografts
4.4. Organotypic Invasion Assay
4.5. Nucleic Acid Extraction
4.6. Gene Expression Analysis
4.7. Whole-Genome Sequencing (WGS) and Bioinformatics Analysis Pipeline
4.8. Chemical Compound Library Assays and Analysis
4.9. Secondary Dose-Response Screening
4.10. Cell Lysate Preparation and Western Blot Analysis
4.11. Data Repository
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cSCC | Cutaneous squamous cell carcinoma |
| NER | Nuclear excision repair |
| SNV | Small nucleotide variant |
| UV | Ultraviolet radiation |
| WGS | Whole genome sequencing |
References
- Vasconcelos, L.; Melo, J.C.; Miot, H.A.; Marques, M.E.A.; Abbade, L.P.F. Invasive head and neck cutaneous squamous cell carcinoma: Clinical and histopathological characteristics, frequency of local recurrence and metastasis. An. Bras. Dermatol. 2014, 89, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.; Garbe, C.; Lebbe, C.; Malvehy, J.; del Marmol, V.; Pehamberger, H.; Peris, K.; Becker, J.C.; Zalaudek, I.; Saiag, P.; et al. Diagnosis and treatment of invasive squamous cell carcinoma of the skin: European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 1989–2007. [Google Scholar] [CrossRef] [PubMed]
- Brougham, N.D.; Dennett, E.R.; Cameron, R.; Tan, S.T. The incidence of metastasis from cutaneous squamous cell carcinoma and the impact of its risk factors. J. Surg. Oncol. 2012, 106, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Makki, F.M.; Mendez, A.I.; Taylor, S.M.; Trites, J.; Bullock, M.; Flowerdew, G.; Hart, R.D. Prognostic factors for metastatic cutaneous squamous cell carcinoma of the parotid. J. Otolaryngol. Head Neck Surg. 2013, 42, 14. [Google Scholar] [CrossRef] [Green Version]
- Burton, K.A.; Ashack, K.A.; Khachemoune, A. Cutaneous Squamous Cell Carcinoma: A Review of High-Risk and Metastatic Disease. Am. J. Clin. Dermatol. 2016, 17, 491–508. [Google Scholar] [CrossRef]
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.G.; Ashton, R.E. Low incidence of metastasis and recurrence from cutaneous squamous cell carcinoma found in a UK population: Do we need to adjust our thinking on this rare but potentially fatal event? J. Surg. Oncol. 2017, 116, 783–788. [Google Scholar] [CrossRef]
- Venables, Z.; Autier, P.; Nijsten, T.; Wong, K.F.; Langan, S.M.; Rous, B.; Broggio, J.; Harwood, C.; Henson, K.; Proby, C.M.; et al. Nationwide Incidence of Metastatic Cutaneous Squamous Cell Carcinoma in England. JAMA Dermatol. 2018, 155, 298–306. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Rischin, D.; Migden, M.R.; Lim, A.M.; Schmults, C.D.; Khushalani, N.I.; Hughes, B.G.M.; Schadendorf, D.; Dunn, L.A.; Hernandez-Aya, L.; Chang, A.L.S.; et al. Phase 2 study of cemiplimab in patients with metastatic cutaneous squamous cell carcinoma: Primary analysis of fixed-dosing, long-term outcome of weight-based dosing. J. Immunother. Cancer 2020, 8, e000775. [Google Scholar] [CrossRef]
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.A.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic Analysis of Metastatic Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilberg, C.; Lee, M.W.; Yu, B.; Ashford, B.; Kraitsek, S.; Ranson, M.; Shannon, K.; Cowley, M.; Iyer, N.G.; Palme, C.E.; et al. Analysis of clinically relevant somatic mutations in high-risk head and neck cutaneous squamous cell carcinoma. Mod. Pathol. 2017, 31, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational Landscape of Aggressive Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [Green Version]
- Badlani, J.; Gupta, R.; Smith, J.; Ashford, B.; Ch’ng, S.; Veness, M.; Clark, J. Metastases to the parotid gland —A review of the clinicopathological evolution, molecular mechanisms and management. Surg. Oncol. 2018, 27, 44–53. [Google Scholar] [CrossRef]
- Mitsui, H.; Suárez-Fariñas, M.; Gulati, N.; Shah, K.R.; Cannizzaro, M.V.; Coats, I.; Felsen, D.; Krueger, J.G.; Carucci, J.A. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J. Investig. Dermatol. 2014, 134, 1418–1427. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.A.; Gauthier, M.-E.A.; Ashford, B.; Gupta, R.; Gayevskiy, V.; Ch’ng, S.; Palme, C.E.; Shannon, K.; Clark, J.R.; Ranson, M.; et al. Mutational Patterns in Metastatic Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Lobl, M.B.; Clarey, D.; Higgins, S.; Sutton, A.; Hansen, L.; Wysong, A. Targeted next-generation sequencing of matched localized and metastatic primary high-risk SCCs identifies driver and co-occurring mutations and novel therapeutic targets. J. Dermatol. Sci. 2020, 99, 30–43. [Google Scholar] [CrossRef]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, J.N. Cell lines battle cancer. Nature 2012, 483, 544. [Google Scholar] [CrossRef] [PubMed]
- Wilding, J.L.; Bodmer, W.F. Cancer Cell Lines for Drug Discovery and Development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Grandis, J.R.; Carey, T.E.; Gollin, S.M.; Whiteside, T.L.; Koch, W.M.; Ferris, R.L.; Lai, S.Y. Head and neck squamous cell carcinoma cell lines: Established models and rationale for selection. Head Neck 2007, 29, 163–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, S.; Aso, K. Establishment of a cell line of human skin squamous cell carcinoma in vitro. Br. J. Dermatol. 1981, 105, 125–132. [Google Scholar] [CrossRef]
- Farshchian, M.; Kivisaari, A.; Ala-aho, R.; Riihilä, P.; Kallajoki, M.; Grénman, R.; Peltonen, J.; Pihlajaniemi, T.; Heljasvaara, R.; Kähäri, V.-M. Serpin Peptidase Inhibitor Clade A Member 1 (SerpinA1) Is a Novel Biomarker for Progression of Cutaneous Squamous Cell Carcinoma. Am. J. Pathol. 2011, 179, 1110–1119. [Google Scholar] [CrossRef]
- Proby, C.M.; Purdie, K.J.; Sexton, C.J.; Purkis, P.; Navsaria, H.A.; Stables, J.N.; Leigh, I.M. Spontaneous keratinocyte cell lines representing early and advanced stages of malignant transformation of the epidermis. Exp. Dermatol. 2000, 9, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Purdie, K.J.; Wang, J.; Harwood, C.A.; Proby, C.M.; Pourreyron, C.; Mladkova, N.; Nagano, A.; Dhayade, S.; Athineos, D.; et al. A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing. Int. J. Mol. Sci. 2019, 20, 3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic Keratinocyte Lines Requiring Anchorage and Fibroblast Support Cultured from Human Squamous Cell Carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar]
- Anderson, A.N.; McClanahan, D.; Jacobs, J.; Jeng, S.; Vigoda, M.; Blucher, A.S.; Zheng, C.; Yoo, Y.J.; Hale, C.; Ouyang, X.; et al. Functional genomic analysis identifies drug targetable pathways in invasive and metastatic cutaneous squamous cell carcinoma. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005439. [Google Scholar] [CrossRef]
- Cañueto, J.; Cardeñoso, E.; García, J.L.; Santos-Briz, Á.; Castellanos-Martín, A.; Fernández-López, E.; Blanco Gómez, A.; Pérez-Losada, J.; Román-Curto, C. Epidermal growth factor receptor expression is associated with poor outcome in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 176, 1279–1287. [Google Scholar] [CrossRef]
- Ashford, B.G.; Clark, J.; Gupta, R.; Iyer, N.G.; Yu, B.; Ranson, M. Reviewing the genetic alterations in high-risk cutaneous squamous cell carcinoma: A search for prognostic markers and therapeutic targets. Head Neck 2017, 39, 1462–1469. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Gao, B.; Minna, J.D. Lung cancer cell lines: Useless artifacts or invaluable tools for medical science? Lung Cancer 2010, 68, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Janus, J.M.; O’Shaughnessy, R.F.L.; Harwood, C.A.; Maffucci, T. Phosphoinositide 3-Kinase-Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas. Cancers 2017, 9, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.-C.N.; Walker, A.L.; Liu, Y.-Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.-P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [Green Version]
- Lukk, M.; Kapushesky, M.; Nikkilä, J.; Parkinson, H.; Goncalves, A.; Huber, W.; Ukkonen, E.; Brazma, A. A global map of human gene expression. Nat. Biotechnol. 2010, 28, 322. [Google Scholar] [CrossRef] [Green Version]
- Ertel, A.; Verghese, A.; Byers, S.W.; Ochs, M.; Tozeren, A. Pathway-specific differences between tumor cell lines and normal and tumor tissue cells. Mol. Cancer 2006, 5, 55. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Capes-Davis, A.; Davis, J.M.; Downward, J.; Freshney, R.I.; Knezevic, I.; Lovell-Badge, R.; Masters, J.R.; Meredith, J.; Stacey, G.N.; et al. Guidelines for the use of cell lines in biomedical research. Br. J. Cancer 2014, 111, 1021–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 2016, 39, 231–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, X.V.; Burgers, P.M. Two modes of FEN1 binding to PCNA regulated by DNA. EMBO J. 2000, 19, 3811–3821. [Google Scholar] [CrossRef]
- Creighton, C.; Kuick, R.; Misek, D.E.; Rickman, D.S.; Brichory, F.M.; Rouillard, J.-M.; Omenn, G.S.; Hanash, S. Profiling of pathway-specific changes in gene expression following growth of human cancer cell lines transplanted into mice. Genome Biol. 2003, 4, R46. [Google Scholar] [CrossRef] [Green Version]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigani, G.; Dorsch, M.; Peacock, C.D.; et al. A Primary Xenograft Model of Small Cell Lung Cancer Reveals Irreversible Changes in Gene Expression Imposed by Culture In-Vitro. Cancer Res. 2009, 69, 3364. [Google Scholar] [CrossRef] [Green Version]
- Melotte, V.; Yi, J.M.; Lentjes, M.H.F.M.; Smits, K.M.; Van Neste, L.; Niessen, H.E.C.; Wouters, K.A.D.; Louwagie, J.; Schuebel, K.E.; Herman, J.G.; et al. Spectrin repeat containing nuclear envelope 1 and forkhead box protein E1 are promising markers for the detection of colorectal cancer in blood. Cancer Prev. Res. 2015, 8, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Ong, M.S.; Deng, S.; Halim, C.E.; Cai, W.; Tan, T.Z.; Huang, R.Y.-J.; Sethi, G.; Hooi, S.C.; Kumar, A.P.; Yap, C.T. Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers 2020, 12, 238. [Google Scholar] [CrossRef] [Green Version]
- Montaudié, H.; Viotti, J.; Combemale, P.; Dutriaux, C.; Dupin, N.; Robert, C.; Mortier, L.; Kaphan, R.; Duval-Modeste, A.-B.; Dalle, S.; et al. Cetuximab is efficient and safe in patients with advanced cutaneous squamous cell carcinoma: A retrospective, multicentre study. Oncotarget 2020, 11, 378–385. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, J.; Zhu, F.; Ma, X.; Han, B.; Wei, X.; Tan, C.; Jiang, Y.; Chen, Y. Analysis of bypass signaling in EGFR pathway and profiling of bypass genes for predicting response to anticancer EGFR tyrosine kinase inhibitors. Mol. Biosyst. 2012, 8, 2645–2656. [Google Scholar] [CrossRef]
- Corchado-Cobos, R.; García-Sancha, N.; González-Sarmiento, R.; Pérez-Losada, J.; Cañueto, J. Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 2956. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.R. Reduction of Contamination of Epithelial Cultures by Fibroblasts. Cold Spring Harb. Protoc. 2008, 2008, pdb.prot4478. [Google Scholar] [CrossRef]
- Timpson, P.; McGhee, E.J.; Erami, Z.; Nobis, M.; Quinn, J.A.; Edward, M.; Anderson, K.I. Organotypic Collagen I Assay: A Malleable Platform to Assess Cell Behaviour in a 3-Dimensional Context. J. Vis. Exp. 2011, 56, e3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Saunders, C.T.; Wong, W.S.W.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef] [Green Version]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole genome analyses of metastatic solid tumors. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favero, F.; Joshi, T.; Marquard, A.M.; Birkbak, N.J.; Krzystanek, M.; Li, Q.; Szallasi, Z.; Eklund, A.C. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 64–70. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2015, 32, 1220–1222. [Google Scholar] [CrossRef]
- Pagel, K.A.; Kim, R.; Moad, K.; Busby, B.; Zheng, L.; Tokheim, C.; Ryan, M.; Karchin, R. Integrated Informatics Analysis of Cancer-Related Variants. JCO Clin. Cancer Inform. 2020, 4, 310–317. [Google Scholar] [CrossRef]
- Gayevskiy, V.; Roscioli, T.; Dinger, M.E.; Cowley, M.J. Seave: A comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics 2018, 35, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Brungs, D.; Minaei, E.; Piper, A.K.; Perry, J.; Splitt, A.; Carolan, M.; Ryan, S.; Wu, X.J.; Corde, S.; Tehei, M.; et al. Establishment of novel long-term cultures from EpCAM positive and negative circulating tumour cells from patients with metastatic gastroesophageal cancer. Sci. Rep. 2020, 10, 539. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Drug Class | UW-CSCC1 | UW-CSCC1-R | UW-CSCC2 |
|---|---|---|---|---|
| IC50 µM | ||||
| Dactosilib | Dual PI3K/mTOR inhibitor | 0.022 ± 0.003 | 0.080 ± 0.023 | 0.028 ± 0.012 |
| PIK-75 | PI3K inhibitor | 0.122 ± 0.012 | 0.204 ± 0.05 | 0.026 ± 0.012 |
| 5-Fluorouracil | Antimetabolite | 4.47 ± 0.4 | 7.56 ± 0.1 | - |
| Carboplatin † | Platinum analogue | 22.40 | 22.90 | 200.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perry, J.; Ashford, B.; Thind, A.S.; Gauthier, M.-E.; Minaei, E.; Major, G.; Iyer, N.G.; Gupta, R.; Clark, J.; Ranson, M. Comprehensive Mutational and Phenotypic Characterization of New Metastatic Cutaneous Squamous Cell Carcinoma Cell Lines Reveal Novel Drug Susceptibilities. Int. J. Mol. Sci. 2020, 21, 9536. https://doi.org/10.3390/ijms21249536
Perry J, Ashford B, Thind AS, Gauthier M-E, Minaei E, Major G, Iyer NG, Gupta R, Clark J, Ranson M. Comprehensive Mutational and Phenotypic Characterization of New Metastatic Cutaneous Squamous Cell Carcinoma Cell Lines Reveal Novel Drug Susceptibilities. International Journal of Molecular Sciences. 2020; 21(24):9536. https://doi.org/10.3390/ijms21249536
Chicago/Turabian StylePerry, Jay, Bruce Ashford, Amarinder Singh Thind, Marie-Emilie Gauthier, Elahe Minaei, Gretel Major, Narayanan Gopalakrishna Iyer, Ruta Gupta, Jonathan Clark, and Marie Ranson. 2020. "Comprehensive Mutational and Phenotypic Characterization of New Metastatic Cutaneous Squamous Cell Carcinoma Cell Lines Reveal Novel Drug Susceptibilities" International Journal of Molecular Sciences 21, no. 24: 9536. https://doi.org/10.3390/ijms21249536