Melanoma Biomarkers and Their Potential Application for In Vivo Diagnostic Imaging Modalities

 , ,
, ,
Abstract
:1. Introduction
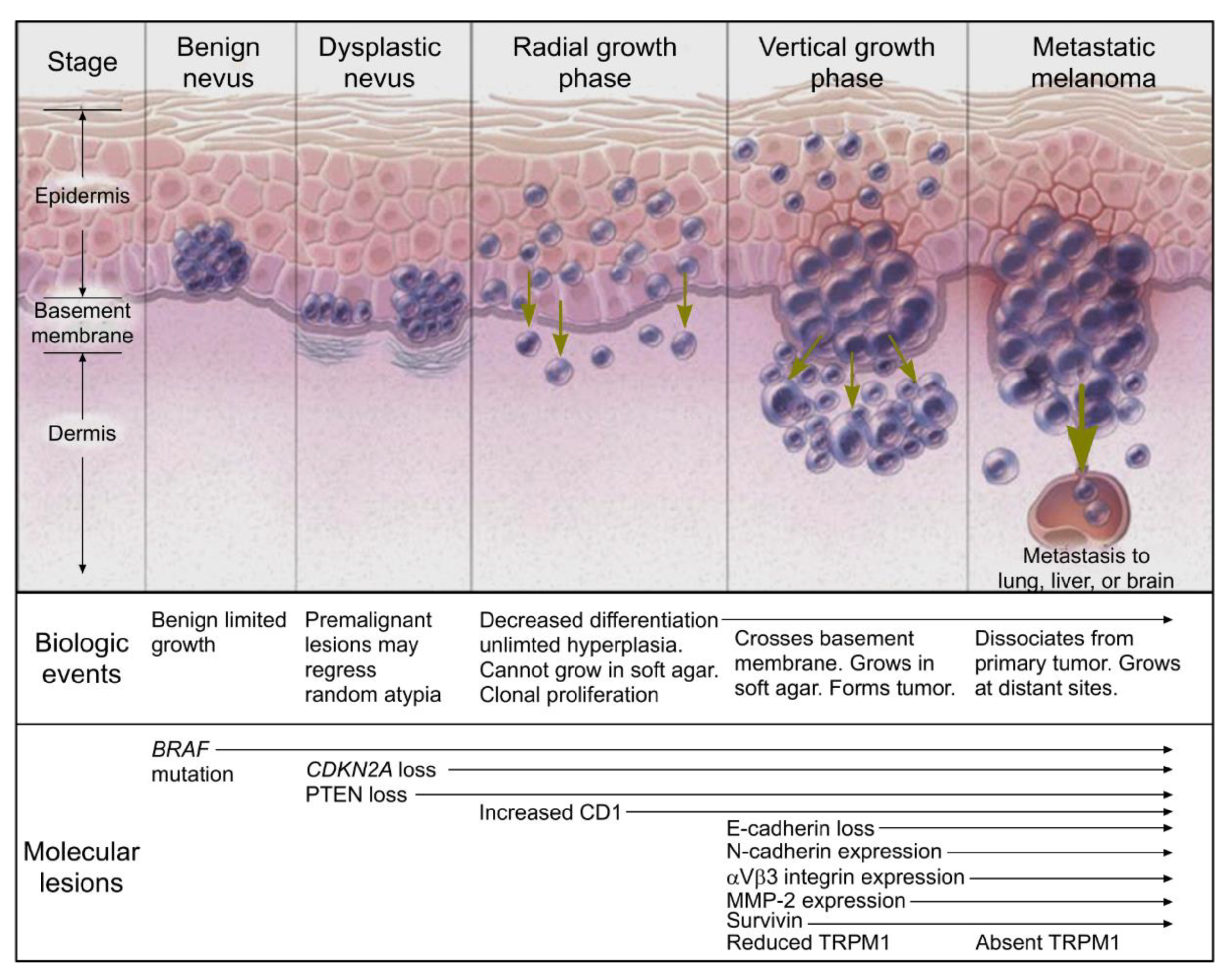
2. Melanoma Progression
3. Method
4. Melanoma Biomarkers
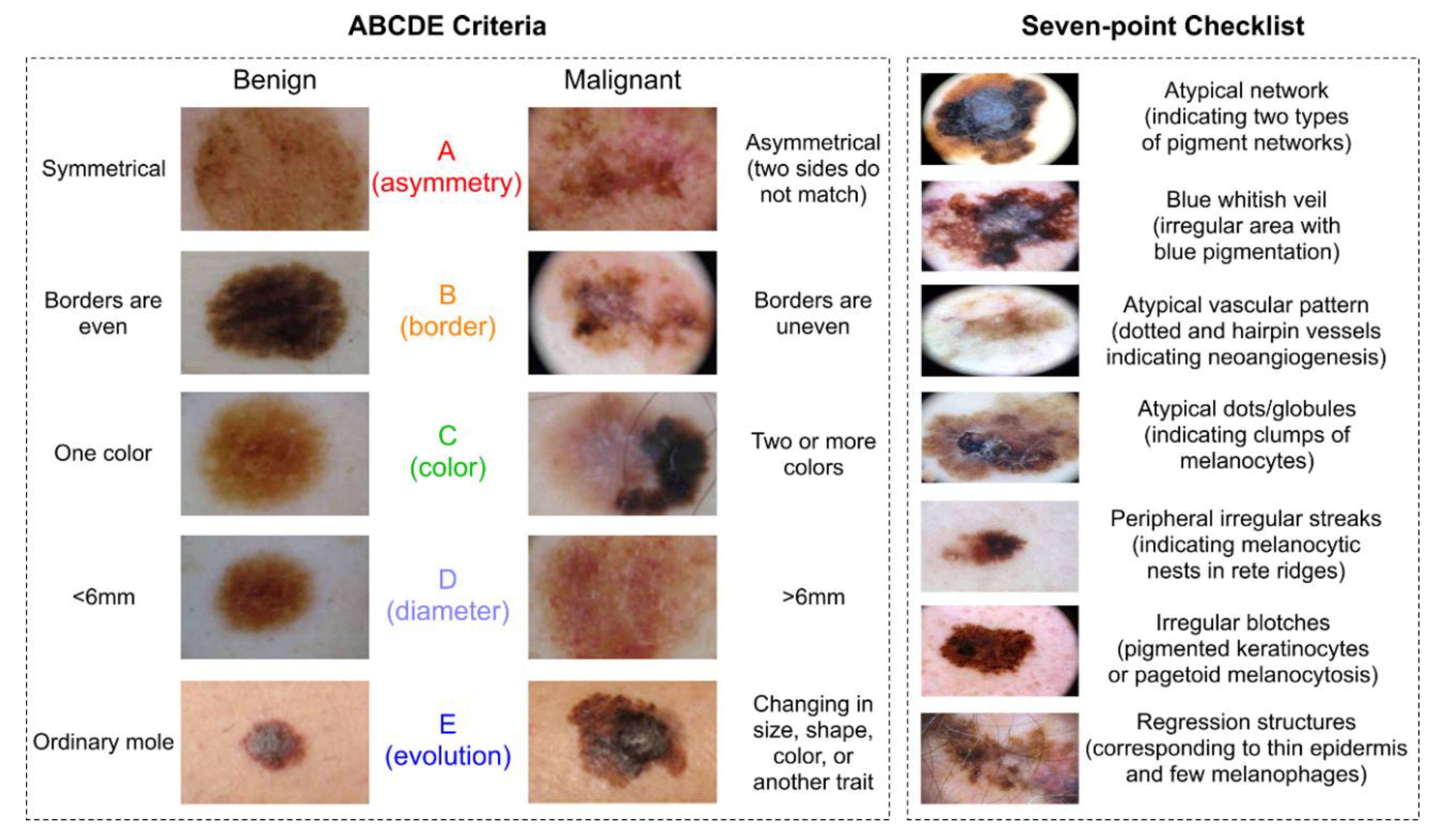
4.1. Visual
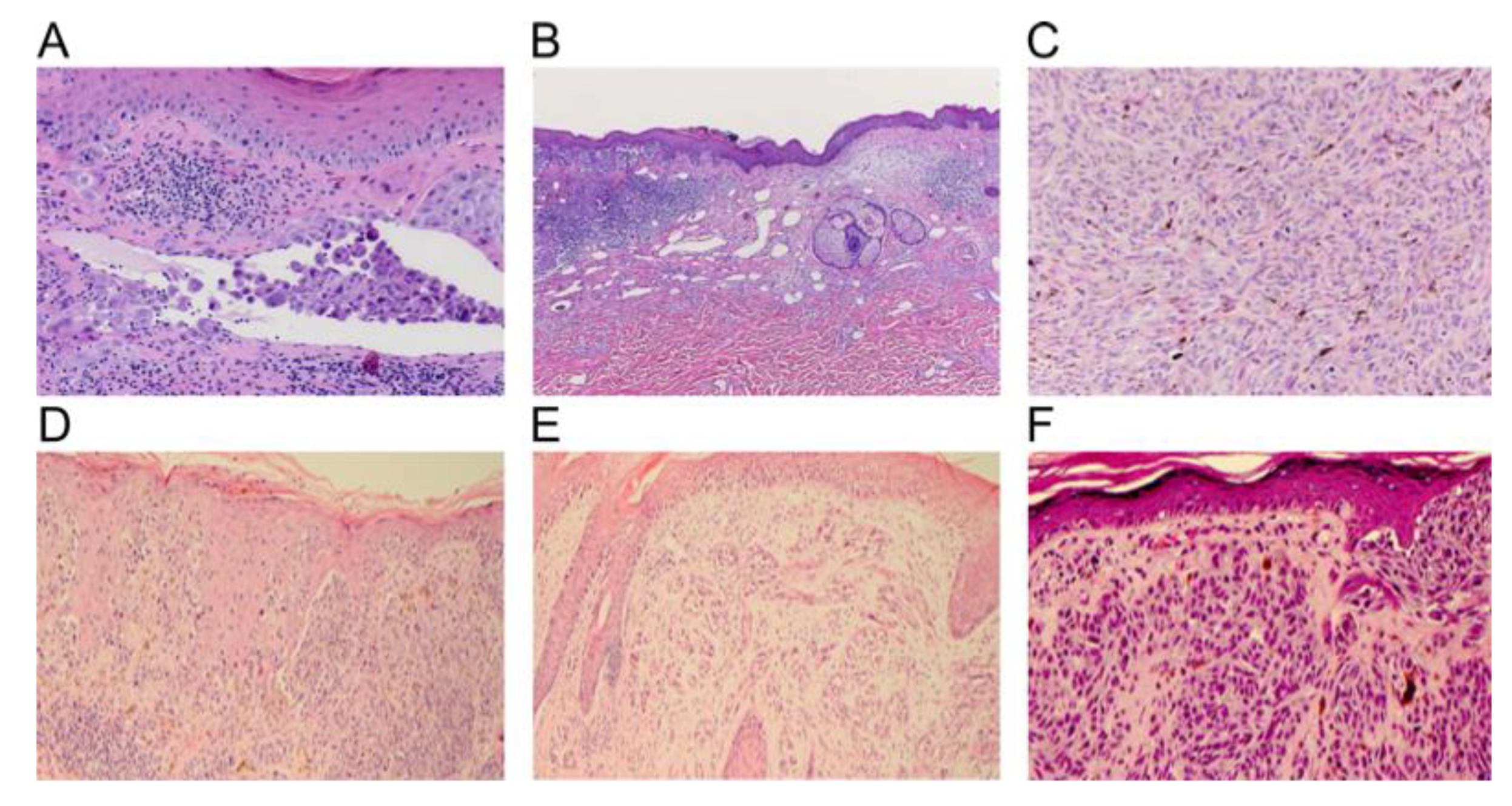
4.2. Histopathology
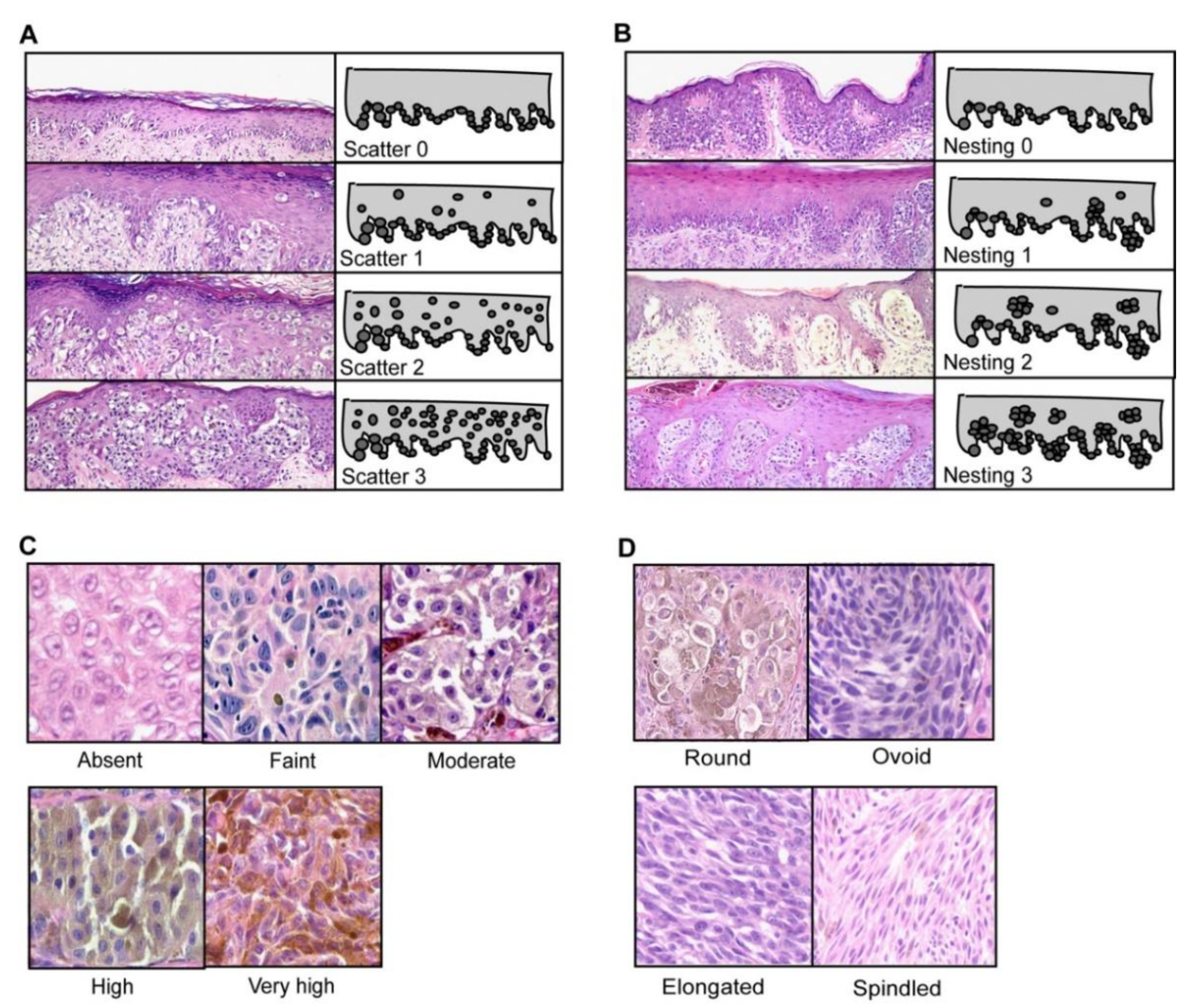
4.3. Morphology
4.4. Immunohistochemical Stains
4.5. Serologic/Molecular Diagnosis
5. Conclusions
Funding
Conflicts of Interest
References
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review. 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2018.
- American Cancer Society. Cancer Facts & Figures 2020; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Stern, R.S. Prevalence of a history of skin cancer in 2007: Results of an incidence-based model. Arch. Dermatol. 2010, 146, 279–282. [Google Scholar] [CrossRef] [PubMed]
- The American Cancer Society Medical and Editorial Content Team. Key Statistics for Melanoma Skin Cancer. Available online: https://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html (accessed on 29 April 2020).
- Shellenberger, R.; Nabhan, M.; Kakaraparthi, S. Melanoma screening: A plan for improving early detection. Ann. Med. 2016, 48, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Garibyan, L.; Fisher, D.E. How sunlight causes melanoma. Curr. Oncol. Rep. 2010, 12, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Cherobin, A.; Wainstein, A.J.A.; Colosimo, E.A.; Goulart, E.M.A.; Bittencourt, F.V. Prognostic factors for metastasis in cutaneous melanoma. An. Bras. Dermatol. 2018, 93, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Santos, I.P.; van Doorn, R.; Caspers, P.J.; Bakker Schut, T.C.; Barroso, E.M.; Nijsten, T.E.C.; Noordhoek Hegt, V.; Koljenović, S.; Puppels, G.J. Improving clinical diagnosis of early-stage cutaneous melanoma based on Raman spectroscopy. Br. J. Cancer 2018, 119, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Trinh, V.A. Current management of metastatic melanoma. Am. J. Health Syst. Pharm 2008, 65, S3–S8. [Google Scholar] [CrossRef]
- Bolognia, J.; Schaffer, J.; Cerroni, L. Dermatology, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Thomas, L.; Tranchand, P.; Berard, F.; Secchi, T.; Colin, C.; Moulin, G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998, 197, 11–17. [Google Scholar] [CrossRef]
- Cashin-Garbutt, A. Genetic Testing in Melanoma: An Interview with Dr. Diane McDowell, US Medical Affairs Lead, Oncology GSK; McDowell, D.D., Ed.; Melanoma Research Victoria: Victoria, Australia, 2014. [Google Scholar]
- Wilson, R.L.; Yentzer, B.A.; Isom, S.P.; Feldman, S.R.; Fleischer, A.B., Jr. How good are US dermatologists at discriminating skin cancers? A number-needed-to-treat analysis. J. Dermatolog. Treat. 2012, 23, 65–69. [Google Scholar] [CrossRef]
- Matsumoto, M.; Secrest, A.; Anderson, A.; Saul, M.I.; Ho, J.; Kirkwood, J.M.; Ferris, L.K. Estimating the cost of skin cancer detection by dermatology providers in a large health care system. J. Am. Acad. Dermatol. 2018, 78, 701–709.e1. [Google Scholar] [CrossRef]
- Alva, A.K.; Udaykumar, V.R. Desmoplastic melanoma: A diagnostic dilemma. J. Clin. Diagn. Res. 2013, 7, 1172–1173. [Google Scholar] [CrossRef]
- Zaenker, P.; Calapre, L.; Clark, M.; Marsavela, G.; Aya-Bonilla, C.; Gray, E.; Ziman, M. Blood-Based Melanoma Detection. Available online: https://dermnetnz.org/topics/blood-based-melanoma-detection/ (accessed on 30 November 2020).
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Hum. Mutat. 2003, 21, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Colebatch, A.J.; Scolyer, R.A. Trajectories of premalignancy during the journey from melanocyte to melanoma. Pathology 2018, 50, 16–23. [Google Scholar] [CrossRef]
- Gumaste, P.V.; Penn, L.A.; Cymerman, R.M.; Kirchhoff, T.; Polsky, D.; McLellan, B. Skin cancer risk in BRCA1/2 mutation carriers. Br. J. Dermatol. 2015, 172, 1498–1506. [Google Scholar] [CrossRef] [Green Version]
- Psaty, E.L.; Scope, A.; Halpern, A.C.; Marghoob, A.A. Defining the patient at high risk for melanoma. Int. J. Dermatol. 2010, 49, 362–376. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Rozeman, E.A.; Dekker, T.J.A.; Haanen, J.; Blank, C.U. Advanced Melanoma: Current Treatment Options, Biomarkers, and Future Perspectives. Am. J. Clin. Dermatol. 2018, 19, 303–317. [Google Scholar] [CrossRef]
- Bastian, B.C. The molecular pathology of melanoma: An integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol. 2014, 9, 239–271. [Google Scholar] [CrossRef] [Green Version]
- Tsao, H.; Bevona, C.; Goggins, W.; Quinn, T. The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: A population-based estimate. Arch. Dermatol. 2003, 139, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Yang, Q.; Wilder, P.T.; Carrier, F.; Weber, D.J. The calcium-binding protein S100B down-regulates p53 and apoptosis in malignant melanoma. J. Biol. Chem. 2010, 285, 27487–27498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo 2014, 28, 1005–1011. [Google Scholar] [CrossRef]
- Guterres, A.; Herlyn, M.; Villanueva, J. Melanoma. In eLs; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2018; pp. 1–10. [Google Scholar] [CrossRef]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef]
- Abbasi, N.R.; Shaw, H.M.; Rigel, D.S.; Friedman, R.J.; McCarthy, W.H.; Osman, I.; Kopf, A.W.; Polsky, D. Early diagnosis of cutaneous melanoma: Revisiting the ABCD criteria. JAMA 2004, 292, 2771–2776. [Google Scholar] [CrossRef]
- Tsao, H.; Olazagasti, J.M.; Cordoro, K.M.; Brewer, J.D.; Taylor, S.C.; Bordeaux, J.S.; Chren, M.M.; Sober, A.J.; Tegeler, C.; Bhushan, R.; et al. Early detection of melanoma: Reviewing the ABCDEs. J. Am. Acad. Dermatol. 2015, 72, 717–723. [Google Scholar] [CrossRef]
- Russo, T.; Piccolo, V.; Ferrara, G.; Agozzino, M.; Alfano, R.; Longo, C.; Argenziano, G. Dermoscopy pathology correlation in melanoma. J. Dermatol. 2017, 44, 507–514. [Google Scholar] [CrossRef]
- Jin, L.; Arai, E.; Anzai, S.; Kimura, T.; Tsuchida, T.; Nagata, K.; Shimizu, M. Reassessment of histopathology and dermoscopy findings in 145 Japanese cases of melanocytic nevus of the sole: Toward a pathological diagnosis of early-stage malignant melanoma in situ. Pathol. Int. 2010, 60, 65–70. [Google Scholar] [CrossRef]
- ABCDE Melanoma. Available online: https://www.hcmarbella.com/wp-content/uploads/2015/07/abcde_melanoma_eng.jpg (accessed on 2 May 2020).
- Mun, J.H.; Ohn, J.; Kim, W.I.; Park, S.M.; Kim, M.B. Dermoscopy of Melanomas on the Trunk and Extremities in Asians. PLoS ONE 2016, 11, e0158374. [Google Scholar] [CrossRef] [Green Version]
- Deinlein, T.; Arzberger, E.; Zalaudek, I.; Massone, C.; Garcias-Ladaria, J.; Oliveira, A.; Schulter, G.; Hofmann-Wellenhof, R. Dermoscopic characteristics of melanoma according to the criteria “ulceration” and “mitotic rate” of the AJCC 2009 staging system for melanoma. PLoS ONE 2017, 12, e0174871. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, M.; Pardhe, N. Dermoscopy and pigmented lesions of oral cavity. Ank. Med. J. 2017, 17, 189–191. [Google Scholar] [CrossRef]
- Iznardo, H.; Garcia-Melendo, C.; Yélamos, O. Lentigo Maligna: Clinical Presentation and Appropriate Management. Clin. Cosmet. Investig. Dermatol. 2020, 13, 837–855. [Google Scholar] [CrossRef] [PubMed]
- Ralph, B.; Katrin, K. Histopathoilogical Correlation (Full Text). Available online: https://dermoscopedia.org/w/index.php?title=Histopathoilogical_correlation_(full_text)&oldid=14521 (accessed on 4 April 2020).
- Smith, L.; Macneil, S. State of the art in non-invasive imaging of cutaneous melanoma. Skin Res. Technol. 2011, 17, 257–269. [Google Scholar] [CrossRef]
- Filosa, A.; Filosa, G. Melanoma Diagnosis: The Importance of Histopathological Report. Dermatopathology 2018, 5, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Piris, A.; Mihm, M.C., Jr. Progress in melanoma histopathology and diagnosis. Hematol. Oncol. Clin. N. Am. 2009, 23, 467–480. [Google Scholar] [CrossRef]
- Duncan, L.M. The Classification of Cutaneous Melanoma. Hematol. Oncol. Clin. N. Am. 2009, 23, 501–513. [Google Scholar] [CrossRef]
- Viros, A.; Fridlyand, J.; Bauer, J.; Lasithiotakis, K.; Garbe, C.; Pinkel, D.; Bastian, B.C. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med. 2008, 5, e120. [Google Scholar] [CrossRef] [Green Version]
- Elmore, J.G.; Barnhill, R.L.; Elder, D.E.; Longton, G.M.; Pepe, M.S.; Reisch, L.M.; Carney, P.A.; Titus, L.J.; Nelson, H.D.; Onega, T.; et al. Pathologists’ diagnosis of invasive melanoma and melanocytic proliferations: Observer accuracy and reproducibility study. BMJ 2017, 357, j2813. [Google Scholar] [CrossRef] [Green Version]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Bronstein, Y.; Ross, M.I.; Askew, R.L.; Lee, J.E.; Gershenwald, J.E.; Royal, R.; Cormier, J.N. Contemporary diagnostic imaging modalities for the staging and surveillance of melanoma patients: A meta-analysis. J. Natl. Cancer Inst. 2011, 103, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ak, I.; Stokkel, M.; Bergman, W.; Pauwels, E. Cutaneous malignant melanoma: Clinical aspects, imaging modalities and treatment. Eur. J. Nucl. Med. 2000, 27, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Wassef, C.; Rao, B.K. Uses of non-invasive imaging in the diagnosis of skin cancer: An overview of the currently available modalities. Int. J. Dermatol. 2013, 52, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Burke-Smith, A.; Collier, J.; Jones, I. A comparison of non-invasive imaging modalities: Infrared thermography, spectrophotometric intracutaneous analysis and laser Doppler imaging for the assessment of adult burns. Burns 2015, 41, 1695–1707. [Google Scholar] [CrossRef]
- Nasiriavanaki, M.; Xia, J.; Wan, H.; Bauer, A.Q.; Culver, J.P.; Wang, L.V. High-resolution photoacoustic tomography of resting-state functional connectivity in the mouse brain. Proc. Natl. Acad. Sci. USA 2014, 111, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Xia, J.; Maslov, K.I.; Nasiriavanaki, M.; Tsytsarev, V.; Demchenko, A.V.; Wang, L.V. Noninvasive photoacoustic computed tomography of mouse brain metabolism in vivo. Neuroimage 2013, 64, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Fatima, A.; Kratkiewicz, K.; Manwar, R.; Zafar, M.; Zhang, R.; Huang, B.; Dadashzadeh, N.; Xia, J.; Avanaki, K.M. Review of cost reduction methods in photoacoustic computed tomography. Photoacoustics 2019, 15, 100137. [Google Scholar] [CrossRef]
- Kratkiewicz, K.; Manwara, R.; Zhou, Y.; Mozaffarzadeh, M.; Avanaki, K. Technical considerations when using verasonics research ultrasound platform for developing a photoacoustic imaging system. arXiv 2020, arXiv:2008.06086. [Google Scholar]
- Manwar, R.; Hosseinzadeh, M.; Hariri, A.; Kratkiewicz, K.; Noei, S.; MR, N.A. Photoacoustic Signal Enhancement: Towards Utilization of Low Energy Laser Diodes in Real-Time Photoacoustic Imaging. Sensors 2018, 18, 3498. [Google Scholar] [CrossRef] [Green Version]
- Manwar, R.; Li, X.; Mahmoodkalayeh, S.; Asano, E.; Zhu, D.; Avanaki, K. Deep learning protocol for improved photoacoustic brain imaging. J. Biophotonics 2020, 13, e202000212. [Google Scholar] [CrossRef]
- Kratkiewicz, K.; Manwar, R.; Rajabi-Estarabadi, A.; Fakhoury, J.; Meiliute, J.; Daveluy, S.; Mehregan, D.; Avanaki, K.M. Photoacoustic/ultrasound/optical coherence tomography evaluation of melanoma lesion and healthy skin in a Swine model. Sensors 2019, 19, 2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanaki, M.R.; Hojjat, A.; Podoleanu, A.G. Investigation of computer-based skin cancer detection using optical coherence tomography. J. Mod. Opt. 2009, 56, 1536–1544. [Google Scholar] [CrossRef]
- Nasiri-Avanaki, M.R.; Sira, M.; Aber, A.; Hojjatoleslami, S.A.; Schofield, J.B.; Jones, C.; Podoleanu, A.G. Improved imaging of basal cell carcinoma using dynamic focus optical coherence tomography. J. Investig. Dermatol. 2011, 131, S38. [Google Scholar]
- Nasiri-Avanaki, M.; Aber, A.; Hojjatoleslami, S.; Sira, M.; Schofield, J.B.; Jones, C.; Podoleanu, A.G. Dynamic focus optical coherence tomography: Feasibility for improved basal cell carcinoma investigation. In Proceedings of the Imaging, Manipulation, and Analysis of Biomolecules, Cells, and Tissues X, San Francisco, CA, USA, 9 February 2012; p. 82252J. [Google Scholar]
- Hojjatoleslami, A.; Avanaki, M.R. OCT skin image enhancement through attenuation compensation. Appl. Opt. 2012, 51, 4927–4935. [Google Scholar] [CrossRef]
- Avanaki, M.R.; Hojjatoleslami, A. Skin layer detection of optical coherence tomography images. Optik 2013, 124, 5665–5668. [Google Scholar] [CrossRef]
- Abad, A.T.K.; Adabi, S.; Soltanizadeh, H.; Daveluy, S.; Clayton, A.; Avanaki, M.R. A novel dermo-epidermal localization algorithm for swept source OCT images of human skin. In Proceedings of the Optical Coherence Tomography and Coherence Domain Optical Methods in Biomedicine XXI, San Francisco, CA, USA, 17 February 2017; p. 100533C. [Google Scholar]
- Adabi, S.; Conforto, S.; Hosseinzadeh, M.; Noe, S.; Daveluy, S.; Mehregan, D.; Nasiriavanaki, M. Textural analysis of optical coherence tomography skin images: Quantitative differentiation between healthy and cancerous tissues. In Proceedings of the Optical Coherence Tomography and Coherence Domain Optical Methods in Biomedicine XXI, San Francisco, CA, USA, 17 February 2017; p. 100533F. [Google Scholar]
- Taghavikhalilbad, A.; Adabi, S.; Clayton, A.; Soltanizadeh, H.; Mehregan, D.; Avanaki, M.R. Semi-automated localization of dermal epidermal junction in optical coherence tomography images of skin. Appl. Opt. 2017, 56, 3116–3121. [Google Scholar] [CrossRef]
- Avanaki, M.R.; Podoleanu, A. En-face time-domain optical coherence tomography with dynamic focus for high-resolution imaging. J. Biomed. Opt. 2017, 22, 056009. [Google Scholar] [CrossRef] [Green Version]
- Adabi, S.; Turani, Z.; Fatemizadeh, E.; Clayton, A.; Nasiriavanaki, M. Optical coherence tomography technology and quality improvement methods for optical coherence tomography images of skin: A short review. Biomed. Eng. Comput. Biol. 2017, 8, 1179597217713475. [Google Scholar] [CrossRef]
- Adabi, S.; Hosseinzadeh, M.; Noei, S.; Conforto, S.; Daveluy, S.; Clayton, A.; Mehregan, D.; Nasiriavanaki, M. Universal in vivo textural model for human skin based on optical coherence tomograms. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tes, D.; Aber, A.; Zafar, M.; Horton, L.; Fotouhi, A.; Xu, Q.; Moiin, A.; Thompson, A.D.; Moraes Pinto Blumetti, T.C.; Daveluy, S. Granular cell tumor imaging using optical coherence tomography. Biomed. Eng. Comput. Biol. 2018, 9, 1179597218790250. [Google Scholar] [CrossRef] [Green Version]
- Adabi, S.; Fotouhi, A.; Xu, Q.; Daveluy, S.; Mehregan, D.; Podoleanu, A.; Nasiriavanaki, M. An overview of methods to mitigate artifacts in optical coherence tomography imaging of the skin. Skin Res. Technol. 2018, 24, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Adabi, S.; Clayton, A.; Daveluy, S.; Mehregan, D.; Nasiriavanaki, M. Swept-Source Optical Coherence Tomography–Supervised Biopsy. Dermatol. Surg. 2018, 44, 768–775. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, S.; Fotouhi, A.; Turk, D.; Sriranga, P.; Rajabi-Estarabadi, A.; Nouri, K.; Daveluy, S.; Mehregan, D.; Nasiriavanaki, M. OCT image atlas of healthy skin on sun-exposed areas. Skin Res. Technol. 2018, 24, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.; Horton, L.; Poozesh, P.; Baqersad, J.; Nasiriavanaki, M. Vibration analysis of healthy skin: Toward a noninvasive skin diagnosis methodology. J. Biomed. Opt. 2019, 24, 015001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turani, Z.; Fatemizadeh, E.; Blumetti, T.; Daveluy, S.; Moraes, A.F.; Chen, W.; Mehregan, D.; Andersen, P.E.; Nasiriavanaki, M. Optical radiomic signatures derived from optical coherence tomography images improve identification of melanoma. Cancer Res. 2019, 79, 2021–2030. [Google Scholar] [CrossRef] [Green Version]
- Turani, Z.; Fatemizadeh, E.; Blumetti, T.; Daveluy, S.; Moraes, A.F.; Chen, W.; Mehregan, D.; Andersen, P.E.; Nasiriavanaki, M. Optical radiomic signatures derived from OCT images to improve identification of melanoma. In Proceedings of the European Conference on Biomedical Optics, Munich, Germany, 23–25 June 2019; p. 11078_23. [Google Scholar]
- Jalilian, E.; Xu, Q.; Horton, L.; Fotouhi, A.; Reddy, S.; Manwar, R.; Daveluy, S.; Mehregan, D.; Gelovani, J.; Avanaki, K. Contrast-enhanced optical coherence tomography for melanoma detection: An in vitro study. J. Biophotonics 2020, 13, e201960097. [Google Scholar] [CrossRef]
- Eybposh, M.H.; Turani, Z.; Mehregan, D.; Nasiriavanaki, M. Cluster-based filtering framework for speckle reduction in OCT images. Biomed. Opt. Express 2018, 9, 6359–6373. [Google Scholar] [CrossRef]
- Avanaki, M.R.; Podoleanu, A.G.; Schofield, J.B.; Jones, C.; Sira, M.; Liu, Y.; Hojjat, A. Quantitative evaluation of scattering in optical coherence tomography skin images using the extended Huygens-Fresnel theorem. Appl. Opt. 2013, 52, 1574–1580. [Google Scholar] [CrossRef]
- Avanaki, M.; Laissue, P.; Hojjat, S. De-Noising Speckled Optical Coherence Tomography Images Using an Algorithm Based on Artificial Neural Network. J. Neurosci. Neuroeng. 2013, 2, 347–352. [Google Scholar] [CrossRef]
- Almasganj, M.; Adabi, S.; Fatemizadeh, E.; Xu, Q.; Sadeghi, H.; Daveluy, S.; Nasiriavanaki, M. A Spatially-Variant Deconvolution Method Based on Total Variation for Optical Coherence Tomography Images; SPIE: Bellingham, WA, USA, 2017; Volume 10137. [Google Scholar]
- Rajabi-Estarabadi, A.; Bittar, J.M.; Zheng, C.; Nascimento, V.; Camacho, I.; Feun, L.G.; Nasiriavanaki, M.; Kunz, M.; Nouri, K. Optical coherence tomography imaging of melanoma skin cancer. Lasers Med. Sci. 2019, 34, 411–420. [Google Scholar] [CrossRef]
- Borsari, S.; Pampena, R.; Lallas, A.; Kyrgidis, A.; Moscarella, E.; Benati, E.; Raucci, M.; Pellacani, G.; Zalaudek, I.; Argenziano, G.; et al. Clinical Indications for Use of Reflectance Confocal Microscopy for Skin Cancer Diagnosis. JAMA Dermatol. 2016, 152, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Waddell, A.; Star, P.; Guitera, P. Advances in the use of reflectance confocal microscopy in melanoma. Melanoma Manag. 2018, 5, Mmt04. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinnes, J.; Bamber, J.; Chuchu, N.; Bayliss, S.E.; Takwoingi, Y.; Davenport, C.; Godfrey, K.; O’Sullivan, C.; Matin, R.N.; Deeks, J.J.; et al. High-frequency ultrasound for diagnosing skin cancer in adults. Cochrane Database Syst. Rev. 2018, 12, Cd013188. [Google Scholar] [CrossRef]
- Botar Jid, C.; Bolboacă, S.D.; Cosgarea, R.; Şenilă, S.; Rogojan, L.; Lenghel, M.; Vasilescu, D.; Dudea, S.M. Doppler ultrasound and strain elastography in the assessment of cutaneous melanoma: Preliminary results. Med. Ultrason. 2015, 17, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Giovagnorio, F.; Andreoli, C.; De Cicco, M.L. Color Doppler sonography of focal lesions of the skin and subcutaneous tissue. J. Ultrasound Med. 1999, 18, 89–93. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Dubois, A.; Levecq, O.; Azimani, H.; Siret, D.; Barut, A.; Suppa, M.; Del Marmol, V.; Malvehy, J.; Cinotti, E.; Rubegni, P.; et al. Line-field confocal optical coherence tomography for high-resolution noninvasive imaging of skin tumors. J. Biomed. Opt. 2018, 23, 106007. [Google Scholar] [CrossRef] [Green Version]
- Wortsman, X. Sonography of the primary cutaneous melanoma: A review. Radiol. Res. Pract. 2012, 2012, 814396. [Google Scholar] [CrossRef] [Green Version]
- Urvanegia, A.C.; Tavoloni Braga, J.C.; Shitara, D.; Fregnani, J.H.; Neves, J.I.; Pinto, C.A.; Marghoob, A.A.; Duprat, J.P.; Rezze, G.G. Reflectance confocal microscopy features of BRAF V600E mutated thin melanomas detected by immunohistochemistry. PLoS ONE 2017, 12, e0179745. [Google Scholar] [CrossRef] [Green Version]
- Eisenstein, A.; Gonzalez, E.C.; Raghunathan, R.; Xu, X.; Wu, M.; McLean, E.O.; McGee, J.; Ryu, B.; Alani, R.M. Emerging Biomarkers in Cutaneous Melanoma. Mol. Diagn. Ther. 2018, 22, 203–218. [Google Scholar] [CrossRef]
- Abbas, O.; Miller, D.D.; Bhawan, J. Cutaneous malignant melanoma: Update on diagnostic and prognostic biomarkers. Am. J. Dermatopathol. 2014, 36, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W.; Fritz, G.; Schäfer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci. 2002, 7, d1356–d1368. [Google Scholar] [PubMed]
- Ordóñez, N.G. Value of melanocytic-associated immunohistochemical markers in the diagnosis of malignant melanoma: A review and update. Hum. Pathol. 2014, 45, 191–205. [Google Scholar] [CrossRef]
- Weinstein, D.; Leininger, J.; Hamby, C.; Safai, B. Diagnostic and prognostic biomarkers in melanoma. J. Clin. Aesthet. Dermatol. 2014, 7, 13–24. [Google Scholar] [PubMed]
- Nonaka, D.; Chiriboga, L.; Rubin, B.P. Differential expression of S100 protein subtypes in malignant melanoma, and benign and malignant peripheral nerve sheath tumors. J. Cutan. Pathol. 2008, 35, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Ribé, A.; McNutt, N.S. S100A6 protein expression is different in Spitz nevi and melanomas. Mod. Pathol. 2003, 16, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Theos, A.C.; Truschel, S.T.; Raposo, G.; Marks, M.S. The Silver locus product Pmel17/gp100/Silv/ME20: Controversial in name and in function. Pigment. Cell Res. 2005, 18, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Prieto, V.G.; Shea, C.R. Use of immunohistochemistry in melanocytic lesions. J. Cutan. Pathol. 2008, 35 (Suppl. 2), 1–10. [Google Scholar] [CrossRef]
- Prieto, V.G.; Shea, C.R. Immunohistochemistry of melanocytic proliferations. Arch. Pathol. Lab. Med. 2011, 135, 853–859. [Google Scholar] [CrossRef]
- Li, L.X.; Crotty, K.A.; McCarthy, S.W.; Palmer, A.A.; Kril, J.J. A zonal comparison of MIB1-Ki67 immunoreactivity in benign and malignant melanocytic lesions. Am. J. Dermatopathol. 2000, 22, 489–495. [Google Scholar] [CrossRef]
- Chorny, J.A.; Barr, R.J.; Kyshtoobayeva, A.; Jakowatz, J.; Reed, R.J. Ki-67 and p53 expression in minimal deviation melanomas as compared with other nevomelanocytic lesions. Mod. Pathol. 2003, 16, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, S.; Argraves, M.; Kristjansson, A.; Ferenczi, K.; Dadras, S.S. A quantitative comparison between SOX10 and MART-1 immunostaining to detect melanocytic hyperplasia in chronically sun-damaged skin. J. Cutan. Pathol. 2018, 45, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, M.; Lamant, L.; Tournier, E.; Valentin, T.; Rochaix, P.; Terrier, P.; Ranchere-Vince, D.; Coindre, J.M.; Filleron, T.; Le Guellec, S. Evaluation of eight melanocytic and neural crest-associated markers in a well-characterised series of 124 malignant peripheral nerve sheath tumours (MPNST): Useful to distinguish MPNST from melanoma? Histopathology 2018, 73, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Rochaix, P.; Lacroix-Triki, M.; Lamant, L.; Pichereaux, C.; Valmary, S.; Puente, E.; Al Saati, T.; Monsarrat, B.; Susini, C.; Buscail, L.; et al. PNL2, a new monoclonal antibody directed against a fixative-resistant melanocyte antigen. Mod. Pathol. 2003, 16, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubovits, J.; Buzney, E.; Yu, L.; Duncan, L.M. HMB-45, S-100, NK1/C3, and MART-1 in metastatic melanoma. Hum. Pathol. 2004, 35, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.; Ferrone, S.; Wang, X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv. Cancer Res. 2010, 109, 73–121. [Google Scholar] [CrossRef]
- Jungbluth, A.A.; Iversen, K.; Coplan, K.; Kolb, D.; Stockert, E.; Chen, Y.T.; Old, L.J.; Busam, K. T311--an anti-tyrosinase monoclonal antibody for the detection of melanocytic lesions in paraffin embedded tissues. Pathol. Res. Pract. 2000, 196, 235–242. [Google Scholar] [CrossRef]
- Busam, K.J.; Kucukgöl, D.; Sato, E.; Frosina, D.; Teruya-Feldstein, J.; Jungbluth, A.A. Immunohistochemical analysis of novel monoclonal antibody PNL2 and comparison with other melanocyte differentiation markers. Am. J. Surg. Pathol. 2005, 29, 400–406. [Google Scholar] [CrossRef]
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411. [Google Scholar] [CrossRef]
- Busam, K.J.; Iversen, K.; Coplan, K.C.; Jungbluth, A.A. Analysis of microphthalmia transcription factor expression in normal tissues and tumors, and comparison of its expression with S-100 protein, gp100, and tyrosinase in desmoplastic malignant melanoma. Am. J. Surg. Pathol. 2001, 25, 197–204. [Google Scholar] [CrossRef]
- Granter, S.R.; Weilbaecher, K.N.; Quigley, C.; Fletcher, C.D.; Fisher, D.E. Microphthalmia transcription factor: Not a sensitive or specific marker for the diagnosis of desmoplastic melanoma and spindle cell (non-desmoplastic) melanoma. Am. J. Dermatopathol. 2001, 23, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Potterf, S.B.; Mollaaghababa, R.; Hou, L.; Southard-Smith, E.M.; Hornyak, T.J.; Arnheiter, H.; Pavan, W.J. Analysis of SOX10 function in neural crest-derived melanocyte development: SOX10-dependent transcriptional control of dopachrome tautomerase. Dev. Biol. 2001, 237, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Kelsh, R.N. Sorting out Sox10 functions in neural crest development. Bioessays 2006, 28, 788–798. [Google Scholar] [CrossRef]
- Blochin, E.; Nonaka, D. Diagnostic value of Sox10 immunohistochemical staining for the detection of metastatic melanoma in sentinel lymph nodes. Histopathology 2009, 55, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Jennings, C.; Kim, J. Identification of nodal metastases in melanoma using sox-10. Am. J. Dermatopathol. 2011, 33, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Onfray, F.; López, M.; Lundqvist, A.; Aguirre, A.; Escobar, A.; Serrano, A.; Korenblit, C.; Petersson, M.; Chhajlani, V.; Larsson, O.; et al. Tissue distribution and differential expression of melanocortin 1 receptor, a malignant melanoma marker. Br. J. Cancer 2002, 87, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Borrón, J.C.; Sánchez-Laorden, B.L.; Jiménez-Cervantes, C. Melanocortin-1 receptor structure and functional regulation. Pigment. Cell Res. 2005, 18, 393–410. [Google Scholar] [CrossRef] [PubMed]
- López, M.N.; Pereda, C.; Ramírez, M.; Mendoza-Naranjo, A.; Serrano, A.; Ferreira, A.; Poblete, R.; Kalergis, A.M.; Kiessling, R.; Salazar-Onfray, F. Melanocortin 1 receptor is expressed by uveal malignant melanoma and can be considered a new target for diagnosis and immunotherapy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1219–1227. [Google Scholar] [CrossRef]
- Goodison, S.; Urquidi, V. The cancer testis antigen PRAME as a biomarker for solid tumor cancer management. Biomark. Med. 2012, 6, 629–632. [Google Scholar] [CrossRef]
- Lezcano, C.; Jungbluth, A.A.; Nehal, K.S.; Hollmann, T.J.; Busam, K.J. PRAME Expression in Melanocytic Tumors. Am. J. Surg. Pathol. 2018, 42, 1456–1465. [Google Scholar] [CrossRef]
- Lezcano, C.; Jungbluth, A.A.; Busam, K.J. Comparison of Immunohistochemistry for PRAME with Cytogenetic Test Results in the Evaluation of Challenging Melanocytic Tumors. Am. J. Surg. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Casper, D.J.; Ross, K.I.; Messina, J.L.; Sondak, V.K.; Bodden, C.N.; McCardle, T.W.; Glass, L.F. Useof anti-phosphohistone H3 immunohistochemistry to determine mitotic rate in thin melanoma. Am. J. Dermatopathol. 2010, 32, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Ferringer, T. Update on immunohistochemistry in melanocytic lesions. Dermatol. Clin. 2012, 30, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Ohsie, S.J.; Sarantopoulos, G.P.; Cochran, A.J.; Binder, S.W. Immunohistochemical characteristics of melanoma. J. Cutan. Pathol. 2008, 35, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Rolih, V.; Barutello, G.; Iussich, S.; De Maria, R.; Quaglino, E.; Buracco, P.; Cavallo, F.; Riccardo, F. CSPG4: A prototype oncoantigen for translational immunotherapy studies. J. Transl. Med. 2017, 15, 151. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Constantin, C.; Tanase, C. Immune-related biomarkers for diagnosis/prognosis and therapy monitoring of cutaneous melanoma. Expert Rev. Mol. Diagn. 2010, 10, 897–919. [Google Scholar] [CrossRef]
- Nielsen, P.S.; Riber-Hansen, R.; Schmidt, H.; Steiniche, T. Automated quantification of proliferation with automated hot-spot selection in phosphohistone H3/MART1 dual-stained stage I/II melanoma. Diagn. Pathol. 2016, 11, 35. [Google Scholar] [CrossRef] [Green Version]
- Compton, L.A.; Murphy, G.F.; Lian, C.G. Diagnostic Immunohistochemistry in Cutaneous Neoplasia: An Update. Dermatopathology 2015, 2, 15–42. [Google Scholar] [CrossRef]
- Muto, Y.; Fujimura, T.; Kambayashi, Y.; Ohuchi, K.; Amagai, R.; Hashimoto, A.; Aiba, S. Metastatic PRAME-Expressing Juvenile Spitzoid Melanoma on the Buttock. Case Rep. Oncol. 2020, 13, 1141–1144. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [Green Version]
- Deichmann, M.; Benner, A.; Bock, M.; Jäckel, A.; Uhl, K.; Waldmann, V.; Näher, H. S100-Beta, melanoma-inhibiting activity, and lactate dehydrogenase discriminate progressive from nonprogressive American Joint Committee on Cancer stage IV melanoma. J. Clin. Oncol. 1999, 17, 1891–1896. [Google Scholar] [CrossRef] [PubMed]
- Philippidou, D.; Schmitt, M.; Moser, D.; Margue, C.; Nazarov, P.V.; Muller, A.; Vallar, L.; Nashan, D.; Behrmann, I.; Kreis, S. Signatures of microRNAs and selected microRNA target genes in human melanoma. Cancer Res. 2010, 70, 4163–4173. [Google Scholar] [CrossRef] [Green Version]
- Armand-Labit, V.; Meyer, N.; Casanova, A.; Bonnabau, H.; Platzer, V.; Tournier, E.; Sansas, B.; Verdun, S.; Thouvenot, B.; Hilselberger, B.; et al. Identification of a Circulating MicroRNA Profile as a Biomarker of Metastatic Cutaneous Melanoma. Acta Derm. Venereol. 2016, 96, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagnart, O.C.; Sindic, C.J.; Laterre, C. Particle counting immunoassay of S100 protein in serum. Possible relevance in tumors and ischemic disorders of the central nervous system. Clin. Chem. 1988, 34, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.B.; Stoffel-Wagner, B.; Bierwirth, T.; Mezger, J.; Klingmüller, D. Clinical significance of serum S100 in metastatic malignant melanoma. Eur. J. Cancer 1995, 31a, 1898–1902. [Google Scholar] [CrossRef]
- Leachman, S.A.; Lucero, O.M.; Sampson, J.E.; Cassidy, P.; Bruno, W.; Queirolo, P.; Ghiorzo, P. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017, 36, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, J.; Wadt, K.A.; Hayward, N.K. Melanoma genetics. J. Med. Genet. 2016, 53, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Pavri, S.N.; Clune, J.; Ariyan, S.; Narayan, D. Malignant Melanoma: Beyond the Basics. Plast. Reconstr. Surg. 2016, 138, 330e–340e. [Google Scholar] [CrossRef]
- Azzola, M.F.; Shaw, H.M.; Thompson, J.F.; Soong, S.J.; Scolyer, R.A.; Watson, G.F.; Colman, M.H.; Zhang, Y. Tumor mitotic rate is a more powerful prognostic indicator than ulceration in patients with primary cutaneous melanoma: An analysis of 3661 patients from a single center. Cancer 2003, 97, 1488–1498. [Google Scholar] [CrossRef]





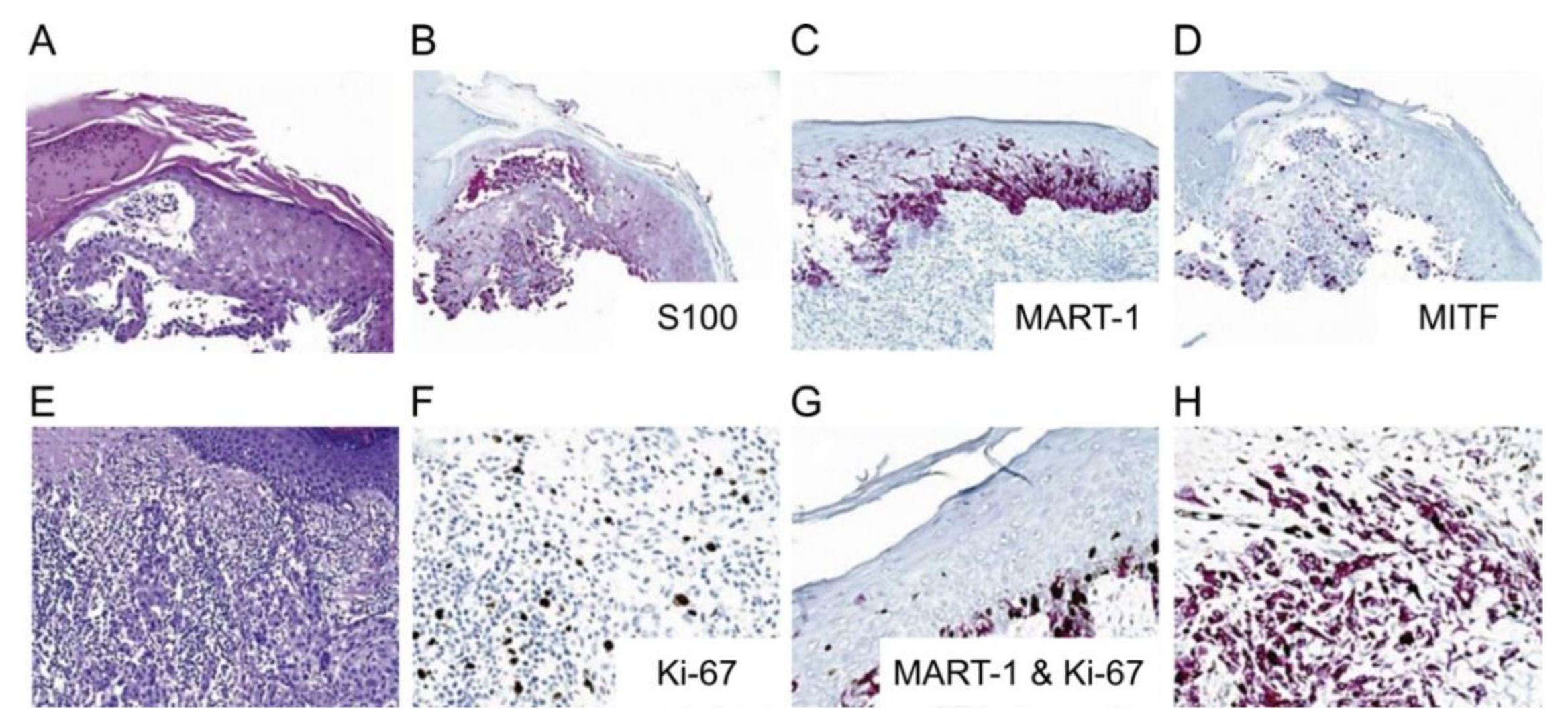
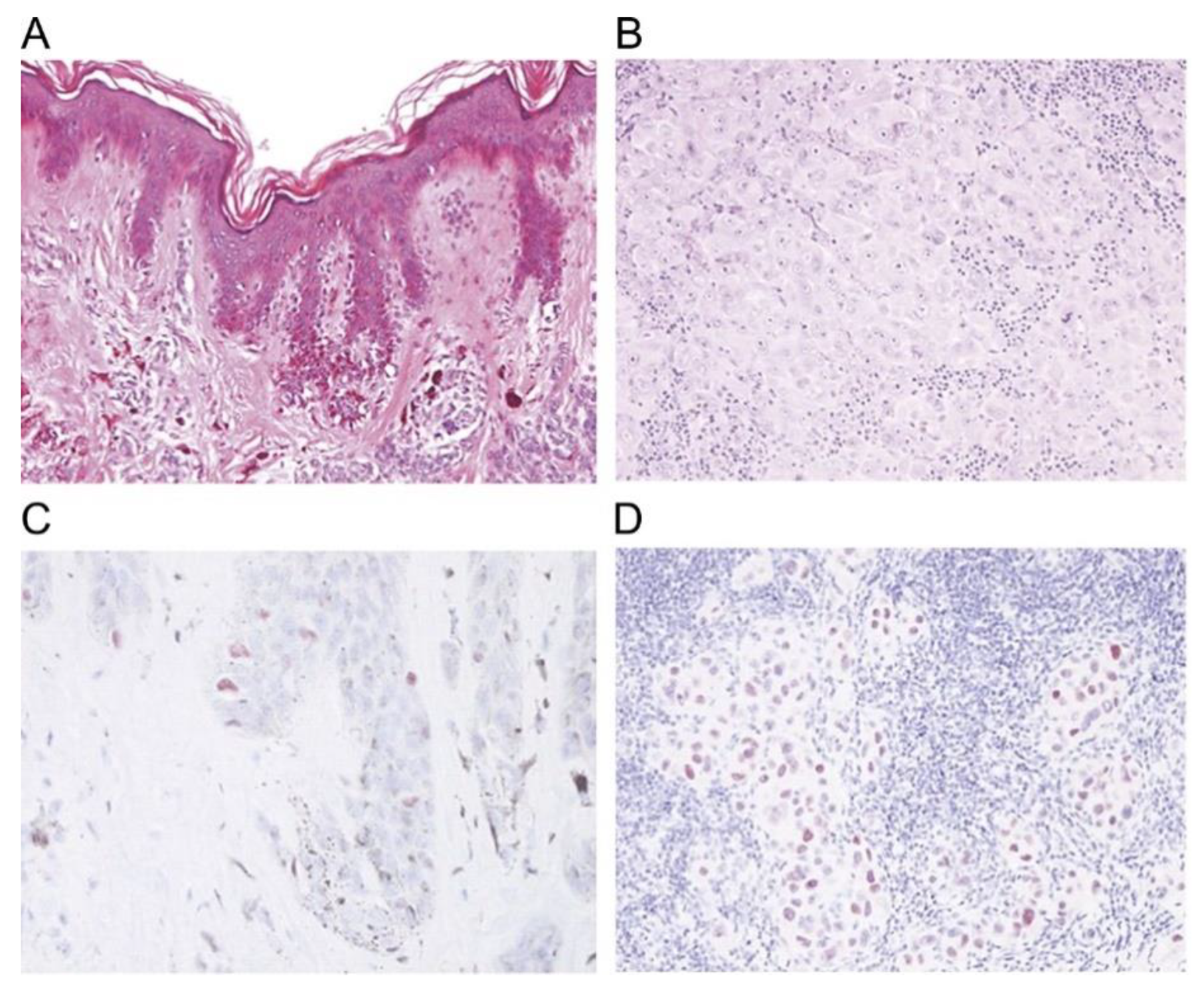


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mandatory Features [44] | Additional Features [36] |
|---|---|
| Ulceration | Melanocytes that are more often arranged as solitary predominating over melanocytes in nests |
| Mitotic rate | Irregular distribution of nests |
| Regression | Nests of melanocytes varying in size and shape |
| Lymphovascular Invasion | Slight cytological atypia, namely mild cellular pleomorphism, larger sized cells than normal cells, abundant cytoplasm, and distinct nucleoli |
| Perineural Invasion | Single cells often extending irregularly far down to the eccrine duct epithelium |
| Breslow Thickness | Pagetoid spread of melanocytes, ascent of melanocytes in the granular layer |
| Satellitosis | |
| Status of Surgical Margin |
| Immunohistochemical Biomarker | Function | Current Use | Staining Pattern |
|---|---|---|---|
| S100 Protein Family | involved in multiple cellular processes such as cellular growth, cell cycle progression, cellular motility, calcium homeostasis, transcription, and protein phosphorylation [97,98] | highly sensitive (93%–100%) and stains most melanocytic lesions, but lacks specificity [97,127] | nuclear and cytoplasmic, strong and diffuse [97,128] |
| HMB 45 | monoclonal antibody against PMEL17 (gp100), plays role in organizational structure of melanoma [97] | very specific, lower sensitivity (70%–90%) than S100 [97] | cytoplasmic, finely granular [97] |
| Ki-67 | non-histone nuclear protein, proliferation marker [95] | useful in differentiating benign nevi from melanoma [95] | nuclear [95] |
| Melan A | assists in processing of PMEL17 for the formation of stage II melanosomes [97] | sensitivity is 85%–97% for primary melanoma, sensitivity is 57%–92% for metastatic melanoma, specificity is 95%–100% [97], not expressed in dendritic cells in lymph nodes [97] | cytoplasmic [97] |
| CSPG4 | tissue development and cell motility [94,98] | more sensitive detection for metastatic melanoma than S100B, HMB 45, and MART-1 [94] | tumor cell membrane [129] |
| Tyrosinase | primary enzyme involved in melanin synthesis [97] | highly specific (97%–100%) [97,98] for primary melanoma [98], expressed in clear cell sarcomas, pigmented neurofibroma, and 20% of angiomyolipomas [97] | cytoplasmic [97] |
| PNL2 | monoclonal antibody with unknown antigen, reacts with neutrophils and melanocytes [97] | positive in 75%–100% of primary metastatic and epithelioid melanomas, can stain positive in PEComas, clear cell sarcoma, and melanocytic schwannomas [97] | cytoplasmic [97] |
| MITF | neural crest cell differentiation [97] | limited use due to lack of specificity for melanoma but may be useful in an immunohistochemical panel [97] | nuclear [97,128,130] |
| SOX10 | embryonic determination of cell fate [97] | sensitive for primary and metastatic melanomas [97], positive in clear cell sarcomas and peripheral nerve sheath tumors [97] | nuclear [97] |
| MC1R | G protein-coupled receptor family that controls pigment and plays role in skin phenotype and sensitivity [97] | sensitive for melanoma, not restricted to only melanocytes. Also present in neurons, hepatocyte, renal tubular cells, adrenal medullary cells, and myocardial cells [97,120] | cell surface and intracellular expression [120] |
| PRAME | part of the cancer testis antigen family, antigen recognition by T lymphocytes [123] | Stains positive in >75% of cells present in non-spindle cell cutaneous melanoma and is positive in 88%–94% of non-spindle cell cutaneous melanoma cases [124] | nuclear, diffuse reactivity [124] |
| pHH3 | correlates with mitosis looking for phosphorylation of histone H3 [95] | may overestimate mitoses due to melanocyte and non-melanocyte melanomas [95] | nuclear [131] |
| p16 | protein product of CDKN2A gene [95] | 50%–98% of melanomas show loss of nuclear staining [95] | nuclear, decreased in melanomas [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hessler, M.; Jalilian, E.; Xu, Q.; Reddy, S.; Horton, L.; Elkin, K.; Manwar, R.; Tsoukas, M.; Mehregan, D.; Avanaki, K. Melanoma Biomarkers and Their Potential Application for In Vivo Diagnostic Imaging Modalities. Int. J. Mol. Sci. 2020, 21, 9583. https://doi.org/10.3390/ijms21249583
Hessler M, Jalilian E, Xu Q, Reddy S, Horton L, Elkin K, Manwar R, Tsoukas M, Mehregan D, Avanaki K. Melanoma Biomarkers and Their Potential Application for In Vivo Diagnostic Imaging Modalities. International Journal of Molecular Sciences. 2020; 21(24):9583. https://doi.org/10.3390/ijms21249583
Chicago/Turabian StyleHessler, Monica, Elmira Jalilian, Qiuyun Xu, Shriya Reddy, Luke Horton, Kenneth Elkin, Rayyan Manwar, Maria Tsoukas, Darius Mehregan, and Kamran Avanaki. 2020. "Melanoma Biomarkers and Their Potential Application for In Vivo Diagnostic Imaging Modalities" International Journal of Molecular Sciences 21, no. 24: 9583. https://doi.org/10.3390/ijms21249583
APA StyleHessler, M., Jalilian, E., Xu, Q., Reddy, S., Horton, L., Elkin, K., Manwar, R., Tsoukas, M., Mehregan, D., & Avanaki, K. (2020). Melanoma Biomarkers and Their Potential Application for In Vivo Diagnostic Imaging Modalities. International Journal of Molecular Sciences, 21(24), 9583. https://doi.org/10.3390/ijms21249583