Identification of Novel Potential Genes Involved in Cancer by Integrated Comparative Analyses



Abstract
:1. Introduction
2. Results
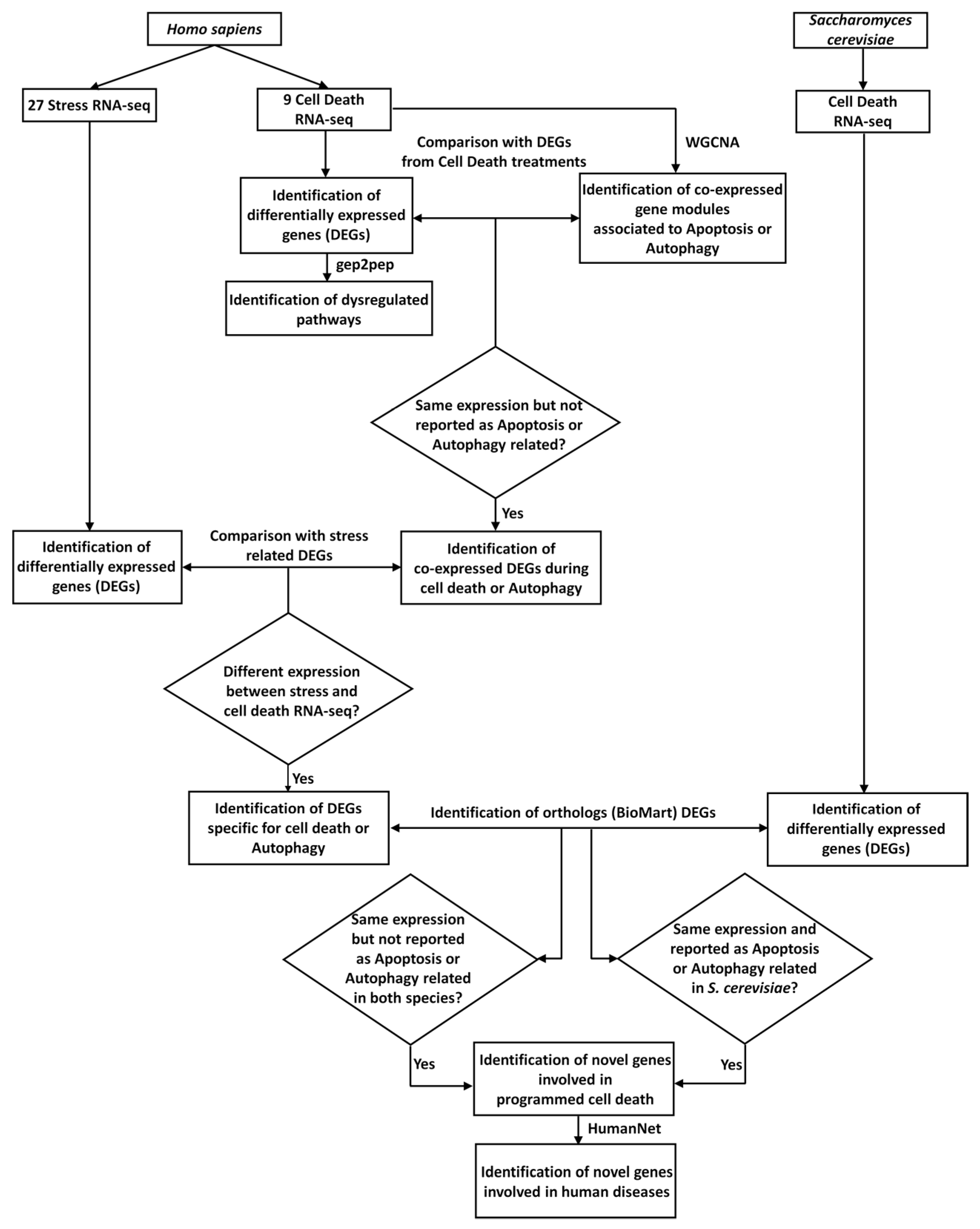
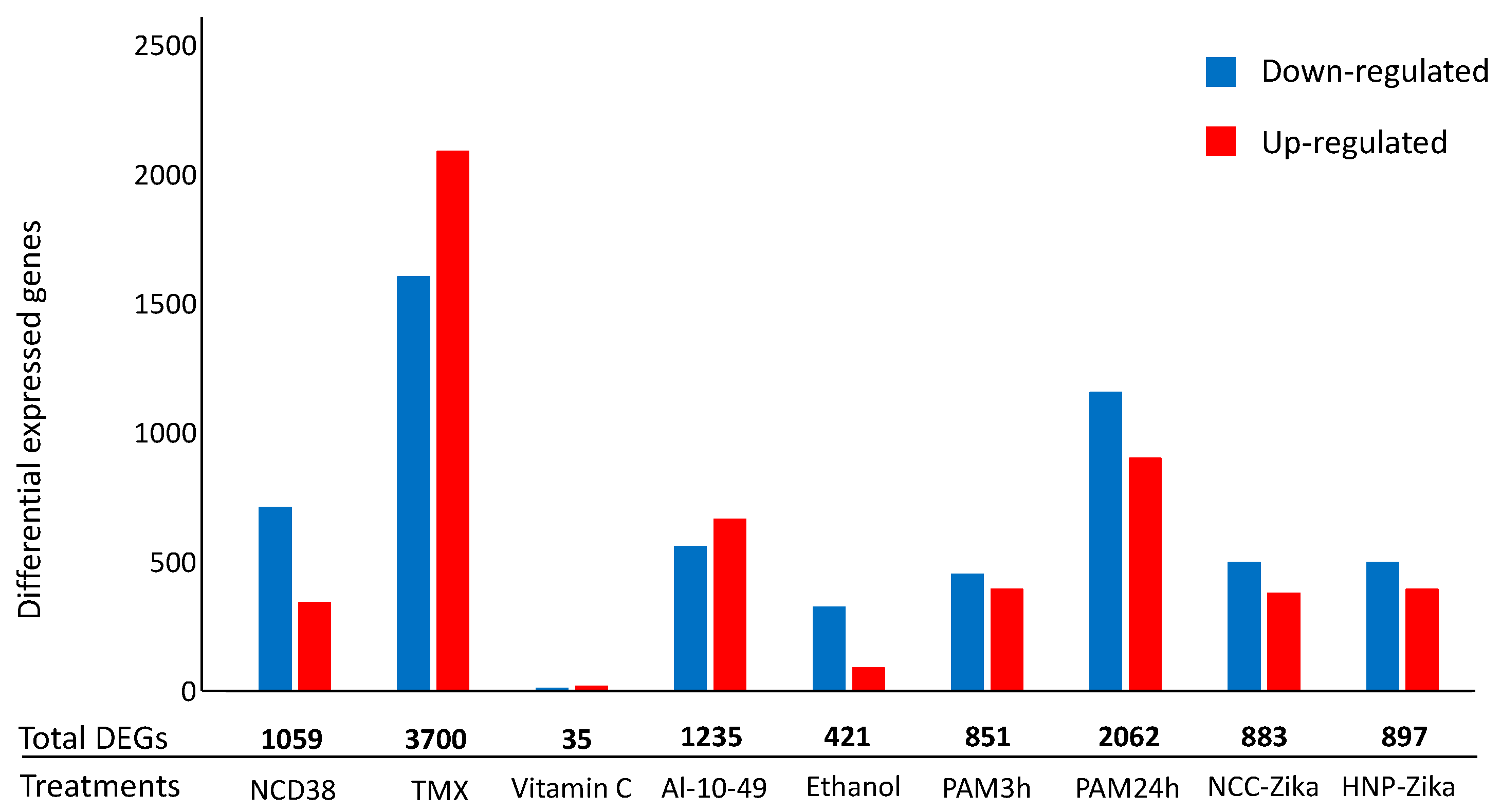
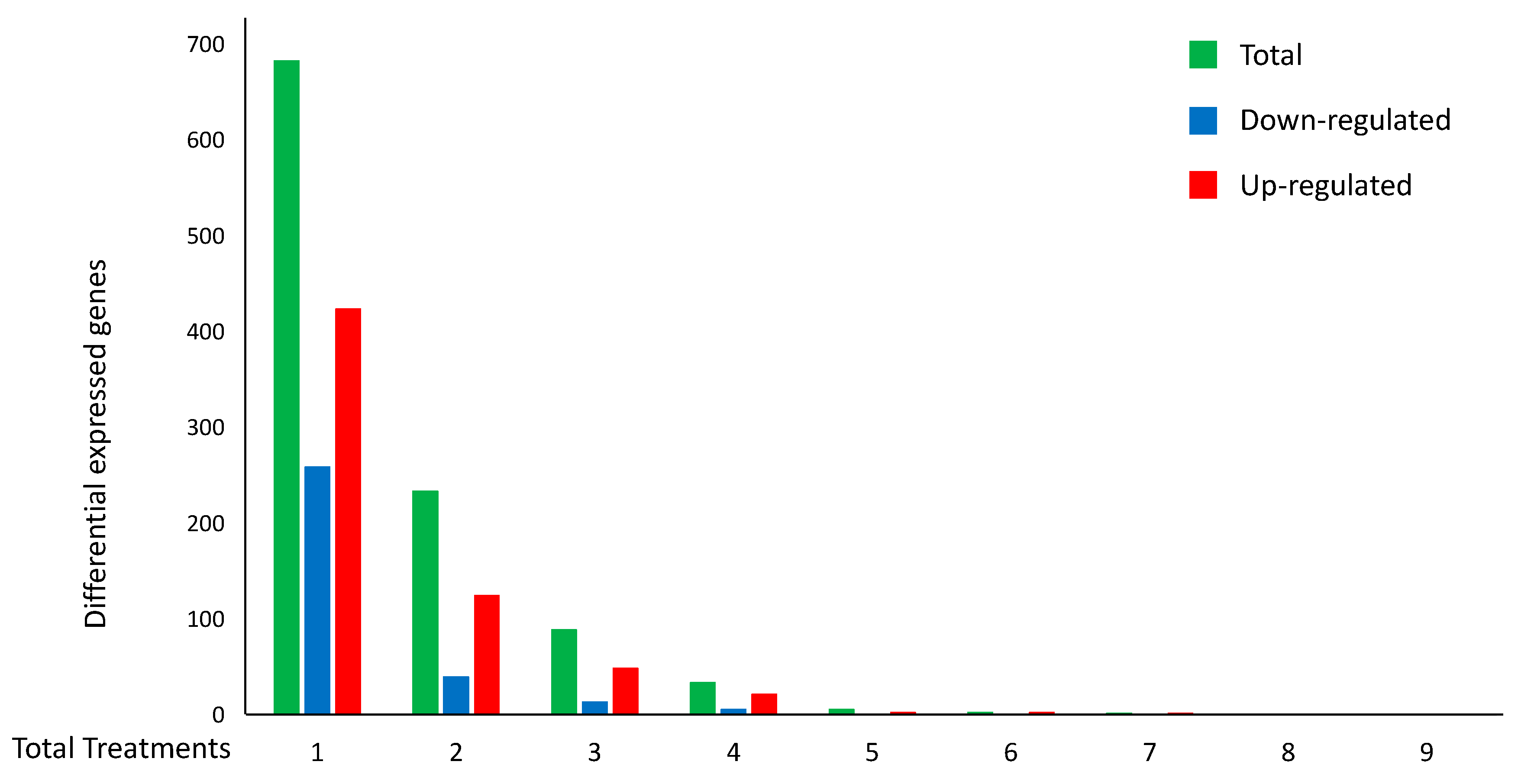
2.1. H. sapiens Cell-Death-Related Differentially-Expressed Genes and Dysregulated Pathways
2.2. H. sapiens Co-Expressed Genes in Cell-Death-Related RNA-Seq Experiments
2.3. Cross-Comparisons between H. sapiens and S. cerevisiae Gene-Expression Profiles in Cell Death and Prediction of Disease-Related Genes.
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibert, B.; Mehlen, P. Dependence receptors and cancer: Addiction to trophic ligands. Cancer Res. 2015, 75, 5171–5175. [Google Scholar] [CrossRef] [Green Version]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial permeability transition: New findings and persisting uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-Linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florey, O.; Kim, S.E.; Overholtzer, M. Entosis: Cell-in-Cell formation that kills through entotic cell death. Curr. Mol. Med. 2015, 15, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.J.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-Regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Azzouz, D.; Palaniyar, N. ApoNETosis: Discovery of a novel form of neutrophil death with concomitant apoptosis and NETosis. Cell Death Dis. 2018, 9, 839. [Google Scholar] [CrossRef] [Green Version]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.A.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-Induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Xie, Y.; Liu, J.; Sun, L.; Zeng, D.; Wang, P.; Ma, X.; Kroemer, G.; Bartlett, D.L.; et al. JTC801 induces ph-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology 2018, 154, 1480–1493. [Google Scholar] [CrossRef]
- Renehan, A.G.; Booth, C.; Potten, C.S. What is apoptosis, and why is it important? BMJ 2001, 322, 1536–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengartner, M.O.; Robert Horvitz, H. Programmed cell death in caenorhabditis elegans. Curr. Opin. Genet. Dev. 1994, 4, 581–586. [Google Scholar] [CrossRef]
- Sulston, J.E.; Horvitz, H.R. Post-Embryonic cell lineages of the nematode, caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; García-Sáez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-Consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-J.; Zhang, X.-D.; Sun, W.; Qi, L.; Wu, J.-C.; Qin, Z.-H. DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis. 2015, 6, e1624. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Deng, G.; Tan, P.; Xing, C.; Guan, C.; Jiang, C.; Zhang, Y.; Ning, B.; Li, C.; Yin, B.; et al. Beclin 2 negatively regulates innate immune signaling and tumor development. J. Clin. Investig. 2020, 130. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kroemer, G. Transient autophagy inhibition precipitates oncogenesis: A red flag for pharmacological autophagy inhibitors? Trends Cell Biol. 2020, 30, 339–340. [Google Scholar] [CrossRef]
- Zimmermann, A.; Kainz, K.; Andryushkova, A.; Hofer, S.; Madeo, F.; Carmona-Gutierrez, D. Autophagy: One more Nobel Prize for yeast. Microb. cell (Graz, Austria) 2016, 3, 579–581. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Aguilera, A.; Austriaco, N.; Ayscough, K.; Balzan, R.; Bar-Nun, S.; Barrientos, A.; Belenky, P.; et al. Guidelines and recommendations on yeast cell death nomenclature. Microb. Cell (Graz, Austria) 2018, 5, 4–31. [Google Scholar] [CrossRef] [Green Version]
- Ludovico, P.; Sousa, M.J.; Silva, M.T.; Leão, C.; Côrte-Real, M. Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology 2001, 147, 2409–2415. [Google Scholar] [CrossRef] [Green Version]
- Falcone, C.; Mazzoni, C. External and internal triggers of cell death in yeast. Cell. Mol. Life Sci. 2016, 73, 2237–2250. [Google Scholar] [CrossRef]
- Madeo, F.; Fröhlich, E.; Ligr, M.; Grey, M.; Sigrist, S.J.; Wolf, D.H.; Fröhlich, K.-U. Oxygen stress: A regulator of apoptosis in yeast. J. Cell Biol. 1999, 145, 757–767. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Cebollero, E.; Reggiori, F. Regulation of autophagy in yeast saccharomyces cerevisiae. Biochim. Biophys. Acta 2009, 1793, 1413–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.; Kim, C.Y.; Yang, S.; Kim, E.; Hart, T.; Marcotte, E.M.; Lee, I. HumanNet v2: Human gene networks for disease research. Nucleic Acids Res. 2019, 47, D573–D580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, F.; Carrella, D.; Gao, X.; di Bernardo, D. Gep2pep: A bioconductor package for the creation and analysis of pathway-based expression profiles. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart-Biological queries made easy. BMC Genom. 2009, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Dong, Y.; Hu, J.; Fan, L.; Chen, Q. RNA-Seq-Based transcriptomic and metabolomic analysis reveal stress responses and programmed cell death induced by acetic acid in saccharomyces cerevisiae. Sci. Rep. 2017, 7, 42659. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease–gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, H.; Cheng, W.; Fu, D.; Xia, T.; Kibbe, W.A.; Lin, S.M. A framework for annotating human genome in disease context. PLoS ONE 2012, 7, e49686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, T.; Büttner, S.; Kroemer, G.; Madeo, F. The mitochondrial pathway in yeast apoptosis. Apoptosis 2007, 12, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nouraini, S.; Reed, J.C. Yeast as a tool for apoptosis research. Curr. Opin. Microbiol. 1999, 2, 618–623. [Google Scholar] [CrossRef]
- Greenwood, M.T.; Ludovico, P. Expressing and functional analysis of mammalian apoptotic regulators in yeast. Cell Death Differ. 2010, 17, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [Green Version]
- Hollander, M.C.; Zhan, Q.; Bae, I.; Fornace, A.J.J. Mammalian GADD34, an apoptosis-and DNA damage-inducible gene. J. Biol. Chem. 1997, 272, 13731–13737. [Google Scholar] [CrossRef] [Green Version]
- Grishin, A.V.; Azhipa, O.; Semenov, I.; Corey, S.J. Interaction between growth arrest-DNA damage protein 34 and Src kinase Lyn negatively regulates genotoxic apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 10172–10177. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, G.; Staiano, L.; Moretti, M.N.; De Cegli, R.; Fagnocchi, L.; Di Tullio, G.; Polletti, S.; Braccia, C.; Armirotti, A.; Zippo, A.; et al. GADD34 is a modulator of autophagy during starvation. Sci. Adv. 2020, 6, eabb0205. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-Stress-Induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Harada, K.; Supriatno; Kawashima, Y.; Itashiki, Y.; Yoshida, H.; Sato, M. Down-Regulation of S-phase kinase associated protein 2 (Skp2) induces apoptosis in oral cancer cells. Oral Oncol. 2005, 41, 623–630. [Google Scholar] [CrossRef]
- Zhu, X.-F.; Li, W.; Ma, J.-Y.; Shao, N.; Zhang, Y.-J.; Liu, R.-M.; Wu, W.-B.; Lin, Y.; Wang, S.-M. Knockdown of heme oxygenase-1 promotes apoptosis and autophagy and enhances the cytotoxicity of doxorubicin in breast cancer cells. Oncol. Lett. 2015, 10, 2974–2980. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Erikson, R.L. Polo-Like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5789–5794. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.S.J.; Ortmann, B.M.; Martinelli, A.W.; Houghton, J.W.; Costa, A.S.H.; Burr, S.P.; Antrobus, R.; Frezza, C.; Nathan, J.A. ABHD11 maintains 2-oxoglutarate metabolism by preserving functional lipoylation of the 2-oxoglutarate dehydrogenase complex. Nat. Commun. 2020, 11, 4046. [Google Scholar] [CrossRef]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef]
- Dell’Agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.C.; Ghalei, H.; Doherty, J.R.; Huang, H.; Culver, R.N.; Karbstein, K. Ribosome biogenesis factor Ltv1 chaperones the assembly of the small subunit head. J. Cell Biol. 2018, 217, 4141–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sant, D.W.; Mustafi, S.; Gustafson, C.B.; Chen, J.; Slingerland, J.M.; Wang, G. Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Sci. Rep. 2018, 8, 5306. [Google Scholar] [CrossRef] [PubMed]
- Rouhimoghadam, M.; Safarian, S.; Carroll, J.S.; Sheibani, N.; Bidkhori, G. Tamoxifen-Induced apoptosis of MCF-7 Cells via GPR30/PI3K/MAPKs interactions: Verification by ODE modeling and RNA sequencing. Front. Physiol. 2018, 9, 907. [Google Scholar] [CrossRef] [Green Version]
- Pulikkan, J.A.; Hegde, M.; Ahmad, H.M.; Belaghzal, H.; Illendula, A.; Yu, J.; O’Hagan, K.; Ou, J.; Muller-Tidow, C.; Wolfe, S.A.; et al. CBFβ-SMMHC inhibition triggers apoptosis by disrupting MYC chromatin dynamics in acute myeloid leukemia. Cell 2018, 174, 1325. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Peña-Martínez, P.; Ramakrishnan, R.; Chapellier, M.; Högberg, C.; Glowacki, G.; Orsmark-Pietras, C.; Velasco-Hernández, T.; Lazarević, V.L.; Juliusson, G.; et al. Agonistic targeting of TLR1/TLR2 induces p38 MAPK-dependent apoptosis and NFκB-dependent differentiation of AML cells. Blood Adv. 2017, 1, 2046–2057. [Google Scholar] [CrossRef] [Green Version]
- Sareddy, G.R.; Viswanadhapalli, S.; Surapaneni, P.; Suzuki, T.; Brenner, A.; Vadlamudi, R.K. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene 2017, 36, 2423–2434. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.; Zhang, F.; Wang, Y.; Lee, E.M.; Choi, I.Y.; Lim, H.; Mirakhori, F.; Li, R.; Huang, L.; Xu, T.; et al. Zika virus directly infects peripheral neurons and induces cell death. Nat. Neurosci. 2017, 20, 1209–1212. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.-L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Rendleman, J.; Cheng, Z.; Maity, S.; Kastelic, N.; Munschauer, M.; Allgoewer, K.; Teo, G.; Zhang, Y.B.M.; Lei, A.; Parker, B.; et al. New insights into the cellular temporal response to proteostatic stress. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Des Marais, T.L.; Kluz, T.; Xu, D.; Zhang, X.; Gesumaria, L.; Matsui, M.S.; Costa, M.; Sun, H. Transcription factors and stress response gene alterations in human keratinocytes following solar simulated ultra violet radiation. Sci. Rep. 2017, 7, 13622. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-Omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Landeras-Bueno, S.; Fernández, Y.; Falcón, A.; Oliveros, J.C.; Ortín, J. Chemical genomics identifies the PERK-mediated unfolded protein stress response as a cellular target for influenza virus inhibition. MBio 2016, 7, e00085-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3. Cell Metab. 2018, 28, 721–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinonen, R.; Sugawara, H.; Shumway, M.; Collaboration, on behalf of the International School of Design (INSD). The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P. anRichment tutorial. 2018. [Google Scholar]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Dysregulated Pathways | ES | PV |
|---|---|---|
| Defense response to other organism | 1 | 0.001 |
| Innate immune response | 1 | 0.001 |
| Cellular response to topologically-incorrect protein | 1 | 0.002 |
| Regulation of response to biotic stimulus | 1 | 0.002 |
| Response to topologically-incorrect protein | 1 | 0.002 |
| FC epsilon receptor-signaling pathway | 1 | 0.003 |
| Natural killer cell-mediated immunity | 1 | 0.003 |
| Golgi organization | 1 | 0.003 |
| Autophagy of mitochondrion | 1 | 0.003 |
| Negative regulation of innate immune response | 1 | 0.004 |
| Protein-k48-linked ubiquitination | 1 | 0.004 |
| Response to cytokine | 0.889 | 0.006 |
| Tumor necrosis-factor-mediated signaling pathway | 0.875 | 0.009 |
| Intrinsic apoptotic signaling pathway | 0.875 | 0.009 |
| Positive regulation of response to biotic stimulus | 0.875 | 0.009 |
| Organelle disassembly | 0.875 | 0.009 |
| Endoplasmic reticulum unfolded-protein response | 0.875 | 0.009 |
| Forebrain development | −0.875 | 0.009 |
| Embryonic morphogenesis | −0.875 | 0.009 |
| Head development | −0.875 | 0.009 |
| Tube formation | −0.875 | 0.009 |
| Urogenital-system development | −0.875 | 0.009 |
| Homophilic-cell adhesion via plasma-membrane-adhesion molecules | −0.875 | 0.009 |
| Reactive-oxygen-species metabolic process | −0.875 | 0.009 |
| Male sex differentiation | −0.875 | 0.009 |
| Development of primary sexual characteristics | −0.875 | 0.009 |
| Regulation of neurotransmitter levels | −0.889 | 0.006 |
| Osteoclast differentiation | −1 | 0.002 |
| Spindle organization | −1 | 0.003 |
| Regulation of membrane-lipid distribution | −1 | 0.008 |
| Phospholipid transport | −1 | 0.008 |
| Occurrence in Cell-Death Treatments | Cell-Death DEGs | Concordant Cell-Death DEGs | Concordant DEGs Specific to Cell Death versus Stress | Orthologs with S. cerevisiae | Confirmed in S. cerevisiae by GOs | Concordant in S. cerevisiae Cell Death |
|---|---|---|---|---|---|---|
| 1 | 683 | 683 | 561 | 155 | 2 | 27 |
| >1 | 368 | 265 | 173 | 61 | 0 | 5 |
| Total | 1051 | 948 | 734 | 216 | 2 | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monticolo, F.; Palomba, E.; Chiusano, M.L. Identification of Novel Potential Genes Involved in Cancer by Integrated Comparative Analyses. Int. J. Mol. Sci. 2020, 21, 9560. https://doi.org/10.3390/ijms21249560
Monticolo F, Palomba E, Chiusano ML. Identification of Novel Potential Genes Involved in Cancer by Integrated Comparative Analyses. International Journal of Molecular Sciences. 2020; 21(24):9560. https://doi.org/10.3390/ijms21249560
Chicago/Turabian StyleMonticolo, Francesco, Emanuela Palomba, and Maria Luisa Chiusano. 2020. "Identification of Novel Potential Genes Involved in Cancer by Integrated Comparative Analyses" International Journal of Molecular Sciences 21, no. 24: 9560. https://doi.org/10.3390/ijms21249560
APA StyleMonticolo, F., Palomba, E., & Chiusano, M. L. (2020). Identification of Novel Potential Genes Involved in Cancer by Integrated Comparative Analyses. International Journal of Molecular Sciences, 21(24), 9560. https://doi.org/10.3390/ijms21249560